
Pili Torti: A Feature of Numerous Congenital and Acquired Conditions

 ,
,  ,
, 
 ,
,
Abstract
1. Introduction
2. Inherited Pili Torti
2.1. Isolated Pili Torti
2.1.1. Early Onset (Ronchese) Type
2.1.2. Late Onset (Beare) Type
2.2. Pili Torti in Genetic Diseases or Syndromes
2.2.1. Menkes Disease
2.2.2. Björnstad Syndrome
2.2.3. Netherton Syndrome
2.2.4. Bazex-Dupré-Christol Syndrome
2.3. Pili Torti in Ectodermal Dysplasias
2.3.1. Rapp-Hodgkin Syndrome
2.3.2. Ankyloblepharon-Ectodermal Defects-Cleft Lip/Palate Syndrome
3. Acquired Pili Torti
3.1. Pili Torti Associated with Cicatricial Alopecias
3.1.1. Pili Torti Associated with Primary Cicatricial Alopecias
Lichen Planopilaris
Frontal Fibrosing Alopecia
Discoid Lupus Erythematosus
Pseudopelade of Brocq
Folliculitis Decalvans
Dissecting Cellulitis
Central Centrifugal Cicatricial Alopecia
3.1.2. Pili Torti Associated with Secondary Cicatricial Alopecias
Traction Alopecia
Linear Scleroderma en Coup de Sabre
3.2. Pili Torti in Non-Cicatricial Alopecias
Pili Torti in Alopecia Areata
3.3. Pili Torti in Malignancies
3.4. Drug-Induced Pili Torti
3.5. Other Secondary Causes of Pili Torti
4. Diagnosis
5. Treatment
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ronchese, F. Twisted Hairs (Pili Torti). Arch. Dermatol. Syphilol. 1932, 26, 98–109. [Google Scholar] [CrossRef]
- Rudnicka, L.; Olszewska, M.; Rakowska, A. Atlas of Trichoscopy, 1st ed.; Springer: London, UK, 2012. [Google Scholar]
- Mirmirani, P.; Samimi, S.S.; Mostow, E. Pili torti: Clinical findings, associated disorders, and new insights into mechanisms of hair twisting. Cutis 2009, 84, 143–147. [Google Scholar]
- Mirmirani, P.; Huang, K.P.; Price, V.H. A practical, algorithmic approach to diagnosing hair shaft disorders. Int. J. Dermatol. 2011, 50, 1–12. [Google Scholar] [CrossRef]
- Yang, J.J.; Cade, K.V.; Rezende, F.C.; Pereira, J.M.; Pegas, J.R. Clinical presentation of pili torti--Case report. An. Bras. Dermatol. 2015, 90, 29–31. [Google Scholar] [CrossRef]
- Maruyama, T.; Toyoda, M.; Kanei, A.; Morohashi, M. Pathogenesis in pili torti: Morphological study. J. Dermatol. Sci. 1994, 7, S5–S12. [Google Scholar] [CrossRef]
- Whiting, D.A. Hair shaft defects. In Disorders of Hair Growth: Diagnosis and Treatment, 2nd ed.; Olsen, E.A., Ed.; McGraw Hill: New York, NY, USA, 2003; pp. 123–175. [Google Scholar]
- Marubashi, Y.; Yanagishita, T.; Muto, J.; Taguchi, N.; Sugiura, K.; Kawamoto, Y.; Akiyama, M.; Watanabe, D. Morphological analyses in fragility of pili torti with Björnstad syndrome. J. Dermatol. 2017, 44, 455–458. [Google Scholar] [CrossRef]
- Rudnicka, L.; Olszewska, M.; Waśkiel, A.; Rakowska, A. Trichoscopy in Hair Shaft Disorders. Dermatol. Clin. 2018, 36, 421–430. [Google Scholar] [CrossRef]
- Rakowska, A.; Slowinska, M.; Kowalska-Oledzka, E.; Rudnicka, L. Trichoscopy in genetic hair shaft abnormalities. J. Dermatol. Case Rep. 2008, 2, 14–20. [Google Scholar] [CrossRef]
