
Targeting Mitochondria and Metabolism in Acute Kidney Injury

Abstract
1. Introduction
2. Overview of Renal Energy Metabolism
3. Mitochondrial Dysfunction and Metabolic Derangements in AKI
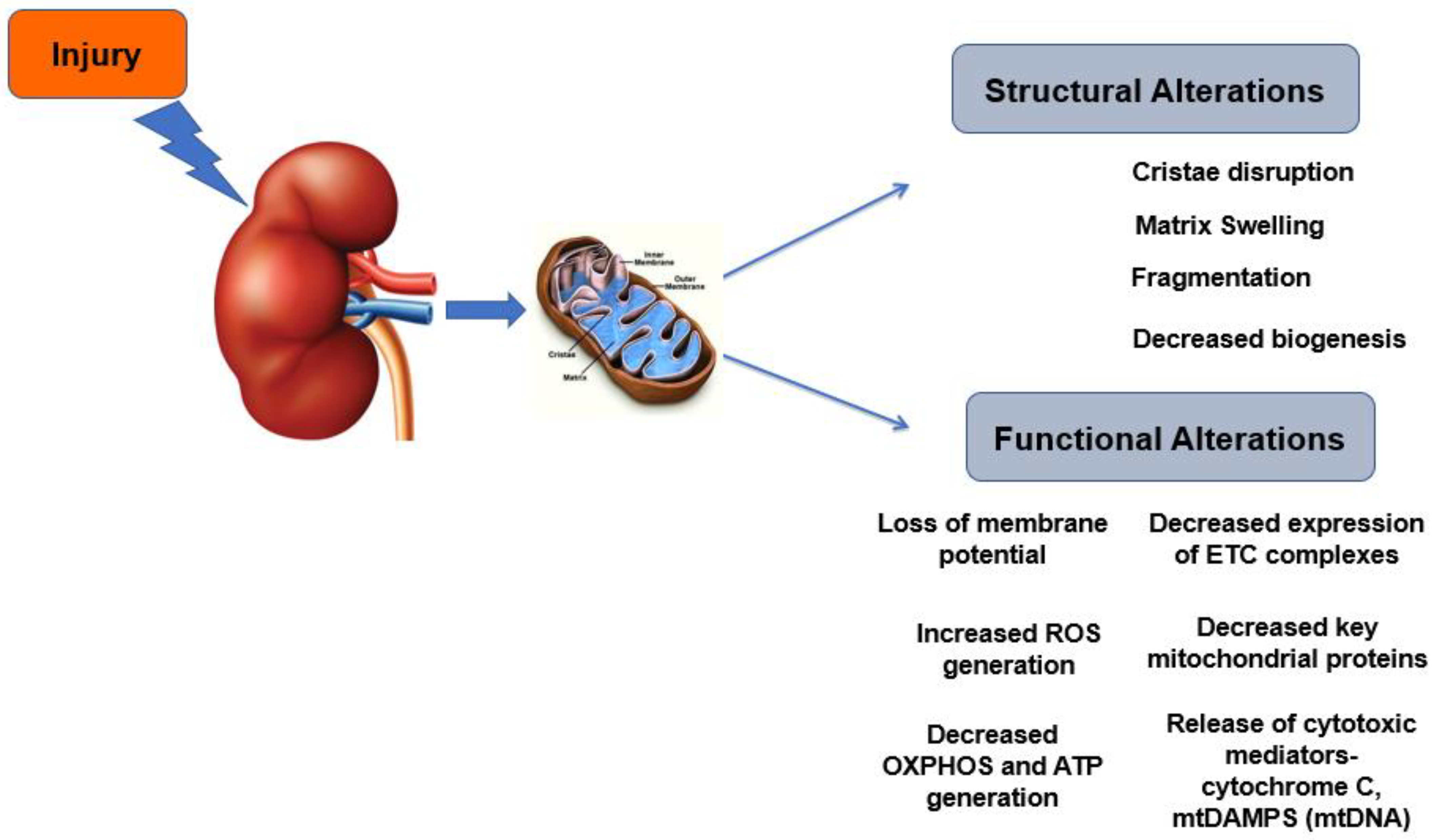
3.1. Mitochondrial Morphology and Biogenesis
3.2. Mitochondrial Functional Alterations
3.3. Metabolic Derangements
4. Therapeutic Targeting of Mitochondrial Dysfunction and Metabolism in AKI
4.1. Mitochondrial Targeted Antioxidants
4.2. Mitochondrial Biogenesis, Function and Quality Control
4.2.1. AMP-Kinase (AMPK)
4.2.2. Sirtuins
4.2.3. Szeto–Schiller Peptides
4.3. Pharmacological Strategies Targeting Metabolism
5. Summary and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Acetyl CoA | Acetyl Coenzyme A |
| ADP | Adenosine Diphosphate |
| AICAR | 5-aminoimidazole-4-carboxamide-1-β-D-riboside |
| AKI | Acute Kidney Injury |
| AMP | Adenosine Monophosphate |
| AMPK | AMP-kinase |
| ATP | Adenosine Triphosphate |
| CKD | Chronic Kidney Disease |
| CLP | Cecal Ligation and Puncture |
| CoQ10 | Coenzyme Q10 |
| Drp1 | Dynamin Related Peptide 1 |
| ETC | Electron Transport Chain |
| FADH2 | Flavin Adenine Dinucleotide (reduced form of FAD) |
| FAO | Fatty Acid Oxidation |
| GFR | Glomerular Filtration Rate |
| IR | Ischemia-Reperfusion |
| KDIGO | Kidney Disease: Improving Global Outcomes |
| LPS | Lipopolysaccharide |
| mtDAMPS | Mitochondrial Damage-Associated Molecular Patterns |
| mtDNA | Mitochondrial DNA |
| NADH | Nicotinamide adenine dinucleotide (reduced form of NAD) |
| NAM | nicotinamide |
| OXPHOS | Oxidative Phosphorylation |
| PDH | Pyruvate Dehydrogenase |
| PDK | Pyruvate Dehydrogenase Kinase |
| PGC1α | Peroxisome Proliferator-activated Receptor γ Coactivator-1α |
| ROS | Reactive Oxygen Species |
| RNAseq | RNA-sequencing |
| s-AKI | In sepsis associated AKI |
| SIRT | Sirtuins |
| SS Peptides | Szeto–Schiller Peptides |
| TCA | Tricarboxylic Acid |
References
- Uchino, S.; Kellum, J.A.; Bellomo, R.; Doig, G.S.; Morimatsu, H.; Morgera, S.; Schetz, M.; Tan, I.; Bouman, C.; Macedo, E.; et al. Acute renal failure in critically ill patients: A multinational, multicenter study. JAMA 2005, 294, 813–818. [Google Scholar] [CrossRef]
- Bagshaw, S.M.; George, C.; Dinu, I.; Bellomo, R. A multi-centre evaluation of the RIFLE criteria for early acute kidney injury in critically ill patients. Nephrol. Dial. Transplant. 2008, 23, 1203–1210. [Google Scholar] [CrossRef]
- Thakar, C.V.; Christianson, A.; Freyberg, R.; Almenoff, P.; Render, M.L. Incidence and outcomes of acute kidney injury in intensive care units: A Veterans Administration study. Crit. Care Med. 2009, 37, 2552–2558. [Google Scholar] [CrossRef]
- Alobaidi, R.; Basu, R.K.; Goldstein, S.L.; Bagshaw, S.M. Sepsis-associated acute kidney injury. Semin. Nephrol. 2015, 35, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Hoste, E.A.; Bagshaw, S.M.; Bellomo, R.; Cely, C.M.; Colman, R.; Cruz, D.N.; Edipidis, K.; Forni, L.G.; Gomersall, C.D.; Govil, D.; et al. Epidemiology of acute kidney injury in critically ill patients: The multinational AKI-EPI study. Intensive Care Med. 2015, 41, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute Kidney Injury and Chronic Kidney Disease as Interconnected Syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Amdur, R.L.; Chawla, L.S.; Amodeo, S.; Kimmel, P.L.; Palant, C.E. Outcomes following diagnosis of acute renal failure in U.S. veterans: Focus on acute tubular necrosis. Kidney Int. 2009, 76, 1089–1097. [Google Scholar]
