
Myeloma Bone Disease: The Osteoblast in the Spotlight


Abstract
:1. Introduction
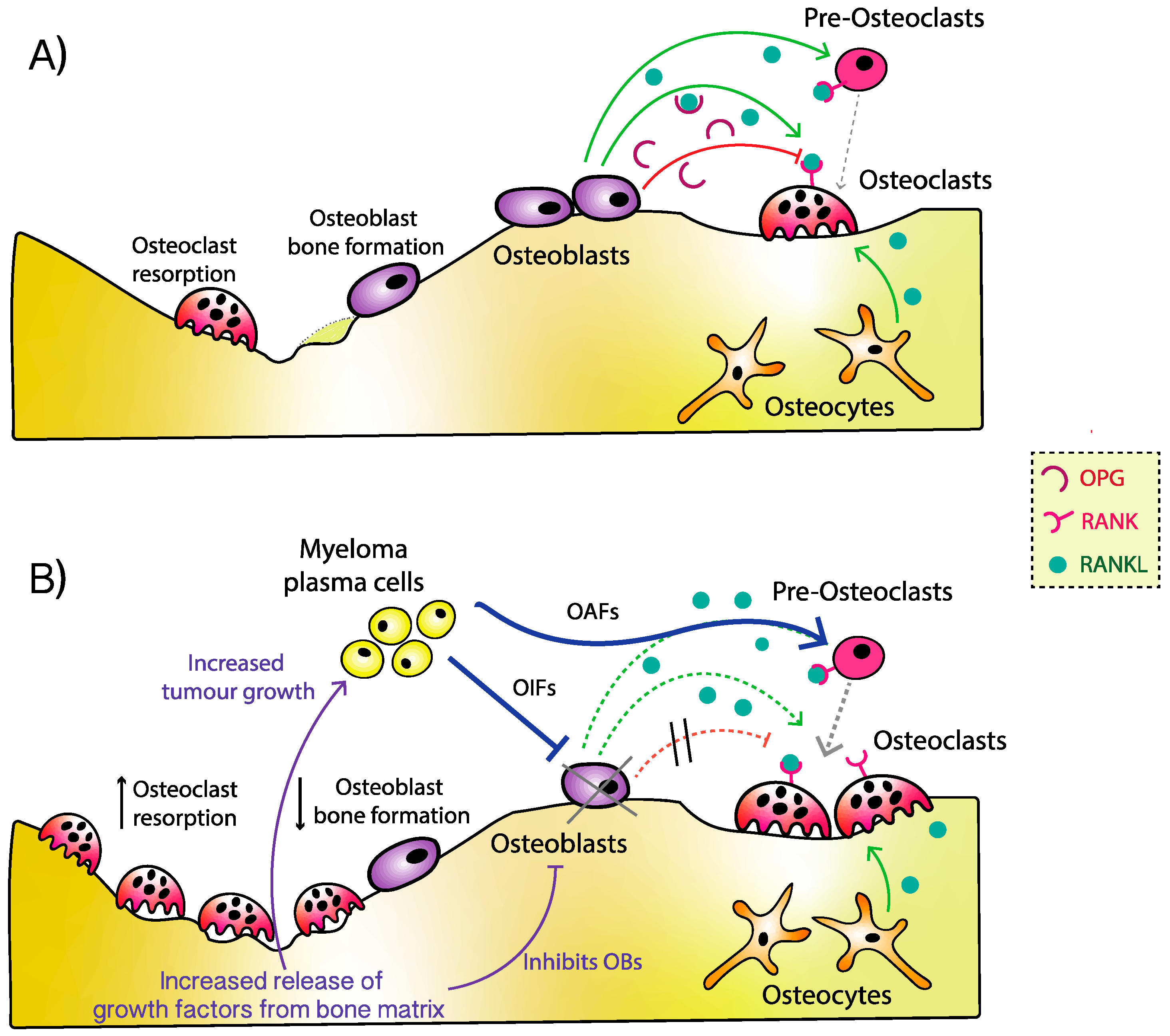
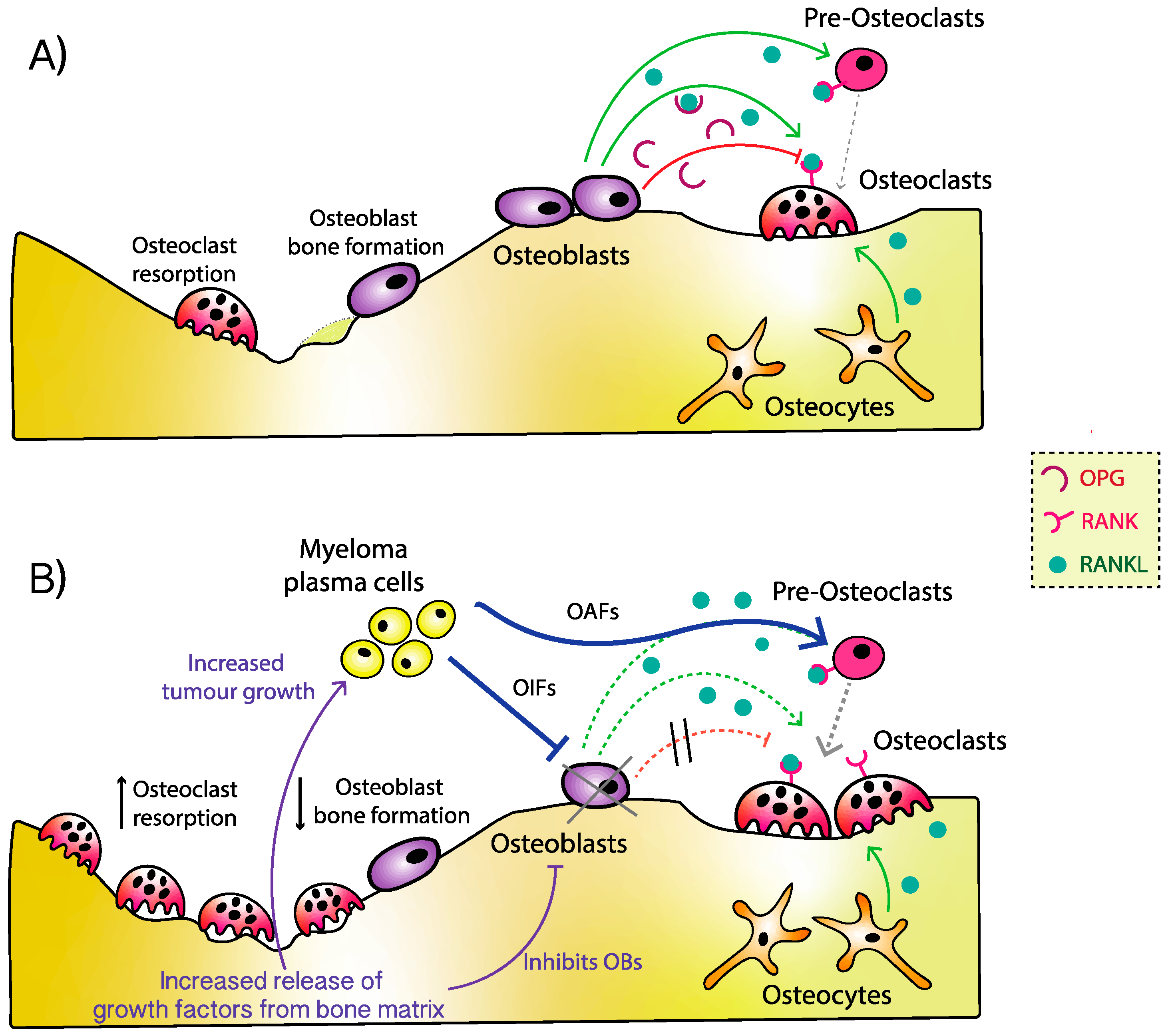
2. Pathophysiology of Myeloma Bone Disease
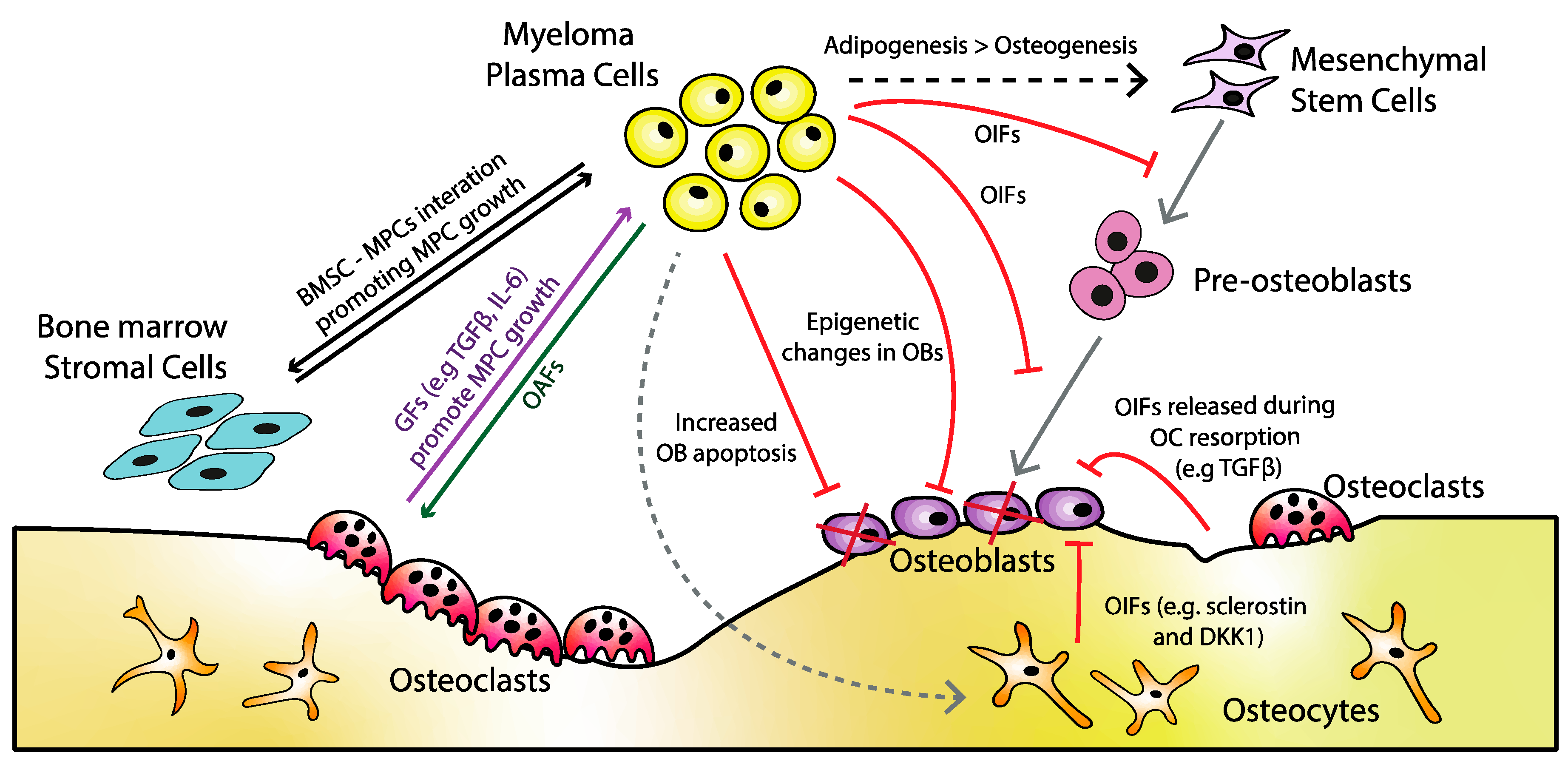
3. Osteoblast Dysfunction
4. Osteoblast Inhibiting Factors
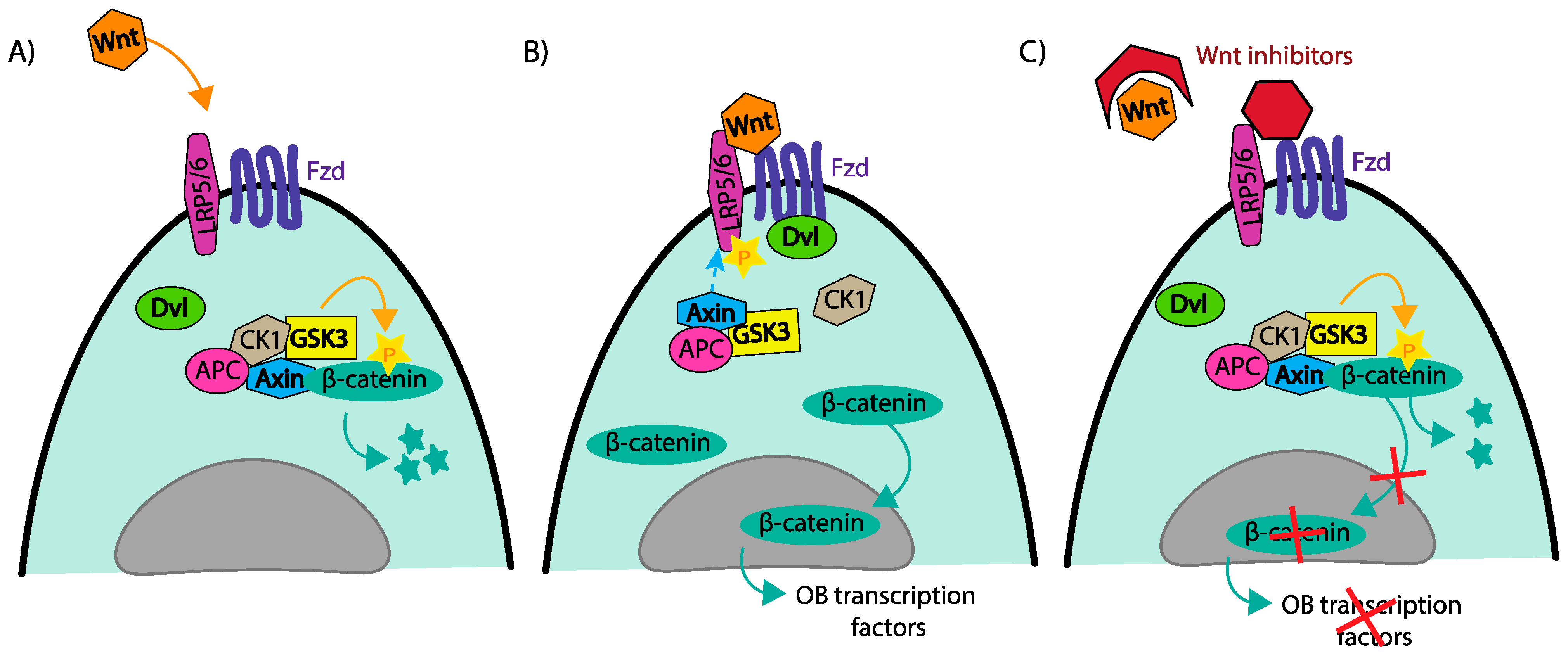
4.1. Wnt/B-Catenin; DKK1, Frizzled Transmembrane Receptors and Sclerostin
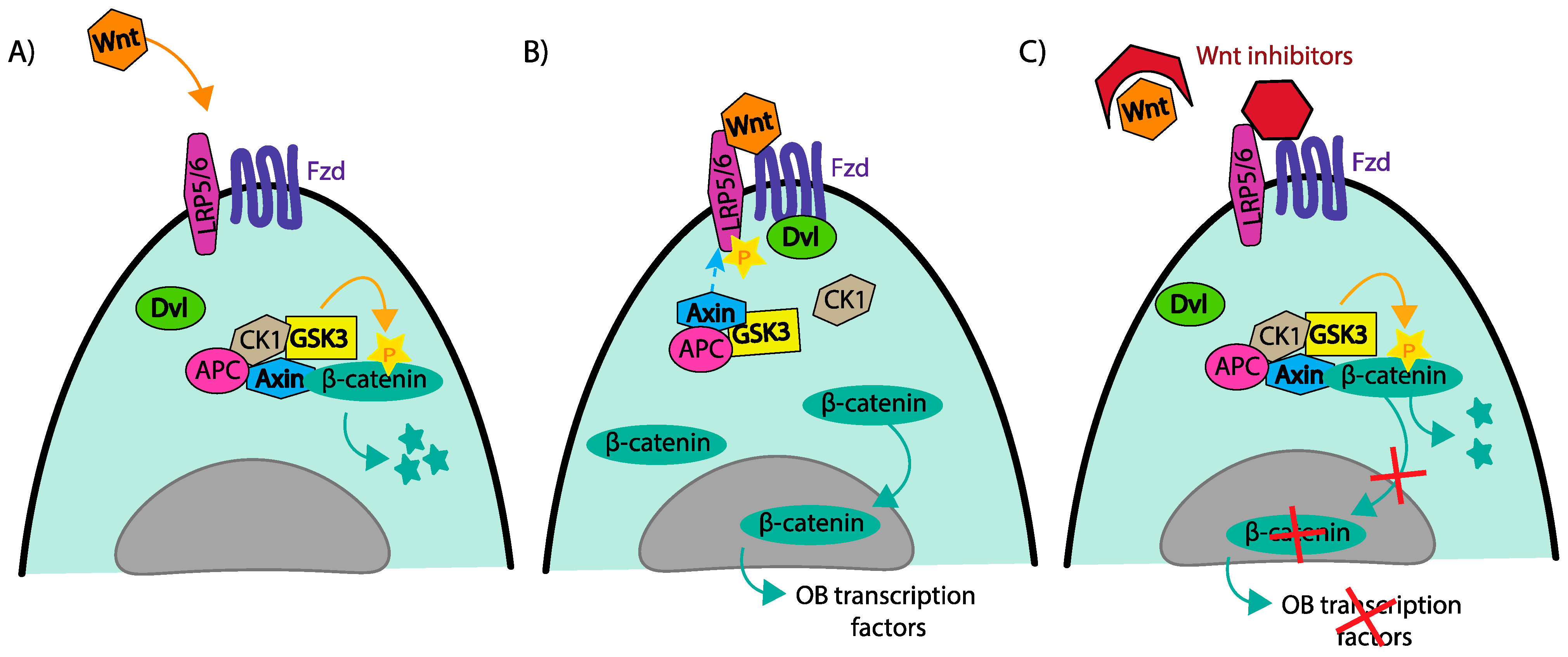
4.2. TGFβ, ACTIVIN A, BMPs, and HGF
4.3. Interleukins
4.4. Other Factors
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Kazandjian, D. Multiple myeloma epidemiology and survival: A unique malignancy. Semin. Oncol. 2016, 43, 676–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Cook, L.; Macdonald, D.H. Management of paraproteinaemia. Postgrad. Med. J. 2007, 83, 217–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowan, A.J.; Allen, C.; Barac, A.; Basaleem, H.; Bensenor, I.; Curado, M.P.; Foreman, K.; Gupta, R.; Harvey, J.; Hosgood, D.; et al. Global Burden of Multiple Myeloma: A Systematic Analysis for the Global Burden of Disease Study 2016. JAMA Oncol. 2018, 4, 1221–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smittenaar, C.R.; Petersen, K.A.; Stewart, K.; Moitt, N. Cancer incidence and mortality projections in the UK until 2035. Br. J. Cancer 2016, 115, 1147–1155. [Google Scholar] [CrossRef]
- Kyle, R.A.; Gertz, M.A.; Witzig, T.E.; Lust, J.A.; Lacy, M.Q.; Dispenzieri, A.; Fonseca, R.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin. Proc. 2003, 78, 21–33. [Google Scholar] [CrossRef]
- Coleman, R.E. Skeletal complications of malignancy. Cancer 1997, 80 (Suppl. 8), 1588–1594. [Google Scholar] [CrossRef]
- Melton, L.J., 3rd; Kyle, R.A.; Achenbach, S.J.; Oberg, A.L.; Rajkumar, S.V. Fracture risk with multiple myeloma: A population-based study. J. Bone Miner. Res. 2005, 20, 487–493. [Google Scholar] [CrossRef]
- Saad, F.; Lipton, A.; Cook, R.; Chen, Y.M.; Smith, M.; Coleman, R. Pathologic fractures correlate with reduced survival in patients with malignant bone disease. Cancer 2007, 110, 1860–1867. [Google Scholar] [CrossRef]
- Novack, D.V.; Mbalaviele, G. Osteoclasts-Key Players in Skeletal Health and Disease. Microbiol Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Terpos, E.; Kastritis, E.; Christoulas, D.; Gkotzamanidou, M.; Eleutherakis-Papaiakovou, E.; Kanellias, N.; Papatheodorou, A.; Dimopoulos, M.A. Circulating activin-A is elevated in patients with advanced multiple myeloma and correlates with extensive bone involvement and inferior survival; no alterations post-lenalidomide and dexamethasone therapy. Ann. Oncol. 2012, 23, 2681–2686. [Google Scholar] [CrossRef]
- Chantry, A.D.; Heath, D.; Mulivor, A.W.; Pearsall, S.; Baud’huin, M.; Coulton, L.; Evans, H.; Abdul, N.; Werner, E.D.; Bouxsein, M.L.; et al. Inhibiting activin-A signaling stimulates bone formation and prevents cancer-induced bone destruction in vivo. J. Bone Miner. Res. 2010, 25, 2633–2646. [Google Scholar] [CrossRef]
- Vallet, S.; Mukherjee, S.; Vaghela, N.; Hideshima, T.; Fulciniti, M.; Pozzi, S.; Santo, L.; Cirstea, D.; Patel, K.; Sohani, A.R.; et al. Activin A promotes multiple myeloma-induced osteolysis and is a promising target for myeloma bone disease. Proc. Natl. Acad. Sci. USA 2010, 107, 5124–5129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, M.M.; Reagan, M.R.; Youlten, S.E.; Mohanty, S.T.; Seckinger, A.; Terry, R.L.; Pettitt, J.A.; Simic, M.K.; Cheng, T.L.; Morse, A.; et al. Inhibiting the osteocyte-specific protein sclerostin increases bone mass and fracture resistance in multiple myeloma. Blood 2017, 129, 3452–3464. [Google Scholar] [CrossRef] [Green Version]
- Tian, E.; Zhan, F.; Walker, R.; Rasmussen, E.; Ma, Y.; Barlogie, B.; Shaughnessy, J.D., Jr. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N. Engl. J. Med. 2003, 349, 2483–2494. [Google Scholar] [CrossRef]
- Politou, M.C.; Heath, D.J.; Rahemtulla, A.; Szydlo, R.; Anagnostopoulos, A.; Dimopoulos, M.A.; Croucher, P.I.; Terpos, E. Serum concentrations of Dickkopf-1 protein are increased in patients with multiple myeloma and reduced after autologous stem cell transplantation. Int. J. Cancer 2006, 119, 1728–1731. [Google Scholar] [CrossRef]