- Tosti, A. Dermoscopy of the Hair and Nails, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Rouse, C.; Siegfried, E.; Breer, W.; Nahass, G. Hair and sweat glands in families with hypohidrotic ectodermal dysplasia: Further characterization. Arch. Dermatol. 2004, 140, 850–855. [Google Scholar] [CrossRef]
- Rogers, M. Hair shaft abnormalities: Part I. Australas J. Dermatol. 1995, 36. [Google Scholar] [CrossRef]
- Shapira, S.K.; Neish, A.S.; Pober, B.R. Unknown syndrome in sibs: Pili torti, growth delay, developmental delay, and mild neurological abnormalities. J. Med. Genet. 1992, 29, 509–510. [Google Scholar] [PubMed]
- Sharma, P.; Reichert, M.; Lu, Y.; Markello, T.C.; Adams, D.R.; Steinbach, P.J.; Fuqua, B.K.; Parisi, X.; Kaler, S.G.; Vulpe, C.D.; et al. Biallelic HEPHL1 variants impair ferroxidase activity and cause an abnormal hair phenotype. PLoS Genet. 2019, 15, e1008143. [Google Scholar] [CrossRef] [PubMed]
- Sorge, G.; Pavone, L.; Polizzi, A.; Mauceri, L.; Leonardi, R.M.; Tripi, T.; Opitz, J.M. Another “new” form, the palagonia type of acrofacial dysostosis in a Sicilian family. Am. J. Med. Genet. 1997, 69, 388–394. [Google Scholar] [CrossRef]
- Phillips, M.E.; Barrie, H.; Cream, J.J. Arginosuccinic aciduria with pili torti. J. R. Soc. Med. 1981, 74, 221–222. [Google Scholar] [CrossRef] [PubMed]
- Basel-Vanagaite, L.; Attia, R.; Ishida-Yamamoto, A.; Rainshtein, L.; Ben Amitai, D.; Lurie, R.; Pasmanik-Chor, M.; Indelman, M.; Zvulunov, A.; Saban, S.; et al. Autosomal recessive ichthyosis with hypotrichosis caused by a mutation in ST14, encoding type II transmembrane serine protease matriptase. Am. J. Hum. Genet. 2007, 80, 467–477. [Google Scholar] [CrossRef]
- Yung, A.; Newton-Bishop, J.A. A case of Bazex-Dupre-Christol syndrome associated with multiple genital trichoepitheliomas. Br. J. Dermatol. 2005, 153, 682–684. [Google Scholar] [CrossRef]
- Richards, K.A.; Mancini, A.J. Three members of a family with pili torti and sensorineural hearing loss: The Bjornstad syndrome. J. Am. Acad. Dermatol. 2002, 46, 301–303. [Google Scholar] [CrossRef]
- Petit, A.; Dontenwille, M.M.; Bardon, C.B.; Civatte, J. Pili torti with congenital deafness (Bjornstad’s syndrome)--report of three cases in one family, suggesting autosomal dominant transmission. Clin. Exp. Dermatol. 1993, 18, 94–95. [Google Scholar] [CrossRef]
- Patel, H.P.; Unis, M.E. Pili torti in association with citrullinemia. J. Am. Acad. Dermatol. 1985, 12, 203–206. [Google Scholar] [CrossRef]
- Silengo, M.; Valenzise, M.; Pagliardini, S.; Spada, M. Hair changes in congenital disorders of glycosylation (CDG type 1). Eur. J. Pediatric 2003, 162, 114–115. [Google Scholar] [CrossRef]
- Kurwa, A.R.; Abdel-Aziz, A.H. Pili torti-congenital and acquired. Acta Dermatol. Venereol. 1973, 53, 385–392. [Google Scholar] [PubMed]
- Hoeger, P.; Kinsler, V.; Yan, A.; Harper, J.; Oranje, A.; Bodemer, C.; Larralde, M.; Luk, D.; Mendiratta, V.; Purvis, D. Harper’s Textbook of Pediatric Dermatology, 1st ed.; Wiley-Blackwell: Oxford, UK, 2019. [Google Scholar]