- Hsu, C.-Y.; Chertow, G.M.; McCulloch, C.E.; Fan, D.; Ordoñez, J.D.; Go, A.S. Nonrecovery of Kidney Function and Death after Acute on Chronic Renal Failure. Clin. J. Am. Soc. Nephrol. 2009, 4, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Zarjou, A.; Agarwal, A. Sepsis and acute kidney injury. J. Am. Soc. Nephrol. 2011, 22, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Ricksten, S.E.; Bragadottir, G.; Redfors, B.; Nordquist, L. Renal oxygenation and haemodynamics in acute kidney injury and chronic kidney disease. Clin. Exp. Pharmacol. Physiol. 2013, 40, 138–147. [Google Scholar] [CrossRef]
- Bullen, A.; Liu, Z.Z.; Hepokoski, M.; Li, Y.; Singh, P. Renal Oxygenation and Hemodynamics in Kidney Injury. Nephron 2017, 137, 260–263. [Google Scholar] [CrossRef]
- Prabhleen Singh, S.C.T.a.A.A.M. Metabolic Basis of Solute Transport. In Brenner and Rector’s The KidneyBrenner and Rector’s The Kidney, 11th ed.; Elsevier: Philadelphia, PA, USA, 2019. [Google Scholar]
- Ralto, K.M.; Parikh, S.M. Mitochondria in Acute Kidney Injury. Semin. Nephrol. 2016, 36, 8–16. [Google Scholar] [CrossRef]
- Sas, K.M.; Kayampilly, P.; Byun, J.; Nair, V.; Hinder, L.M.; Hur, J.; Zhang, H.; Lin, C.; Qi, N.R.; Michailidis, G.; et al. Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight 2016, 1, e86976. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Rowe, I.; Chiaravalli, M.; Mannella, V.; Ulisse, V.; Quilici, G.; Pema, M.; Song, X.W.; Xu, H.; Mari, S.; Qian, F.; et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat. Med. 2013, 19, 488–493. [Google Scholar] [CrossRef]
- Chiaravalli, M.; Rowe, I.; Mannella, V.; Quilici, G.; Canu, T.; Bianchi, V.; Gurgone, A.; Antunes, S.; D’Adamo, P.; Esposito, A.; et al. 2-Deoxy-d-Glucose Ameliorates PKD Progression. J. Am. Soc. Nephrol. 2016, 27, 1958–1969. [Google Scholar] [CrossRef]
- Dimmer, K.S.; Scorrano, L. (De)constructing mitochondria: What for? Physiology 2006, 21, 233–241. [Google Scholar] [CrossRef]
- Mandel, L.J. Metabolic substrates, cellular energy production, and the regulation of proximal tubular transport. Annu. Rev. Physiol. 1985, 47, 85–101. [Google Scholar] [CrossRef]
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trezeguet, V.; Lauquin, G.J.; Brandolin, G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003, 426, 39–44. [Google Scholar] [CrossRef]
- Mather, A.; Pollock, C. Glucose handling by the kidney. Kidney Int. Suppl. 2011, 79, S1–S6. [Google Scholar] [CrossRef]
- Rich, P.R. The molecular machinery of Keilin’s respiratory chain. Biochem. Soc. Trans. 2003, 31, 1095–1105. [Google Scholar] [CrossRef]
- Guder, W.G.; Wagner, S.; Wirthensohn, G. Metabolic fuels along the nephron: Pathways and intracellular mechanisms of interaction. Kidney Int. 1986, 29, 41–45. [Google Scholar] [CrossRef]
- Gerich, J.E.; Meyer, C.; Woerle, H.J.; Stumvoll, M. Renal gluconeogenesis: Its importance in human glucose homeostasis. Diabetes Care 2001, 24, 382–391. [Google Scholar] [CrossRef]
- Forbes, J.M.; Thorburn, D.R. Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [Google Scholar] [CrossRef]
- Guder, W.G.; Ross, B.D. Enzyme distribution along the nephron. Kidney Int. 1984, 26, 101–111. [Google Scholar] [CrossRef]
- Cargill, K.; Sims-Lucas, S. Metabolic requirements of the nephron. Pediatr. Nephrol. 2020, 35, 1–8. [Google Scholar] [CrossRef]
- Brooks, C.; Wei, Q.; Cho, S.G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef]