- D’Souza, S.; del Prete, D.; Jin, S.; Sun, Q.; Huston, A.J.; Kostov, F.E.; Sammut, B.; Hong, C.-S.; Anderson, J.L.; Patrene, K.D.; et al. Gfi1 expressed in bone marrow stromal cells is a novel osteoblast suppressor in patients with multiple myeloma bone disease. Blood 2011, 118, 6871–6880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Standal, T.; Abildgaard, N.; Fagerli, U.M.; Stordal, B.; Hjertner, O.; Borset, M.; Algarín, E.M.; Martín-Sánchez, M.; Corbacho-González, D.; Ortiz-De-Solorzano, C.; et al. HGF inhibits BMP-induced osteoblastogenesis: Possible implications for the bone disease of multiple myeloma. Blood 2007, 109, 3024–3030. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, I.B.; Christensen, J.H.; Lyng, M.B.; Moller, M.B.; Pedersen, L.; Rasmussen, L.M.; Ditzel, H.J.; Abildgaard, N. Hepatocyte growth factor pathway upregulation in the bone marrow microenvironment in multiple myeloma is associated with lytic bone disease. Br. J. Haematol. 2013, 161, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Trotter, T.N.; Nan, L.; Luo, R.; Javed, A.; Sanderson, R.D.; Suva, L.J.; Yang, Y. Heparanase inhibits osteoblastogenesis and shifts bone marrow progenitor cell fate in myeloma bone disease. Bone 2013, 57, 10–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Chung, H.Y.; Ehrlich, L.A.; Jelinek, D.F.; Callander, N.S.; Roodman, G.D.; Choi, S.J. IL-3 expression by myeloma cells increases both osteoclast formation and growth of myeloma cells. Blood 2004, 103, 2308–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrlich, L.A.; Chung, H.Y.; Ghobrial, I.; Choi, S.J.; Morandi, F.; Colla, S.; Rizzoli, V.; David Roodman, G.; Giuliani, N. IL-3 is a potential inhibitor of osteoblast differentiation in multiple myeloma. Blood 2005, 106, 1407–1414. [Google Scholar] [CrossRef]
- Giuliani, N.; Colla, S.; Morandi, F.; Lazzaretti, M.; Sala, R.; Bonomini, S.; Grano, M.; Colucci, S.; Svaldi, M.; Rizzoli, V. Myeloma cells block RUNX2/CBFA1 activity in human bone marrow osteoblast progenitors and inhibit osteoblast formation and differentiation. Blood 2005, 106, 2472–2483. [Google Scholar] [CrossRef]
- Weitzmann, M.N.; Roggia, C.; Toraldo, G.; Weitzmann, L.; Pacifici, R. Increased production of IL-7 uncouples bone formation from bone resorption during estrogen deficiency. J. Clin. Investig. 2002, 110, 1643–1650. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, N.; Rizzoli, V. Myeloma cells and bone marrow osteoblast interactions: Role in the development of osteolytic lesions in multiple myeloma. Leuk. Lymphoma 2007, 48, 2323–2329. [Google Scholar] [CrossRef] [PubMed]
- Hjertner, O.; Torgersen, M.L.; Seidel, C.; Hjorth-Hansen, H.; Waage, A.; Borset, M.; Sundan, A. Hepatocyte growth factor (HGF) induces interleukin-11 secretion from osteoblasts: A possible role for HGF in myeloma-associated osteolytic bone disease. Blood 1999, 94, 3883–3888. [Google Scholar] [CrossRef] [PubMed]
- Vallet, S.; Pozzi, S.; Patel, K.; Vaghela, N.; Fulciniti, M.T.; Veiby, P.; Hideshima, T.; Santo, L.; Cirstea, D.; Scadden, D.T.; et al. A novel role for CCL3 (MIP-1alpha) in myeloma-induced bone disease via osteocalcin downregulation and inhibition of osteoblast function. Leukemia 2011, 25, 1174–1181. [Google Scholar] [CrossRef]
- Fu, R.; Liu, H.; Zhao, S.; Wang, Y.; Li, L.; Gao, S.; Ruan, E.; Wang, G.; Wang, H.Q.; Song, J.; et al. Osteoblast inhibition by chemokine cytokine ligand3 in myeloma-induced bone disease. Cancer Cell Int. 2014, 14, 132. [Google Scholar] [CrossRef] [Green Version]
- Groen, R.W.; de Rooij, M.F.; Kocemba, K.A.; Reijmers, R.M.; de Haan-Kramer, A.; Overdijk, M.B.; Aalders, L.; Rozemuller, H.; Martens, A.C.; Bergsagel, P.L.; et al. N-cadherin-mediated interaction with multiple myeloma cells inhibits osteoblast differentiation. Haematologica 2011, 96, 1653–1661. [Google Scholar] [CrossRef] [Green Version]
- Hiasa, M.; Teramachi, J.; Oda, A.; Amachi, R.; Harada, T.; Nakamura, S.; Miki, H.; Fujii, S.; Kagawa, K.; Watanabe, K.; et al. Pim-2 kinase is an important target of treatment for tumor progression and bone loss in myeloma. Leukemia 2015, 29, 207–217. [Google Scholar] [CrossRef]