- McMichael, A.J.; Hordinsky, M.K. Hair and Scalp Disorders: Medical, Surgical, and Cosmetic Treatments, 2nd ed.; CRC Press: London, UK, 2018; p. 325. [Google Scholar]
- Crandall, B.F.; Samec, L.; Sparkes, R.S.; Wright, S.W. A familial syndrome of deafness, alopecia, and hypogonadism. J. Pediatric 1973, 82, 461–465. [Google Scholar] [CrossRef]
- Rybojad, M.; Moraillon, I.; Bonafé, J.L.; Cambon, L.; Evrard, P. Pilar dysplasia: An early marker of giant axonal neuropathy. Ann. Dermatol. Venereol. 1998, 125, 892–893. [Google Scholar]
- Schaffer, J.V.; Bazzi, H.; Vitebsky, A.; Witkiewicz, A.; Kovich, O.I.; Kamino, H.; Shapiro, L.S.; Amin, S.P.; Orlow, S.J.; Christiano, A.M. Mutations in the desmoglein 4 gene underlie localized autosomal recessive hypotrichosis with monilethrix hairs and congenital scalp erosions. J. Investig. Dermatol. 2006, 126, 1286–1291. [Google Scholar] [CrossRef]
- Zlotogorski, A.; Marek, D.; Horev, L.; Abu, A.; Ben-Amitai, D.; Gerad, L.; Ingber, A.; Frydman, M.; Reznik-Wolf, H.; Vardy, D.A.; et al. An autosomal recessive form of monilethrix is caused by mutations in DSG4: Clinical overlap with localized autosomal recessive hypotrichosis. J. Investig. Dermatol. 2006, 126, 1292–1296. [Google Scholar] [CrossRef]
- Lurie, R.; Ben-Amitai, D.; Laron, Z. Laron syndrome (primary growth hormone insensitivity): A unique model to explore the effect of insulin-like growth factor 1 deficiency on human hair. Dermatology 2004, 208, 314–318. [Google Scholar] [CrossRef]
- Spiegl, B.; Hundeiker, M. Congenital hereditary hypotrychosis. Generalized autosomal dominant hypotrichosis with pili torti (hypotrichosis congenita hereditaria Marie Unna). Fortschr. Med. 1979, 97, 2018–2022. [Google Scholar]
- Pierini, A.M.; Ortonne, J.P.; Floret, D. Cutaneous manifestations of McCune-Albright syndrome: Report of a case (author’s transl). Ann. Dermatol. Venereol. 1981, 108, 969–976. [Google Scholar]
- Bodemer, C.; Rotig, A.; Rustin, P.; Cormier, V.; Niaudet, P.; Saudubray, J.M.; Rabier, D.; Munnich, A.; de Prost, Y. Hair and skin disorders as signs of mitochondrial disease. Pediatrics 1999, 103, 428–433. [Google Scholar] [CrossRef]
- Srinivas, S.M.; Hiremagalore, R.; Suryanarayan, S.; Budamakuntala, L. Netherton syndrome with pili torti. Int. J. Trichol. 2013, 5, 225–226. [Google Scholar] [CrossRef]
- Kharge, P.; Shanmukhappa, A.; Shivaram, B.; Budamakuntala, L. Comèl–Netherton’s syndrome in siblings. Indian J. Paediatr. Dermatol. 2016, 17. [Google Scholar] [CrossRef]
- Ronce, N.; Moizard, M.P.; Robb, L.; Toutain, A.; Villard, L.; Moraine, C. A C2055T transition in exon 8 of the ATP7A gene is associated with exon skipping in an occipital horn syndrome family. Am. J. Hum. Genet. 1997, 61, 233–238. [Google Scholar] [CrossRef]
- Mevorah, B.; Goldberg, I.; Sprecher, E.; Bergman, R.; Metzker, A.; Luria, R.; Gat, A.; Brenner, S. Olmsted syndrome: Mutilating palmoplantar keratoderma with periorificial keratotic plaques. J. Am. Acad. Dermatol. 2005, 53, S266–S272. [Google Scholar] [CrossRef] [PubMed]