- Hall, A.M. Maintaining mitochondrial morphology in AKI: Looks matter. J. Am. Soc. Nephrol. 2013, 24, 1185–1187. [Google Scholar] [CrossRef]
- Hall, A.M.; Rhodes, G.J.; Sandoval, R.M.; Corridon, P.R.; Molitoris, B.A. In vivo multiphoton imaging of mitochondrial structure and function during acute kidney injury. Kidney Int. 2013, 83, 72–83. [Google Scholar] [CrossRef]
- Funk, J.A.; Schnellmann, R.G. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am. J. Physiol. Renal Physiol. 2012, 302, F853–F864. [Google Scholar] [CrossRef]
- Zhan, M.; Brooks, C.; Liu, F.; Sun, L.; Dong, Z. Mitochondrial dynamics: Regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013, 83, 568–581. [Google Scholar] [CrossRef]
- Tang, W.X.; Wu, W.H.; Qiu, H.Y.; Bo, H.; Huang, S.M. Amelioration of rhabdomyolysis-induced renal mitochondrial injury and apoptosis through suppression of Drp-1 translocation. J. Nephrol. 2013, 26, 1073–1082. [Google Scholar] [CrossRef]
- Perry, H.M.; Huang, L.; Wilson, R.J.; Bajwa, A.; Sesaki, H.; Yan, Z.; Rosin, D.L.; Kashatus, D.F.; Okusa, M.D. Dynamin-Related Protein 1 Deficiency Promotes Recovery from AKI. J. Am. Soc. Nephrol. 2018, 29, 194–206. [Google Scholar] [CrossRef]
- Tang, C.; Cai, J.; Yin, X.M.; Weinberg, J.M.; Venkatachalam, M.A.; Dong, Z. Mitochondrial quality control in kidney injury and repair. Nat. Rev. Nephrol. 2021, 17, 299–318. [Google Scholar] [CrossRef]
- Takasu, O.; Gaut, J.P.; Watanabe, E.; To, K.; Fagley, R.E.; Sato, B.; Jarman, S.; Efimov, I.R.; Janks, D.L.; Srivastava, A.; et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am. J. Respir. Crit. Care Med. 2013, 187, 509–517. [Google Scholar] [CrossRef]
- Parekh, D.J.; Weinberg, J.M.; Ercole, B.; Torkko, K.C.; Hilton, W.; Bennett, M.; Devarajan, P.; Venkatachalam, M.A. Tolerance of the human kidney to isolated controlled ischemia. J. Am. Soc. Nephrol. 2013, 24, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Jesinkey, S.R.; Funk, J.A.; Stallons, L.J.; Wills, L.P.; Megyesi, J.K.; Beeson, C.C.; Schnellmann, R.G. Formoterol restores mitochondrial and renal function after ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2014, 25, 1157–1162. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.M. Mitochondrial biogenesis in kidney disease. J. Am. Soc. Nephrol. 2011, 22, 431–436. [Google Scholar] [CrossRef]
- Tran, M.; Tam, D.; Bardia, A.; Bhasin, M.; Rowe, G.C.; Kher, A.; Zsengeller, Z.K.; Akhavan-Sharif, M.R.; Khankin, E.V.; Saintgeniez, M.; et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Investig. 2011, 121, 4003–4014. [Google Scholar] [CrossRef]
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef]
- Drury, E.R.; Zsengeller, Z.K.; Stillman, I.E.; Khankin, E.V.; Pavlakis, M.; Parikh, S.M. Renal PGC1alpha May Be Associated with Recovery after Delayed Graft Function. Nephron 2018, 138, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Feldkamp, T.; Kribben, A.; Weinberg, J.M. Assessment of mitochondrial membrane potential in proximal tubules after hypoxia-reoxygenation. Am. J. Physiol. Renal Physiol. 2005, 288, F1092–F1102. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Hu, Y.; Quiros, P.M.; Wei, Q.; Lopez-Otin, C.; Dong, Z. OMA1 mediates OPA1 proteolysis and mitochondrial fragmentation in experimental models of ischemic kidney injury. Am. J. Physiol. Renal Physiol. 2014, 306, F1318–F1326. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Liu, S.; Soong, Y.; Wu, D.; Darrah, S.F.; Cheng, F.Y.; Zhao, Z.; Ganger, M.; Tow, C.Y.; Seshan, S.V. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J. Am. Soc. Nephrol. 2011, 22, 1041–1052. [Google Scholar] [CrossRef]