- Brunetti, G.; Oranger, A.; Mori, G.; Specchia, G.; Rinaldi, E.; Curci, P.; Zallone, A.; Rizzi, R.; Grano, M.; Colucci, S. Sclerostin is overexpressed by plasma cells from multiple myeloma patients. Ann. N. Y. Acad. Sci. 2011, 1237, 19–23. [Google Scholar] [CrossRef]
- Terpos, E.; Christoulas, D.; Katodritou, E.; Bratengeier, C.; Gkotzamanidou, M.; Michalis, E.; Delimpasi, S.; Pouli, A.; Meletis, J.; Kastritis, E.; et al. Elevated circulating sclerostin correlates with advanced disease features and abnormal bone remodeling in symptomatic myeloma: Reduction post-bortezomib monotherapy. Int. J. Cancer 2012, 131, 1466–1471. [Google Scholar] [CrossRef]
- Oshima, T.; Abe, M.; Asano, J.; Hara, T.; Kitazoe, K.; Sekimoto, E.; Tanaka, Y.; Shibata, H.; Hashimoto, T.; Ozaki, S.; et al. Myeloma cells suppress bone formation by secreting a soluble Wnt inhibitor, sFRP-2. Blood 2005, 106, 3160–3165. [Google Scholar] [CrossRef] [Green Version]
- Mukai, T.; Otsuka, F.; Otani, H.; Yamashita, M.; Takasugi, K.; Inagaki, K.; Yamamura, M.; Makino, H. TNF-alpha inhibits BMP-induced osteoblast differentiation through activating SAPK/JNK signaling. Biochem. Biophys. Res. Commun. 2007, 356, 1004–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamik, J.; Jin, S.; Sun, Q.; Zhang, P.; Weiss, K.R.; Anderson, J.L.; Silbermann, R.; Roodman, G.D.; Galson, D.L. EZH2 or HDAC1 Inhibition Reverses Multiple Myeloma-Induced Epigenetic Suppression of Osteoblast Differentiation. Mol. Cancer Res. 2017, 15, 405–417. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, K.; Abe, M.; Hiasa, M.; Oda, A.; Amou, H.; Kido, S.; Harada, T.; Tanaka, O.; Miki, H.; Nakamura, S.; et al. Tgf-Beta inhibition restores terminal osteoblast differentiation to suppress myeloma growth. PLoS ONE 2010, 5, e9870. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, R.D.; Yang, Y. Syndecan-1: A dynamic regulator of the myeloma microenvironment. Clin. Exp. Metastasis. 2008, 25, 149–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, B.Z. Adhesion molecules--The lifelines of multiple myeloma cells. Semin. Cancer Biol. 2010, 20, 186–195. [Google Scholar] [CrossRef]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Dimopoulos, M.A. Pathogenesis of bone disease in multiple myeloma: From bench to bedside. Blood Cancer J. 2018, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Schwarzer, R.; Nickel, N.; Godau, J.; Willie, B.M.; Duda, G.N.; Schwarzer, R.; Cirovic, B.; Leutz, A.; Manz, R.; Bogen, B.; et al. Notch pathway inhibition controls myeloma bone disease in the murine MOPC315.BM model. Blood Cancer J. 2014, 4, e217. [Google Scholar] [CrossRef]
- Terpos, E.; Zamagni, E.; Lentzsch, S.; Drake, M.T.; Garcia-Sanz, R.; Abildgaard, N.; Ntanasis-Stathopoulos, I.; Schjesvold, F.; de la Rubia, J.; Kyriakou, C.; et al. Treatment of multiple myeloma-related bone disease: Recommendations from the Bone Working Group of the International Myeloma Working Group. Lancet Oncol. 2021, 22, e119–e130. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, C.; Trotter, T.N.; Gowda, P.S.; Lu, Y.; Ponnazhagan, S.; Javed, A.; Li, J.; Yang, Y. Runx2 Deficiency in Osteoblasts Promotes Myeloma Progression by Altering the Bone Microenvironment at New Bone Sites. Cancer Res. 2020, 80, 1036–1048. [Google Scholar] [CrossRef] [Green Version]
- Trotter, T.N.; Li, M.; Pan, Q.; Peker, D.; Rowan, P.D.; Li, J.; Zhan, F.; Suva, L.J.; Javed, A.; Yang, Y. Myeloma cell-derived Runx2 promotes myeloma progression in bone. Blood 2015, 125, 3598–3608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Liu, H.; Li, Y.; Shao, Q.; Chen, J.; Song, J.; Fu, R. Multiple myeloma-derived exosomes inhibit osteoblastic differentiation and improve IL-6 secretion of BMSCs from multiple myeloma. J. Investig. Med. 2020, 68, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, H.; He, J.; Lin, P.; Tong, Q.; Yang, J. Myeloma cells shift osteoblastogenesis to adipogenesis by inhibiting the ubiquitin ligase MURF1 in mesenchymal stem cells. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef]
- Adamik, J.; Roodman, G.D.; Galson, D.L. Epigenetic-Based Mechanisms of Osteoblast Suppression in Multiple Myeloma Bone Disease. JBMR Plus. 2019, 3, e10183. [Google Scholar] [CrossRef] [PubMed]
- Adamik, J.; Galson, D.L.; Roodman, G.D. Osteoblast suppression in multiple myeloma bone disease. J. Bone Oncol. 2018, 13, 62–70. [Google Scholar] [CrossRef]