- Mevorah, B.; Orion, E.; De Viragh, P.; Bergman, R.; Gat, A.; Legume, C.; Van Neste, D.J.J.; Brenner, S. Peeling skin syndrome with hair changes. Dermatology 1998, 197, 373–376. [Google Scholar] [CrossRef]
- Miteva, M.; Tosti, A. Dermatoscopy of hair shaft disorders. J. Am. Acad. Dermatol. 2013, 68, 473–481. [Google Scholar] [CrossRef]
- Pietrzak, A.; Bartosinska, J.; Filip, A.A.; Rakowska, A.; Adamczyk, M.; Szumilo, J.; Kanitakis, J. Steatocystoma multiplex with hair shaft abnormalities. J. Dermatol. 2015, 42, 521–523. [Google Scholar] [CrossRef]
- Goulet, O.; Vinson, C.; Roquelaure, B.; Brousse, N.; Bodemer, C.; Cezard, J.P. Syndromic (phenotypic) diarrhea in early infancy. Orphanet J. Rare Dis. 2008, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Tay, C.H. Ichthyosiform erythroderma, hair shaft abnormalities, and mental and growth retardation. A new recessive disorder. Arch. Dermatol. 1971, 104, 4–13. [Google Scholar] [CrossRef]
- Botta, E.; Nardo, T.; Broughton, B.C.; Marinoni, S.; Lehmann, A.R.; Stefanini, M. Analysis of mutations in the XPD gene in Italian patients with trichothiodystrophy: Site of mutation correlates with repair deficiency, but gene dosage appears to determine clinical severity. Am. J. Hum. Genet. 1998, 63, 1036–1048. [Google Scholar] [CrossRef]
- Tonnesen, T.; Kleijer, W.J.; Horn, N. Incidence of Menkes disease. Hum. Genet. 1991, 86, 408–410. [Google Scholar] [CrossRef]
- Horn, N.; Wittung-Stafshede, P. ATP7A-Regulated Enzyme Metalation and Trafficking in the Menkes Disease Puzzle. Biomedicines 2021, 9, 391. [Google Scholar] [CrossRef] [PubMed]
- Hinson, J.T.; Fantin, V.R.; Schönberger, J.; Breivik, N.; Siem, G.; McDonough, B.; Sharma, P.; Keogh, I.; Godinho, R.; Santos, F.; et al. Missense mutations in the BCS1L gene as a cause of the Björnstad syndrome. N. Engl. J. Med. 2007, 356, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Chavanas, S.; Bodemer, C.; Rochat, A.; Hamel-Teillac, D.; Ali, M.; Irvine, A.D.; Bonafe, J.L.; Wilkinson, J.; Taieb, A.; Barrandon, Y.; et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat. Genet. 2000, 25, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Sprecher, E.; Chavanas, S.; DiGiovanna, J.J.; Amin, S.; Nielsen, K.; Prendiville, J.S.; Silverman, R.; Esterly, N.B.; Spraker, M.K.; Guelig, E.; et al. The spectrum of pathogenic mutations in SPINK5 in 19 families with Netherton syndrome: Implications for mutation detection and first case of prenatal diagnosis. J. Investig. Dermatol. 2001, 117, 179–187. [Google Scholar] [CrossRef]
- Krafchik, B.R.; Toole, J.W. What is Netherton’s syndrome? Int. J. Dermatol. 1983, 22, 459–462. [Google Scholar] [CrossRef]
- Smith, D.L.; Smith, J.G.; Wong, S.W.; deShazo, R.D. Netherton’s syndrome: A syndrome of elevated IgE and characteristic skin and hair findings. J. Allergy Clin. Immunol. 1995, 95, 116–123. [Google Scholar] [CrossRef]