- Feldkamp, T.; Kribben, A.; Roeser, N.F.; Senter, R.A.; Weinberg, J.M. Accumulation of nonesterified fatty acids causes the sustained energetic deficit in kidney proximal tubules after hypoxia-reoxygenation. Am. J. Physiol. Renal Physiol. 2006, 290, F465–F477. [Google Scholar] [CrossRef]
- Tanabe, K.; Tamura, Y.; Lanaspa, M.A.; Miyazaki, M.; Suzuki, N.; Sato, W.; Maeshima, Y.; Schreiner, G.F.; Villarreal, F.J.; Johnson, R.J.; et al. Epicatechin limits renal injury by mitochondrial protection in cisplatin nephropathy. Am. J. Physiol. Renal Physiol. 2012, 303, F1264–F1274. [Google Scholar] [CrossRef]
- Clark, A.J.; Parikh, S.M. Mitochondrial Metabolism in Acute Kidney Injury. Semin. Nephrol. 2020, 40, 101–113. [Google Scholar] [CrossRef]
- Patil, N.K.; Parajuli, N.; MacMillan-Crow, L.A.; Mayeux, P.R. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: Mitochondria-targeted antioxidant mitigates injury. Am. J. Physiol. Renal Physiol. 2014, 306, F734–F743. [Google Scholar] [CrossRef]
- Rojas-Morales, P.; Leon-Contreras, J.C.; Aparicio-Trejo, O.E.; Reyes-Ocampo, J.G.; Medina-Campos, O.N.; Jimenez-Osorio, A.S.; Gonzalez-Reyes, S.; Marquina-Castillo, B.; Hernandez-Pando, R.; Barrera-Oviedo, D.; et al. Fasting reduces oxidative stress, mitochondrial dysfunction and fibrosis induced by renal ischemia-reperfusion injury. Free Radic. Biol. Med. 2019, 135, 60–67. [Google Scholar] [CrossRef]
- Aparicio-Trejo, O.E.; Reyes-Fermin, L.M.; Briones-Herrera, A.; Tapia, E.; Leon-Contreras, J.C.; Hernandez-Pando, R.; Sanchez-Lozada, L.G.; Pedraza-Chaverri, J. Protective effects of N-acetyl-cysteine in mitochondria bioenergetics, oxidative stress, dynamics and S-glutathionylation alterations in acute kidney damage induced by folic acid. Free. Radic. Biol. Med. 2019, 130, 379–396. [Google Scholar] [CrossRef]
- Nath, K.A.; Norby, S.M. Reactive oxygen species and acute renal failure. Am. J. Med. 2000, 109, 665–678. [Google Scholar] [CrossRef]
- Li, Y.; Nourbakhsh, N.; Pham, H.; Tham, R.; Zuckerman, J.E.; Singh, P. Evolution of altered tubular metabolism and mitochondrial function in sepsis-associated acute kidney injury. Am. J. Physiol. Renal Physiol. 2020, 319, F229–F244. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, N.; Tsuji, T.; Ohashi, N.; Kato, A.; Fujigaki, Y.; Yasuda, H. Role of Mitochondrial DNA in Septic AKI via Toll-Like Receptor 9. J. Am. Soc. Nephrol. 2016, 27, 2009–2020. [Google Scholar] [CrossRef] [PubMed]
- Faust, H.E.; Reilly, J.P.; Anderson, B.J.; Ittner, C.A.G.; Forker, C.M.; Zhang, P.; Weaver, B.A.; Holena, D.N.; Lanken, P.N.; Christie, J.D.; et al. Plasma Mitochondrial DNA Levels Are Associated With ARDS in Trauma and Sepsis Patients. Chest 2020, 157, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Hepokoski, M.; Wang, J.; Li, K.; Li, Y.; Gupta, P.; Mai, T.; Moshensky, A.; Alotaibi, M.; Crotty Alexander, L.E.; Malhotra, A.; et al. Altered lung metabolism and mitochondrial DAMPs in lung injury due to acute kidney injury. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2021, 320, L821–L831. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell. Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Stacpoole, P.W. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. J. Natl. Cancer Inst. 2017, 109, djx071. [Google Scholar] [CrossRef]