- Silvestris, F.; Cafforio, P.; Tucci, M.; Grinello, D.; Dammacco, F. Upregulation of osteoblast apoptosis by malignant plasma cells: A role in myeloma bone disease. Br. J. Haematol. 2003, 122, 39–52. [Google Scholar] [CrossRef]
- Croucher, P.I.; Shipman, C.M.; Lippitt, J.; Perry, M.; Asosingh, K.; Hijzen, A.; Brabbs, A.C.; van Beek, E.J.R.; Holen, I.; Skerry, T.M.; et al. Osteoprotegerin inhibits the development of osteolytic bone disease in multiple myeloma. Blood 2001, 98, 3534–3540. [Google Scholar] [CrossRef] [Green Version]
- Vanderkerken, K.; De Leenheer, E.; Shipman, C.; Asosingh, K.; Willems, A.; Van Camp, B.; Croucher, P. Recombinant osteoprotegerin decreases tumor burden and increases survival in a murine model of multiple myeloma. Cancer Res. 2003, 63, 287–289. [Google Scholar]
- Seidel, C.; Hjertner, O.; Abildgaard, N.; Heickendorff, L.; Hjorth, M.; Westin, J.; Lanng Nielsen, J. Serum osteoprotegerin levels are reduced in patients with multiple myeloma with lytic bone disease. Blood 2001, 98, 2269–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albers, J.; Keller, J.; Baranowsky, A.; Beil, F.T.; Catala-Lehnen, P.; Schulze, J.; Amling, M.; Schinke, T. Canonical Wnt signaling inhibits osteoclastogenesis independent of osteoprotegerin. J. Cell Biol. 2013, 200, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Spaan, I.; Raymakers, R.A.; van de Stolpe, A.; Peperzak, V. Wnt signaling in multiple myeloma: A central player in disease with therapeutic potential. J. Hematol. Oncol. 2018, 11, 67. [Google Scholar] [CrossRef]
- Van Andel, H.; Kocemba, K.A.; Spaargaren, M.; Pals, S.T. Aberrant Wnt signaling in multiple myeloma: Molecular mechanisms and targeting options. Leukemia 2019, 33, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Bolzoni, M.; Donofrio, G.; Storti, P.; Guasco, D.; Toscani, D.; Lazzaretti, M.; Bonomini, S.; Agnelli, L.; Capocefalo, A.; Dalla Palma, B.; et al. Myeloma cells inhibit non-canonical wnt co-receptor ror2 expression in human bone marrow osteoprogenitor cells: Effect of wnt5a/ror2 pathway activation on the osteogenic differentiation impairment induced by myeloma cells. Leukemia 2013, 27, 451–463. [Google Scholar] [CrossRef]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef]
- Kenkre, J.S.; Bassett, J. The bone remodelling cycle. Ann. Clin. Biochem. 2018, 55, 308–327. [Google Scholar] [CrossRef] [PubMed]
- Westendorf, J.J.; Kahler, R.A.; Schroeder, T.M. Wnt signaling in osteoblasts and bone diseases. Gene 2004, 341, 19–39. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Kypta, R. Secreted antagonists of the Wnt signalling pathway. J. Cell Sci. 2003, 116 Pt 13, 2627–2634. [Google Scholar] [CrossRef] [Green Version]
- Itasaki, N.; Jones, C.M.; Mercurio, S.; Rowe, A.; Domingos, P.M.; Smith, J.C.; Krumlauf, B. Wise, a context-dependent activator and inhibitor of Wnt signalling. Development 2003, 130, 4295–4305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Meng, S.; Song, H.; Claret, F.X. Dickkopf-1 is a key regulator of myeloma bone disease: Opportunities and challenges for therapeutic intervention. Blood Rev. 2013, 27, 261–267. [Google Scholar] [CrossRef] [Green Version]
- Mao, B.; Wu, W.; Davidson, G.; Marhold, J.; Li, M.; Mechler, B.M.; Delius, H.; Hoppe, D.; Stannek, P.; Walter, C.; et al. Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature 2002, 417, 664–667. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Joiner, D.M.; Oyserman, S.M.; Sharma, P.; Goldstein, S.A.; He, X.; Hauschka, P.V. Bone mass is inversely proportional to Dkk1 levels in mice. Bone 2007, 41, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Robbiani, D.F.; Chesi, M.; Bergsagel, P.L. Bone lesions in molecular subtypes of multiple myeloma. N. Engl. J. Med. 2004, 351, 197–198. [Google Scholar] [CrossRef]
- Qiang, Y.W.; Chen, Y.; Stephens, O.; Brown, N.; Chen, B.; Epstein, J.; Barlogie, B.; Shaughnessy, J.D., Jr. Myeloma-derived Dickkopf-1 disrupts Wnt-regulated osteoprotegerin and RANKL production by osteoblasts: A potential mechanism underlying osteolytic bone lesions in multiple myeloma. Blood 2008, 112, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Yaccoby, S.; Ling, W.; Zhan, F.; Walker, R.; Barlogie, B.; Shaughnessy, J.D., Jr. Antibody-based inhibition of DKK1 suppresses tumor-induced bone resorption and multiple myeloma growth in vivo. Blood 2007, 109, 2106–2111. [Google Scholar] [CrossRef] [PubMed]