- Ancuta, N.; Persa, G.; Caius, S. Netherton syndrome—A small series study. Is there a correlation between atopy manifestations and the presence of multiple hair shaft dystrophies? RoJCED 2017, 4, 28–32. [Google Scholar]
- Schmuth, M.; Martinz, V.; Janecke, A.R.; Fauth, C.; Schossig, A.; Zschocke, J.; Gruber, R. Inherited ichthyoses/generalized Mendelian disorders of cornification. Eur. J. Hum. Genet. 2013, 21, 123–133. [Google Scholar] [CrossRef]
- Bazex, A.; Dupre, A.; Christol, B. Follicular atrophoderma, baso-cellular proliferations and hypotrichosis. Ann. Dermatol. Syphiligr. 1966, 93, 241–254. [Google Scholar]
- Viksnins, P.; Berlin, A. Follicular atrophoderma and basal cell carcinomas: The Bazex syndrome. Arch. Dermatol. 1977, 113, 948–951. [Google Scholar] [CrossRef]
- Bal, E.; Park, H.S.; Belaid-Choucair, Z.; Kayserili, H.; Naville, M.; Madrange, M.; Chiticariu, E.; Hadj-Rabia, S.; Cagnard, N.; Kuonen, F.; et al. Mutations in ACTRT1 and its enhancer RNA elements lead to aberrant activation of Hedgehog signaling in inherited and sporadic basal cell carcinomas. Nat. Med. 2017, 23, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Moreau-Cabarrot, A.; Bonafe, J.L.; Hachich, N.; Jalby, B.C.; Aubert, G.; Rolland, M.; Bazex, J. Follicular atrophoderma, basal cell proliferation and hypotrichosis (Bazex-Dupre-Christol syndrome). A study in 2 families. Ann. Dermatol. Venereol. 1994, 121, 297–301. [Google Scholar] [PubMed]
- Tiodorovic-Zivkovic, D.; Zalaudek, I.; Ferrara, G.; Giorgio, C.M.; Di Nola, K.; Procaccini, E.M.; Argenziano, G. Clinical and dermatoscopic findings in Bazex-Dupre-Christol and Gorlin-Goltz syndromes. J. Am. Acad. Dermatol. 2010, 63, 722–724. [Google Scholar] [CrossRef]
- Itin, P.H. Etiology and pathogenesis of ectodermal dysplasias. Am. J. Med. Genet. A 2014, 164A, 2472–2477. [Google Scholar] [CrossRef]
- Dishop, M.K.; Bree, A.F.; Hicks, M.J. Pathologic changes of skin and hair in ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome. Am. J. Med. Genet. A 2009, 149a, 1935–1941. [Google Scholar] [CrossRef]
- Campos-Domínguez, M.; Feito-Rodríguez, M.; Molina-López, I.; Lucas-Laguna, R.D.; Martínez-Glez, V.; Suárez-Fernández, R. A newmutation of smarcad1 in a case of basan syndrome (congenital milia and lack of fingerprints). In Proceedings of the 13th World Congress of Pediatric Dermatology, Chicago, IL, USA, 6–9 July 2017; p. S18. [Google Scholar]
- Calzavara-Pinton, P.; Carlino, A.; Benetti, A.; De Panfilis, G. Pili torti and onychodysplasia. Report of a previously undescribed hidrotic ectodermal dysplasia. Dermatologica 1991, 182, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Abramovits-Ackerman, W.; Bustos, T.; Simosa-Leon, V.; Fernandez, L.; Ramella, M. Cutaneous findings in a new syndrome of autosomal recessive ectodermal dysplasia with corkscrew hairs. J. Am. Acad. Dermatol. 1992, 27, 917–921. [Google Scholar] [CrossRef]