- Liu, J.; Edgington-Giordano, F.; Dugas, C.; Abrams, A.; Katakam, P.; Satou, R.; Saifudeen, Z. Regulation of Nephron Progenitor Cell Self-Renewal by Intermediary Metabolism. J. Am. Soc. Nephrol. 2017, 28, 3323–3335. [Google Scholar] [CrossRef]
- Portilla, D. Role of fatty acid beta-oxidation and calcium-independent phospholipase A2 in ischemic acute renal failure. Curr. Opin. Nephrol. Hypertens. 1999, 8, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Nagothu, K.K.; Desai, V.; Lee, T.; Branham, W.; Moland, C.; Megyesi, J.K.; Crew, M.D.; Portilla, D. Transgenic expression of proximal tubule peroxisome proliferator-activated receptor-alpha in mice confers protection during acute kidney injury. Kidney Int. 2009, 76, 1049–1062. [Google Scholar] [CrossRef]
- Fukuhara, Y.; Yamamoto, S.; Yano, F.; Orita, Y.; Fujiwara, Y.; Ueda, N.; Kamada, T.; Noguchi, T.; Tanaka, T. Changes in activities and mRNA levels of glycolytic enzymes of ischemia-reperfused rat kidney. Contrib. Nephrol. 1991, 95, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Lan, R.; Geng, H.; Singha, P.K.; Saikumar, P.; Bottinger, E.P.; Weinberg, J.M.; Venkatachalam, M.A. Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J. Am. Soc. Nephrol. 2016, 27, 3356–3367. [Google Scholar] [CrossRef]
- Legouis, D.; Ricksten, S.E.; Faivre, A.; Verissimo, T.; Gariani, K.; Verney, C.; Galichon, P.; Berchtold, L.; Feraille, E.; Fernandez, M.; et al. Altered proximal tubular cell glucose metabolism during acute kidney injury is associated with mortality. Nat. Metab. 2020, 2, 732–743. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Stallons, L.J.; Schnellmann, R.G. Renal cortical hexokinase and pentose phosphate pathway activation through the EGFR/Akt signaling pathway in endotoxin-induced acute kidney injury. Am. J. Physiol. Renal Physiol. 2014, 307, F435–F444. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.; Chen, W.; Wang, Y.; Gong, F.; Huang, S.; Zhong, M.; Liu, Z.; Chen, Y.; Ma, L.; Yang, Z.; et al. The Warburg Effect Promotes Mitochondrial Injury Regulated by Uncoupling Protein-2 in Septic Acute Kidney Injury. Shock 2021, 55, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Craciun, F.L.; Bijol, V.; Ajay, A.K.; Rao, P.; Kumar, R.K.; Hutchinson, J.; Hofmann, O.; Joshi, N.; Luyendyk, J.P.; Kusebauch, U.; et al. RNA Sequencing Identifies Novel Translational Biomarkers of Kidney Fibrosis. J. Am. Soc. Nephrol. 2016, 27, 1702–1713. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kumar, S.; Dolzhenko, E.; Alvarado, G.F.; Guo, J.; Lu, C.; Chen, Y.; Li, M.; Dessing, M.C.; Parvez, R.K.; et al. Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Kwon, C.H.; Ha, H.K.; Han, M.; Song, S.H. RNA-Seq identifies condition-specific biological signatures of ischemia-reperfusion injury in the human kidney. BMC Nephrol. 2020, 21, 398. [Google Scholar] [CrossRef] [PubMed]
- Kirita, Y.; Wu, H.; Uchimura, K.; Wilson, P.C.; Humphreys, B.D. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc. Natl. Acad. Sci. USA 2020, 117, 15874–15883. [Google Scholar] [CrossRef]
- Nilsson, L.M.; Castresana-Aguirre, M.; Scott, L.; Brismar, H. RNA-seq reveals altered gene expression levels in proximal tubular cell cultures compared to renal cortex but not during early glucotoxicity. Sci. Rep. 2020, 10, 10390. [Google Scholar] [CrossRef]