- Yaccoby, S.; Wezeman, M.J.; Zangari, M.; Walker, R.; Cottler-Fox, M.; Gaddy, D.; Ling, W.; Saha, R.; Barlogie, B.; Tricot, G.; et al. Inhibitory effects of osteoblasts and increased bone formation on myeloma in novel culture systems and a myelomatous mouse model. Haematologica 2006, 91, 192–199. [Google Scholar]
- Heath, D.J.; Chantry, A.D.; Buckle, C.H.; Coulton, L.; Shaughnessy, J.D., Jr.; Evans, H.R.; Snowden, J.A.; Stover, D.R.; Vanderkerken, K.; Croucher, P.I. Inhibiting Dickkopf-1 (Dkk1) removes suppression of bone formation and prevents the development of osteolytic bone disease in multiple myeloma. J. Bone Miner. Res. 2009, 24, 425–436. [Google Scholar] [CrossRef]
- Fulciniti, M.; Tassone, P.; Hideshima, T.; Vallet, S.; Nanjappa, P.; Ettenberg, S.A.; Shen, Z.X.; Patel, N.; Tai, Y.-T.; Chauhan, D.; et al. Anti-DKK1 mAb (BHQ880) as a potential therapeutic agent for multiple myeloma. Blood 2009, 114, 371–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, S.P.; Beck, J.T.; Stewart, A.K.; Shah, J.; Kelly, K.R.; Isaacs, R.; Bilic, S.; Sen, S.; Munshi, N.C. A Phase IB multicentre dose-determination study of BHQ880 in combination with anti-myeloma therapy and zoledronic acid in patients with relapsed or refractory multiple myeloma and prior skeletal-related events. Br. J. Haematol. 2014, 167, 366–375. [Google Scholar] [CrossRef]
- Munshi, N. Early Evidence of Anabolic Bone Activity of BHQ880, a Fully Human Anti-DKK1 Neutralizing Antibody: Results of a Phase 2 Study in Previously Untreated Patients with Smoldering Multiple Myeloma At Risk for Progression. Blood 2012, 120, 331. [Google Scholar] [CrossRef]
- Ring, E.S.; Lawson, M.A.; Snowden, J.A.; Jolley, I.; Chantry, A.D. New agents in the Treatment of Myeloma Bone Disease. Calcif. Tissue Int. 2018, 102, 196–209. [Google Scholar] [CrossRef] [Green Version]
- Terpos, E.; Christoulas, D.; Kastritis, E.; Bagratuni, T.; Gavriatopoulou, M.; Roussou, M.; Papatheodorou, A.; Eleutherakis-Papaiakovou, E.; Kanellias, N.; Liakou, C.; et al. High levels of periostin correlate with increased fracture rate, diffuse MRI pattern, abnormal bone remodeling and advanced disease stage in patients with newly diagnosed symptomatic multiple myeloma. Blood Cancer J. 2016, 6, e482. [Google Scholar] [CrossRef]
- Spatz, J.M.; Wein, M.N.; Gooi, J.H.; Qu, Y.; Garr, J.L.; Liu, S.; Barry, K.J.; Uda, Y.; Lai, F.; Dedic, C.; et al. The Wnt Inhibitor Sclerostin Is Up-regulated by Mechanical Unloading in Osteocytes in Vitro. J. Biol. Chem. 2015, 290, 16744–16758. [Google Scholar] [CrossRef] [Green Version]
- Winkler, D.G.; Sutherland, M.K.; Geoghegan, J.C.; Yu, C.; Hayes, T.; Skonier, J.E.; Shpektor, D.; Jonas, M.; Kovacevich, B.R.; Staehling-Hampton, K.; et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003, 22, 6267–6276. [Google Scholar] [CrossRef] [Green Version]
- Colucci, S.; Brunetti, G.; Oranger, A.; Mori, G.; Sardone, F.; Specchia, G.; Rinaldi, E.; Curci, P.; Liso, V.; Passeri, G.; et al. Myeloma cells suppress osteoblasts through sclerostin secretion. Blood Cancer J. 2011, 1, e27. [Google Scholar] [CrossRef] [PubMed]
- McClung, M.R.; Grauer, A.; Boonen, S.; Bolognese, M.A.; Brown, J.P.; Diez-Perez, A.; Langdahl, B.L.; Reginster, J.-Y.; Zanchetta, J.R.; Wasserman, S.M.; et al. Romosozumab in postmenopausal women with low bone mineral density. N. Engl. J. Med. 2014, 370, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Langdahl, B.L.; Libanati, C.; Crittenden, D.B.; Bolognese, M.A.; Brown, J.P.; Daizadeh, N.S.; Dokoupilova, E.; Engelke, K.; Finkelstein, J.S.; Genant, H.K.; et al. Romosozumab (sclerostin monoclonal antibody) versus teriparatide in postmenopausal women with osteoporosis transitioning from oral bisphosphonate therapy: A randomised, open-label, phase 3 trial. Lancet 2017, 390, 1585–1594. [Google Scholar] [CrossRef]
- Lewiecki, E.M.; Blicharski, T.; Goemaere, S.; Lippuner, K.; Meisner, P.D.; Miller, P.D.; Miyauchi, A.; Maddox, J.; Chen, L.; Horlait, S. A Phase 3 Randomized Placebo-controlled Trial to Evaluate Efficacy and Safety of Romosozumab in Men with Osteoporosis. J. Clin. Endocrinol. Metab. 2018, 103, 3183–3193. [Google Scholar] [CrossRef] [Green Version]