- Wawrzycki, B.; Pietrzak, A.; Chodorowska, G.; Filip, A.A.; Petit, V.; Rudnicka, L.; Dybiec, E.; Rakowska, A.; Sobczynska-Tomaszewska, A.; Kanitakis, J. Ectrodactyly-ectodermal dysplasia-clefting syndrome with unusual cutaneous vitiligoid and psoriasiform lesions due to a novel single point TP63 gene mutation. Postepy Dermatol. Alergol. 2019, 36, 358–364. [Google Scholar] [CrossRef]
- Bree, A.F.; Grange, D.K.; Hicks, M.J.; Goltz, R.W. Dermatologic findings of focal dermal hypoplasia (Goltz syndrome). Am. J. Med. Genet. C Semin. Med. Genet. 2016, 172c, 44–51. [Google Scholar] [CrossRef]
- Kantaputra, P.; Intachai, W.; Kawasaki, K.; Ohazama, A.; Carlson, B.; Quarto, N.; Pruksachatkun, C.; Chuamanochan, M. Clouston syndrome with pili canaliculi, pili torti, overgrown hyponychium, onycholysis, taurodontism and absence of palmoplantar keratoderma. J. Dermatol. 2020, 47, e230–e232. [Google Scholar] [CrossRef]
- Hirano, S.A.; Mason, A.R.; Salkey, K.; Williams, J.V.; Pariser, D.M. Light microscopic hair shaft analysis in ectodermal dysplasia syndromes. Pediatr Dermatol. 2012, 29, 414–420. [Google Scholar] [CrossRef]
- Sanches, S.; Rebellato, P.R.O.; Fabre, A.B.; Campos, G.L.M. Do you know this syndrome? Clouston syndrome. An. Bras. Dermatol. 2017, 92, 417–418. [Google Scholar] [CrossRef]
- Thoden, C.J.; Ryoppy, S.; Kuitunen, P. Oculodentodigital dysplasia syndrome. Report of four cases. Acta Paediatr. Scand. 1977, 66, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Adamski, H.; Chevrant-Breton, J.; Odent, S.; Patoux-Pibouin, M.; Le Marec, B.; Laudren, A.; Urvoy, M. Hair shaft dysplasia in oculo-dento-digital syndrome. Report of a mother-daughter case. Ann. Dermatol. Venereol. 1994, 121, 694–699. [Google Scholar]
- Leachman, S.A.; Kaspar, R.L.; Fleckman, P.; Florell, S.R.; Smith, F.J.; McLean, W.H.; Lunny, D.P.; Milstone, L.M.; van Steensel, M.A.; Munro, C.S.; et al. Clinical and pathological features of pachyonychia congenita. J. Investig. Dermatol. Symp. Proc. 2005, 10, 3–17. [Google Scholar] [CrossRef]
- Irvine, A.D.; McLean, W.H. Human keratin diseases: The increasing spectrum of disease and subtlety of the phenotype-genotype correlation. Br. J. Dermatol. 1999, 140, 815–828. [Google Scholar] [CrossRef] [PubMed]
- Munro, C.S. Pachyonychia congenita: Mutations and clinical presentations. Br. J. Dermatol. 2001, 144, 929–930. [Google Scholar] [CrossRef] [PubMed]
- Silengo, M.C.; Davi, G.F.; Bianco, R.; Costa, M.; DeMarco, A.; Verona, R.; Franceschini, P. Distinctive hair changes (pili torti) in Rapp-Hodgkin ectodermal dysplasia syndrome. Clin. Genet. 1982, 21, 297–300. [Google Scholar] [CrossRef]
- Salinas, C.F.; Montes, G.M. Rapp-Hodgkin syndrome: Observations on ten cases and characteristic hair changes (pili canaliculi). Birth Defects Orig. Artic. Ser. 1988, 24, 149–168. [Google Scholar] [PubMed]
- Reed, W.B.; Brown, A.C.; Sugarman, G.I.; Schlesinger, L. The REEDS syndrome. Birth Defects Orig. Artic. Ser. 1975, 11, 61–73. [Google Scholar]