- Dare, A.J.; Bolton, E.A.; Pettigrew, G.J.; Bradley, J.A.; Saeb-Parsy, K.; Murphy, M.P. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol. 2015, 5, 163–168. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Horvath, B.; Zsengeller, Z.; Zielonka, J.; Tanchian, G.; Holovac, E.; Kechrid, M.; Patel, V.; Stillman, I.E.; Parikh, S.M.; et al. Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radic. Biol. Med. 2012, 52, 497–506. [Google Scholar] [CrossRef]
- Plotnikov, E.Y.; Chupyrkina, A.A.; Jankauskas, S.S.; Pevzner, I.B.; Silachev, D.N.; Skulachev, V.P.; Zorov, D.B. Mechanisms of nephroprotective effect of mitochondria-targeted antioxidants under rhabdomyolysis and ischemia/reperfusion. Biochim. Biophys. Acta 2011, 1812, 77–86. [Google Scholar] [CrossRef]
- Mapuskar, K.A.; Wen, H.; Holanda, D.G.; Rastogi, P.; Steinbach, E.; Han, R.; Coleman, M.C.; Attanasio, M.; Riley, D.P.; Spitz, D.R.; et al. Persistent increase in mitochondrial superoxide mediates cisplatin-induced chronic kidney disease. Redox Biol. 2019, 20, 98–106. [Google Scholar] [CrossRef]
- Garrett, S.M.; Whitaker, R.M.; Beeson, C.C.; Schnellmann, R.G. Agonism of the 5-hydroxytryptamine 1F receptor promotes mitochondrial biogenesis and recovery from acute kidney injury. J. Pharmacol. Exp. Ther. 2014, 350, 257–264. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamaguchi, H.; Kikusato, M.; Hashizume, O.; Nagatoishi, S.; Matsuo, A.; Sato, T.; Kudo, T.; Matsuhashi, T.; Murayama, K.; et al. Mitochonic Acid 5 Binds Mitochondria and Ameliorates Renal Tubular and Cardiac Myocyte Damage. J. Am. Soc. Nephrol. 2016, 27, 1925–1932. [Google Scholar] [CrossRef]
- Reznick, R.M.; Shulman, G.I. The role of AMP-activated protein kinase in mitochondrial biogenesis. J. Physiol. 2006, 574, 33–39. [Google Scholar] [CrossRef]
- Zhang, C.S.; Lin, S.C. AMPK Promotes Autophagy by Facilitating Mitochondrial Fission. Cell Metab. 2016, 23, 399–401. [Google Scholar] [CrossRef]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Loson, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Lempiainen, J.; Finckenberg, P.; Levijoki, J.; Mervaala, E. AMPK activator AICAR ameliorates ischaemia reperfusion injury in the rat kidney. Br. J. Pharmacol. 2012, 166, 1905–1915. [Google Scholar] [CrossRef]
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015, 125, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, R.M.; Corum, D.; Beeson, C.C.; Schnellmann, R.G. Mitochondrial Biogenesis as a Pharmacological Target: A New Approach to Acute and Chronic Diseases. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 229–249. [Google Scholar] [CrossRef]
- Fan, H.; Yang, H.C.; You, L.; Wang, Y.Y.; He, W.J.; Hao, C.M. The histone deacetylase, SIRT1, contributes to the resistance of young mice to ischemia/reperfusion-induced acute kidney injury. Kidney Int. 2013, 83, 404–413. [Google Scholar] [CrossRef]
- Funk, J.A.; Schnellmann, R.G. Accelerated recovery of renal mitochondrial and tubule homeostasis with SIRT1/PGC-1alpha activation following ischemia-reperfusion injury. Toxicol. Appl. Pharmacol. 2013, 273, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Deng, C.X.; Finley, L.W.; Kim, H.S.; Gius, D. SIRT3 is a mitochondrial tumor suppressor: A scientific tale that connects aberrant cellular ROS, the Warburg effect, and carcinogenesis. Cancer Res. 2012, 72, 2468–2472. [Google Scholar] [CrossRef]
- Osman, M.M.; Lulic, D.; Glover, L.; Stahl, C.E.; Lau, T.; van Loveren, H.; Borlongan, C.V. Cyclosporine-A as a neuroprotective agent against stroke: Its translation from laboratory research to clinical application. Neuropeptides 2011, 45, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Y.; Zhang, L.; Sui, M.X.; Zhu, Y.H.; Zeng, L. Protective effects of sirtuin 3 in a murine model of sepsis-induced acute kidney injury. Sci. Rep. 2016, 6, 33201. [Google Scholar] [CrossRef]