- Saag, K.G.; Petersen, J.; Brandi, M.L.; Karaplis, A.C.; Lorentzon, M.; Thomas, T.; Maddox, J.; Fan, M.; Meisner, P.D.; Grauer, A. Romosozumab or Alendronate for Fracture Prevention in Women with Osteoporosis. N. Engl. J. Med. 2017, 377, 1417–1427. [Google Scholar] [CrossRef] [Green Version]
- McColm, J.; Hu, L.; Womack, T.; Tang, C.C.; Chiang, A.Y. Single- and multiple-dose randomized studies of blosozumab, a monoclonal antibody against sclerostin, in healthy postmenopausal women. J. Bone Miner. Res. 2014, 29, 935–943. [Google Scholar] [CrossRef]
- Matsumoto, T.; Abe, M. TGF-beta-related mechanisms of bone destruction in multiple myeloma. Bone 2011, 48, 129–134. [Google Scholar] [CrossRef]
- Urashima, M.; Ogata, A.; Chauhan, D.; Hatziyanni, M.; Vidriales, M.B.; Dedera, D.A.; Schlossman, R.L.; Anderson, K.C. Transforming growth factor-beta1: Differential effects on multiple myeloma versus normal B cells. Blood 1996, 87, 1928–1938. [Google Scholar] [CrossRef] [Green Version]
- Bruns, I.; Cadeddu, R.P.; Brueckmann, I.; Frobel, J.; Geyh, S.; Bust, S.; Fischer, J.C.; Roels, F.; Wilk, C.M.; Schildberg, F.A.; et al. Multiple myeloma-related deregulation of bone marrow-derived CD34(+) hematopoietic stem and progenitor cells. Blood 2012, 120, 2620–2630. [Google Scholar] [CrossRef] [Green Version]
- Geiser, A.G.; Zeng, Q.Q.; Sato, M.; Helvering, L.M.; Hirano, T.; Turner, C.H. Decreased bone mass and bone elasticity in mice lacking the transforming growth factor-beta1 gene. Bone 1998, 23, 87–93. [Google Scholar] [CrossRef]
- Paton-Hough, J.; Tazzyman, S.; Evans, H.; Lath, D.; Down, J.M.; Green, A.C.; Snowden, J.A.; Chantry, A.; Lawson, M.A. Preventing and Repairing Myeloma Bone Disease by Combining Conventional Antiresorptive Treatment with a Bone Anabolic Agent in Murine Models. J. Bone Miner. Res. 2019, 34, 783–796. [Google Scholar] [CrossRef]
- Nyman, J.S.; Merkel, A.R.; Uppuganti, S.; Nayak, B.; Rowland, B.; Makowski, A.J.; Oyajobi, B.O.; Sterling, J.A. Combined treatment with a transforming growth factor beta inhibitor (1D11) and bortezomib improves bone architecture in a mouse model of myeloma-induced bone disease. Bone 2016, 91, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, K.S.; Chen, C.G.; Balooch, G.; Stebbins, E.; McKenna, C.R.; Davis, H.; Niewolna, M.; Peng, X.H.; Nguyen, D.H.N.; Ionova-Martin, S.S.; et al. Pharmacologic inhibition of the TGF-beta type I receptor kinase has anabolic and anti-catabolic effects on bone. PLoS ONE 2009, 4, e5275. [Google Scholar] [CrossRef] [Green Version]
- Green, A.C.; Lath, D.; Hudson, K.; Walkley, B.; Down, J.M.; Owen, R.; Evans, H.R.; Paton-Hough, J.; Reilly, G.C.; Lawson, M.A.; et al. TGFbeta Inhibition Stimulates Collagen Maturation to Enhance Bone Repair and Fracture Resistance in a Murine Myeloma Model. J. Bone Miner. Res. 2019, 34, 2311–2326. [Google Scholar] [CrossRef] [Green Version]
- Lu, A.; Pallero, M.A.; Lei, W.; Hong, H.; Yang, Y.; Suto, M.J.; Murphy-Ullrich, J.E. Inhibition of Transforming Growth Factor-beta Activation Diminishes Tumor Progression and Osteolytic Bone Disease in Mouse Models of Multiple Myeloma. Am. J. Pathol. 2016, 186, 678–690. [Google Scholar] [CrossRef] [Green Version]
- Raje, N.; Vallet, S. Sotatercept, a soluble activin receptor type 2A IgG-Fc fusion protein for the treatment of anemia and bone loss. Curr. Opin. Mol. Ther. 2010, 12, 586–597. [Google Scholar] [PubMed]
- Komrokji, R.; Garcia-Manero, G.; Ades, L.; Prebet, T.; Steensma, D.P.; Jurcic, J.G.; Sekeres, M.; Berdeja, J.; Savona, M.R.; Beyne-Rauzy, O.; et al. Sotatercept with long-term extension for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes: A phase 2, dose-ranging trial. Lancet Haematol. 2018, 5, e63–e72. [Google Scholar] [CrossRef]
- Abdulkadyrov, K.M.; Salogub, G.N.; Khuazheva, N.K.; Sherman, M.L.; Laadem, A.; Barger, R.; Knight, R.; Srinivasan, S.; Terpos, T. Sotatercept in patients with osteolytic lesions of multiple myeloma. Br. J. Haematol. 2014, 165, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, C.; Kizaki, M.; Ikeda, Y. Bone morphogenetic protein (BMP)-2 induces apoptosis in human myeloma cells. Leuk. Lymphoma. 2002, 43, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Seher, A.; Lagler, C.; Stuhmer, T.; Muller-Richter, U.D.A.; Kubler, A.C.; Sebald, W.; Müller, T.D.; Nickel, J. Utilizing BMP-2 muteins for treatment of multiple myeloma. PLoS ONE 2017, 12, e0174884. [Google Scholar] [CrossRef] [Green Version]