- Giorgini, S.; Battini, M.L.; Martinelli, C.; Melli, M.C.; Farella, V.; Policarpi, F. Salamon syndrome: A case report. Ann. Ital. Dermatol. Clin. E Sper. 1992, 46, 217–220. [Google Scholar]
- Szepetiuk, G.; Vanhooteghem, O.; Muller, G.; Stene, J.J.; Nikkels, A.F. Schöpf-Schulz-Passarge syndrome with pili torti: A new association? Eur. J. Dermatol. 2009, 19, 517–518. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, M.; Freire-Maia, N. Trichodysplasia-xeroderma: An autosomal dominant condition. Clin. Genet. 1987, 31, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Rapp, R.S.; Hodgkin, W.E. Anhidrotic ectodermal dysplasia: Autosomal dominant inheritance with palate and lip anomalies. J. Med. Genet. 1968, 5, 269–272. [Google Scholar] [CrossRef]
- Vera-Carbonell, A.; Moya-Quiles, M.R.; Ballesta-Martinez, M.; Lopez-Gonzalez, V.; Bafalliu, J.A.; Guillen-Navarro, E.; Lopez-Exposito, I. Rapp-Hodgkin syndrome and SHFM1 patients: Delineating the p63-Dlx5/Dlx6 pathway. Gene 2012, 497, 292–297. [Google Scholar] [CrossRef]
- Park, S.W.; Yong, S.L.; Martinka, M.; Shapiro, J. Rapp-Hodgkin syndrome: A review of the aspects of hair and hair color. J. Am. Acad. Dermatol. 2005, 53, 729–735. [Google Scholar] [CrossRef]
- Moerman, P.; Fryns, J.P. Ectodermal dysplasia, Rapp-Hodgkin type in a mother and severe ectrodactyly-ectodermal dysplasia-clefting syndrome (EEC) in her child. Am. J. Med. Genet. 1996, 63, 479–481. [Google Scholar] [CrossRef]
- Breslau-Siderius, E.J.; Lavrijsen, A.P.; Otten, F.W.; van der Schroeff, J.G.; Swart, J.G. The Rapp-Hodgkin syndrome. Am. J. Med. Genet. 1991, 38, 107–110. [Google Scholar] [CrossRef]
- Crawford, P.J.M.; Aldred, M.J.; Clarke, A.; Tso, M.S.Y. Rapp-Hodgkin syndrome: An ectodermal dysplasia involving the teeth, hair, nails, and palate. Oral Surg. Oral Med. Oral Pathol. 1989, 67, 50–62. [Google Scholar] [CrossRef]
- Witkop, C.J., Jr.; Brearley, L.J.; Gentry, W.C., Jr. Hypoplastic enamel, onycholysis, and hypohidrosis inherited as an autosomal dominant trait. A review of ectodermal dysplasia syndromes. Oral Surg. Oral Med. Oral Pathol. 1975, 39, 71–86. [Google Scholar] [CrossRef]
- Tosun, G.; Elbay, U. Rapp-Hodgkin syndrome: Clinical and dental findings. J. Clin. Pediatric Dent. 2009, 34, 71–75. [Google Scholar] [CrossRef]
- Fete, M.; vanBokhoven, H.; Clements, S.E.; McKeon, F.; Roop, D.R.; Koster, M.I.; Missero, C.; Attardi, L.D.; Lombillo, V.A.; Ratovitski, E.; et al. International Research Symposium on Ankyloblepharon-Ectodermal Defects-Cleft Lip/Palate (AEC) syndrome. Am. J. Med. Genet. A 2009, 149A, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Karadag Kose, O.; Gulec, A.T. Evaluation of a Handheld Dermatoscope in Clinical Diagnosis of Primary Cicatricial Alopecias. Dermatol. Ther. 2019, 9, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, J.I.; Kim, H.U.; Yun, S.K.; Kim, S.J. Trichoscopic Findings of Hair Loss in Koreans. Ann. Dermatol. 2015, 27, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Batrani, M.; Kubba, A.; Kubba, R. Central centrifugal cicatricial alopecia in Asian scalp: Beyond boundaries and race. In Proceedings of the American Academy of Dermatology 2019 Annual Meeting, Washington, DC, USA, 1–5 March 2019; p. AB179. [Google Scholar]