- Zhao, W.; Zhang, L.; Chen, R.; Lu, H.; Sui, M.; Zhu, Y.; Zeng, L. SIRT3 Protects Against Acute Kidney Injury via AMPK/mTOR-Regulated Autophagy. Front. Physiol. 2018, 9, 1526. [Google Scholar] [CrossRef]
- Szeto, H.H. Pharmacologic Approaches to Improve Mitochondrial Function in AKI and CKD. J. Am. Soc. Nephrol. 2017, 28, 2856–2865. [Google Scholar] [CrossRef]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Szeto, H.H.; Liu, S.; Soong, Y.; Birk, A.V. Improving mitochondrial bioenergetics under ischemic conditions increases warm ischemia tolerance in the kidney. Am. J. Physiol. Renal Physiol. 2015, 308, F11–F21. [Google Scholar] [CrossRef]
- Oh, C.J.; Ha, C.M.; Choi, Y.K.; Park, S.; Choe, M.S.; Jeoung, N.H.; Huh, Y.H.; Kim, H.J.; Kweon, H.S.; Lee, J.M.; et al. Pyruvate dehydrogenase kinase 4 deficiency attenuates cisplatin-induced acute kidney injury. Kidney Int. 2017, 91, 880–895. [Google Scholar] [CrossRef] [PubMed]
- Galgamuwa, R.; Hardy, K.; Dahlstrom, J.E.; Blackburn, A.C.; Wium, E.; Rooke, M.; Cappello, J.Y.; Tummala, P.; Patel, H.R.; Chuah, A.; et al. Dichloroacetate Prevents Cisplatin-Induced Nephrotoxicity without Compromising Cisplatin Anticancer Properties. J. Am. Soc. Nephrol. JASN 2016, 27, 3331–3344. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.M.; Venkatachalam, M.A.; Roeser, N.F.; Saikumar, P.; Dong, Z.; Senter, R.A.; Nissim, I. Anaerobic and aerobic pathways for salvage of proximal tubules from hypoxia-induced mitochondrial injury. Am. J. Physiol. Renal Physiol. 2000, 279, F927–F943. [Google Scholar] [CrossRef]
- Weinberg, J.M.; Venkatachalam, M.A.; Roeser, N.F.; Nissim, I. Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc. Natl. Acad. Sci. USA 2000, 97, 2826–2831. [Google Scholar] [CrossRef] [PubMed]
- Ralto, K.M.; Rhee, E.P.; Parikh, S.M. NAD(+) homeostasis in renal health and disease. Nat. Rev. Nephrol. 2020, 16, 99–111. [Google Scholar] [CrossRef]
- Guan, Y.; Wang, S.R.; Huang, X.Z.; Xie, Q.H.; Xu, Y.Y.; Shang, D.; Hao, C.M. Nicotinamide Mononucleotide, an NAD(+) Precursor, Rescues Age-Associated Susceptibility to AKI in a Sirtuin 1-Dependent Manner. J. Am. Soc. Nephrol. 2017, 28, 2337–2352. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, E.; Mottis, A.; Zietak, M.; De Franco, F.; van der Velpen, V.; Gariani, K.; Ryu, D.; Cialabrini, L.; Matilainen, O.; Liscio, P.; et al. De novo NAD(+) synthesis enhances mitochondrial function and improves health. Nature 2018, 563, 354–359. [Google Scholar] [CrossRef]
- Poyan Mehr, A.; Tran, M.T.; Ralto, K.M.; Leaf, D.E.; Washco, V.; Messmer, J.; Lerner, A.; Kher, A.; Kim, S.H.; Khoury, C.C.; et al. De novo NAD(+) biosynthetic impairment in acute kidney injury in humans. Nat. Med. 2018, 24, 1351–1359. [Google Scholar] [CrossRef]


{kind=link}
| AKI Definition | Serum Creatinine or GFR Criteria | Urine Output Criteria |
| Increase in serum creatinine by ≥0.3 mg/dL (≥26.5 micromol/L) within 48 h, or Increase in serum creatinine to ≥1.5 times baseline, which is known or presumed to have occurred within the prior seven days, or | Urine output <0.5 mL/kg/hour for 6 h | |
| AKI Staging | Serum Creatinine or GFR Criteria | Urine Output Criteria |