- Gooding, S.; Olechnowicz, S.W.Z.; Morris, E.V.; Armitage, A.E.; Arezes, J.; Frost, J.; Repapi, E.; Edwards, J.R.; Ashley, N.; Waugh, C.; et al. Transcriptomic profiling of the myeloma bone-lining niche reveals BMP signalling inhibition to improve bone disease. Nat. Commun. 2019, 10, 4533. [Google Scholar] [CrossRef] [PubMed]
- Paino, T.; Garcia-Gomez, A.; Gonzalez-Mendez, L.; San-Segundo, L.; Hernandez-Garcia, S.; Lopez-Iglesias, A.A.; Algarín, E.M.; Martín-Sánchez, M.; Corbacho, D.; Ortiz-de-Solorzano, C.; et al. The Novel Pan-PIM Kinase Inhibitor, PIM447, Displays Dual Antimyeloma and Bone-Protective Effects, and Potently Synergizes with Current Standards of Care. Clin. Cancer Res. 2017, 23, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Raab, M.S.; Thomas, S.K.; Ocio, E.M.; Guenther, A.; Goh, Y.T.; Talpaz, M.; Hohmann, N.; Zhao, S.; Xiang, F.; Simon, C.; et al. The first-in-human study of the pan-PIM kinase inhibitor PIM447 in patients with relapsed and/or refractory multiple myeloma. Leukemia 2019, 33, 2924–2933. [Google Scholar] [CrossRef] [PubMed]
- Takai, K.; Hara, J.; Matsumoto, K.; Hosoi, G.; Osugi, Y.; Tawa, A.; Okada, S.; Nakamura, T. Hepatocyte growth factor is constitutively produced by human bone marrow stromal cells and indirectly promotes hematopoiesis. Blood 1997, 89, 1560–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grano, M.; Galimi, F.; Zambonin, G.; Colucci, S.; Cottone, E.; Zallone, A.Z.; Comoglio, P.M. Hepatocyte growth factor is a coupling factor for osteoclasts and osteoblasts in vitro. Proc. Natl. Acad. Sci. USA 1996, 93, 7644–7648. [Google Scholar] [CrossRef] [Green Version]
- Seidel, C.; Borset, M.; Turesson, I.; Abildgaard, N.; Sundan, A.; Waage, A. Elevated serum concentrations of hepatocyte growth factor in patients with multiple myeloma. Nord. Myeloma Study Group Blood 1998, 91, 806–812. [Google Scholar]
- Derksen, P.W.; de Gorter, D.J.; Meijer, H.P.; Bende, R.J.; van Dijk, M.; Lokhorst, H.M.; Bloem, A.C.; Spaargaren, M.; Pals, S.T. The hepatocyte growth factor/Met pathway controls proliferation and apoptosis in multiple myeloma. Leukemia 2003, 17, 764–774. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA 1998, 95, 3597–3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, O.; Sabokbar, A.; Pocock, A.; Itonaga, I.; Fujikawa, Y.; Athanasou, N.A. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone 2003, 32, 1–7. [Google Scholar] [CrossRef]
- Giuliani, N.; Colla, S.; Sala, R.; Moroni, M.; Lazzaretti, M.; La Monica, S.; Bonomini, S.; Hojden, M.; Sammarelli, G.; Barillè, S.; et al. Human myeloma cells stimulate the receptor activator of nuclear factor-kappa B ligand (RANKL) in T lymphocytes: A potential role in multiple myeloma bone disease. Blood 2002, 100, 4615–4621. [Google Scholar] [CrossRef] [Green Version]
- Alkharabsheh, O.; Frankel, A.E. Clinical Activity and Tolerability of SL-401 (Tagraxofusp): Recombinant Diphtheria Toxin and Interleukin-3 in Hematologic Malignancies. Biomedicines 2019, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; Das, D.S.; Song, Y.; Macri, V.; Richardson, P.; Brooks, C.L.; Chauhan, D.; Anderson, K.C. A novel agent SL-401 induces anti-myeloma activity by targeting plasmacytoid dendritic cells, osteoclastogenesis and cancer stem-like cells. Leukemia 2017, 31, 2652–2660. [Google Scholar] [CrossRef]
- Hughes, F.J.; Howells, G.L. Interleukin-11 inhibits bone formation in vitro. Calcif. Tissue Int. 1993, 53, 362–364. [Google Scholar] [CrossRef]
- Stromme, O.; Psonka-Antonczyk, K.M.; Stokke, B.T.; Sundan, A.; Arum, C.J.; Brede, G. Myeloma-derived extracellular vesicles mediate HGF/c-Met signaling in osteoblast-like cells. Exp. Cell Res. 2019, 383, 111490. [Google Scholar] [CrossRef]
- Nierste, B.A.; Glackin, C.A.; Kirshner, J. Dkk-1 and IL-7 in plasma of patients with multiple myeloma prevent differentiation of mesenchymal stem cells into osteoblasts. Am. J. Blood Res. 2014, 4, 73–85. [Google Scholar] [PubMed]