| Stage 1 | Increase in serum creatinine to 1.5 to 1.9 times baseline, or Increase in serum creatinine by ≥0.3 mg/dL (≥26.5 micromol/L), or | Reduction in urine output to <0.5 mL/kg/hour for 6 to 12 h. |
| Stage 2 | Increase in serum creatinine to 2.0 to 2.9 times baseline, or | Reduction in urine output to <0.5 mL/kg/hour for ≥12 h. |
| Stage 3 | Increase in serum creatinine to 3.0 times baseline, or Increase in serum creatinine to ≥4.0 mg/dL (≥353.6 micromol/L), or Initiation of kidney replacement therapy, or In patients < 18 years, decrease in estimated glomerular filtration rate (eGFR) to <35 mL/min/1.73 m2. | Reduction in urine output to <0.3 mL/kg/hour for ≥24 h, or Anuria for ≥12 h |
| Therapy | Mechanism | Clinical Trial | Details |
|---|---|---|---|
| Elamipretide (MTP-131) | Prevents the peroxidation of cardiolipin by cytochrome c | Phase I NCT02436447The Safety and Pharmacokinetics of Repeat-dose Intravenous Infusion of MTP-131 in Subjects with Impaired Renal Function (2015) | Open-label, parallel group, multiple dose study to evaluate the safety, tolerability, and pharmacokinetics of one-hour intravenous infusion of MTP-131 administered for 7 consecutive days. |
| ASP1128 | Selective PPARδ modulator, promotes fatty acid oxidation | Phase II NCT03941483Evaluate the Efficacy of ASP1128 (MA-0217) in Subjects at Risk for Acute Kidney Injury Following Coronary Artery Bypass Graft (CABG) and/or Valve Surgery (2019) | Double-blind study to investigate the safety and tolerability of postsurgery treatment with ASP1128, and pharmacokinetic characteristics of ASP1128 in subjects at risk for AKI following CABG and/or valve surgery. |
| Nicotinamide | Incorporates into nicotinamide adeninedinucleotide (NAD)+ and NADP+,coenzymes in enzymaticoxidation-reduction reactions. | Phase II NTC04342975Evaluate the Efficacy of BASIS™ (Nicotinamide Riboside and Pterostilbene) Treatment for Kidney Protection in Patients Treated by Complex Aortic Aneurysm Repair and Aortic Arch Reconstruction (2020) | Single-center, prospective, randomized, double-blinded, placebo-controlled phase II clinical trial to evaluate the efficacy of “NAD+ supplementation” in preventing AKI in patients undergoing complex aortic aneurysm repair and open aortic arch reconstruction. |
| MitoQ | Antioxidant, derivative of CoQ10 with increased mitochondrial uptake | Phase IV NCT02364648Mitochondrial Oxidative Stress and Vascular Health in Chronic Kidney Disease (2017) | Controlled, double blinded trial, Stage 3–5 chronic kidney disease (CKD) patients will be randomly assigned to receive a 4-week daily dose of a mitochondria targeted antioxidant (MitoQ) or a placebo. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Hepokoski, M.; Gu, W.; Simonson, T.; Singh, P. Targeting Mitochondria and Metabolism in Acute Kidney Injury. J. Clin. Med. 2021, 10, 3991. https://doi.org/10.3390/jcm10173991
Li Y, Hepokoski M, Gu W, Simonson T, Singh P. Targeting Mitochondria and Metabolism in Acute Kidney Injury. Journal of Clinical Medicine. 2021; 10(17):3991. https://doi.org/10.3390/jcm10173991
Chicago/Turabian StyleLi, Ying, Mark Hepokoski, Wanjun Gu, Tatum Simonson, and Prabhleen Singh. 2021. "Targeting Mitochondria and Metabolism in Acute Kidney Injury" Journal of Clinical Medicine 10, no. 17: 3991. https://doi.org/10.3390/jcm10173991
APA StyleLi, Y., Hepokoski, M., Gu, W., Simonson, T., & Singh, P. (2021). Targeting Mitochondria and Metabolism in Acute Kidney Injury. Journal of Clinical Medicine, 10(17), 3991. https://doi.org/10.3390/jcm10173991