- Brunetti, G.; Oranger, A.; Mori, G.; Centonze, M.; Colaianni, G.; Rizzi, R.; Liso, V.; Zallone, A.; Grano, M.; Colucci, S. The formation of osteoclasts in multiple myeloma bone disease patients involves the secretion of soluble decoy receptor 3. Ann. N. Y. Acad. Sci. 2010, 1192, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.; Takeshita, S.; Barker, J.E.; Kanagawa, O.; Ross, F.P.; Teitelbaum, S.L. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Investig. 2000, 106, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, G.; Rizzi, R.; Oranger, A.; Gigante, I.; Mori, G.; Taurino, G.; Mongelli, T.; Colaianni, G.; Di Benedetto, A.; Tamma, R.; et al. LIGHT/TNFSF14 increases osteoclastogenesis and decreases osteoblastogenesis in multiple myeloma-bone disease. Oncotarget 2014, 5, 12950–12967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terpos, E.; Christoulas, D.; Gavriatopoulou, M.; Dimopoulos, M.A. Mechanisms of bone destruction in multiple myeloma. Eur. J. Cancer Care 2017, 26, e12761. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| OIFs | Expressed/Released by | Action | References |
|---|---|---|---|
| Activin A | OBs | Inhibits OB differentiation via SMAD2 | [11,12,13] |
| Dkk-1 | OB, BMSCs, and MPCs | Inhibits Wnt/β-catenin via LRP5/6 binding, increases osteoclastogenesis by reducing OPG | [14,15,16] |
| Gfi1 | BMSC | Inhibits Runx2 expression | [17] |
| HGF | MPCs | Inhibits BMP signalling | [18,19] |
| HPSE | MPCs | Increases DKK1 (to inhibit Wnt signalling) and inhibits Runx2 expression | [20] |
| IL-3 | BM T cells | Inhibits BMP-2 initiated OB differentiation | [21,22] |
| IL-7 | BM T cells in MM | Decreases Runx2/Cbfa1 activity, inhibits OB differentiation/maturation | [23,24,25] |
| IL-11 | Likely BMSCs | Dual role as OIF and OAF | [26] |
| MIP-1α (CCL3) | MPCs and macrophages | Inhibits Runx2 and downregulates Osterix | [27,28] |
| N-cadherin | MPCs | Over expressed in 50% MM patients, inhibits OB differentiation via inhibited Wnt signalling | [29] |
| PIM2 | MPCs, MBSCs, and pre-OBs | Associated with reduced OB function, possibly via BMP2 | [30] |
| Sclerostin | MPCs and OCYs | Inhibits Wnt/β-catenin via LRP5/6 binding, leading to inhibited osteoblastogenesis | [14,31,32] |
| sFRP-2 | MPCs | Inhibits Wnt/β-catenin by altering Wnt/Frizzled binding (decoy receptor), inhibits BMP-2 induced OB differentiation | [33] |
| sFRP-3 | MPCs | Inhibits OB differentiation via BMP-2 | [33] |
| TNF-α | MPCs | Increases rates of mature OB apoptosis, possible due to interactions with Runx2 | [34,35] |
| TGFβ | Bone matrix | Inhibits OB differentiation via Runx and DLX-5 | [36] |
| Pharmaceutical Agents | Development Status | Mechanism | Action |
|---|---|---|---|
| Nitrogen-containing bisphosphonates (e.g., Zoledronate) | Approved in MBD | Inhibit farnesyl diphosphate synthase | Inhibit OCs |
| Non-nitrogen-containing bisphosphonates (e.g., Clodronate) | Approved in MBD | Inhibit ATP-dependent enzymes | Inhibit OCs |
| Denosumab | Approved in MBD | Anti-RANKL monoclonal antibody | Inhibit OCs |
| Romosozumab | Approved in OP, preclinical investigation in MBD | Anti-sclerostin monoclonal antibody | Promote OBs |
| Sotatercept | Phase IIa clinical trial in MBD | Recombinant activin type IIa receptor ligand trap | Promote OBs |
| BHQ880 | Phase Ia and II clinical trials in MBD | Anti-Dkk-1 neutralising antibody | Promote OBs |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrews, R.E.; Brown, J.E.; Lawson, M.A.; Chantry, A.D. Myeloma Bone Disease: The Osteoblast in the Spotlight. J. Clin. Med. 2021, 10, 3973. https://doi.org/10.3390/jcm10173973
Andrews RE, Brown JE, Lawson MA, Chantry AD. Myeloma Bone Disease: The Osteoblast in the Spotlight. Journal of Clinical Medicine. 2021; 10(17):3973. https://doi.org/10.3390/jcm10173973
Chicago/Turabian StyleAndrews, Rebecca E., Janet E. Brown, Michelle A. Lawson, and Andrew D. Chantry. 2021. "Myeloma Bone Disease: The Osteoblast in the Spotlight" Journal of Clinical Medicine 10, no. 17: 3973. https://doi.org/10.3390/jcm10173973
APA StyleAndrews, R. E., Brown, J. E., Lawson, M. A., & Chantry, A. D. (2021). Myeloma Bone Disease: The Osteoblast in the Spotlight. Journal of Clinical Medicine, 10(17), 3973. https://doi.org/10.3390/jcm10173973