- Gomez-Quispe, H.; Elena de Las Heras-Alonso, M.; Lobato-Berezo, A.; Velasco-Tamariz, V.; Pindado-Ortega, C.; Moreno-Arrones, O.M.; Vano-Galvan, S.; Saceda-Corralo, D. Trichoscopic findings of discoid lupus erythematosus alopecia: A cross-sectional study. J. Am. Acad. Dermatol. 2021, 84, 804–806. [Google Scholar] [CrossRef]
- Karadağ Köse, Ö.; Borlu, M. Evaluation of trichoscopic findings of tractional alopecia. Türkiye Klin. Derm. Derg. 2019, 29, 7–14. [Google Scholar] [CrossRef]
- Montoya, C.L.; Calvache, N. Linear Morphea Alopecia: New Trichoscopy Findings. Int. J. Trichol. 2017, 9, 92–93. [Google Scholar] [CrossRef]
- Saceda-Corralo, D.; Tosti, A. Trichoscopic Features of Linear Morphea on the Scalp. Ski. Appendage Disord. 2018, 4, 31–33. [Google Scholar] [CrossRef]
- Svigos, K.; Criscito, M.; Marji, J.; Brinster, N.K.; Lo Sicco, K. Linear morphea with evidence of hair regrowth. In Proceedings of the SID 2020 Annual Meeting, Virtual Meeting, 13–16 May 2020; p. S9. [Google Scholar]
- Gajda, P.; Rakowska, A.; Czuwara, J.; Samochocki, Z. Ogniska łysienia jako pierwszy objaw wznowy raka sutka? Dermatol. Rev. 2018, 105, 682–683. [Google Scholar] [CrossRef]
- Rakowska, A.; Jasińska, M.; Sikora, M.; Czuwara, J.; Gajda-Mróz, P.; Warszawik-Hendzel, O.; Kwiatkowska, M.; Waśkiel-Burnat, A.; Olszewska, M.; Rudnicka, L. Cutaneous T-cell lymphoma in erythrodermic cases may be suspected on the basis of scalp examination with dermoscopy. Sci. Rep. 2021, 11, 282. [Google Scholar] [CrossRef]
- Gold, S.C.; Delaney, T.J. Familial acne conglobata, hidradenitis suppurativa, pili torti and cataracts*. Br. J. Dermatol. 1974, 91, 54–57. [Google Scholar] [CrossRef]
- Lurie, R.; Danziger, Y.; Kaplan, Y.; Sulkes, J.; Abramson, E.; Mimouni, M. Acquired pili torti—A structural hair shaft defect in anorexia nervosa. Cutis 1996, 57, 151–156. [Google Scholar]
- Strumia, R.; Borghi, A.; Colombo, E.; Manzato, E.; Gualandi, M. Low prevalence of twisted hair in anorexia nervosa. Clin. Exp. Dermatol. 2005, 30, 349–350. [Google Scholar] [CrossRef] [PubMed]
- Penzi, L.R.; Saavedra, A.; Senna, M.M. Long-standing pili torti in 2 patients with chronic graft-vs.-host disease. Jaad Case Rep. 2018, 4, 44–46. [Google Scholar] [CrossRef]
- Evans, J.B.; Hastings, J.G.; Kaffenberger, B.H. Acquired Pili Torti. Jama Dermatol. 2019, 155, 488. [Google Scholar] [CrossRef] [PubMed]
- Kremer, N.; Martinez, H.; Leshem, Y.A.; Hodak, E.; Zer, A.; Brenner, B.; Amitay-Laish, I. The trichoscopic features of hair shaft anomalies induced by epidermal growth factor receptor inhibitors: A case series. J. Am. Acad. Dermatol. 2020, (in press). [CrossRef]
- Pirmez, R.; Piñeiro-Maceira, J.; Gonzalez, C.G.; Miteva, M. Loose Anchoring of Anagen Hairs and Pili Torti due to Erlotinib. Int. J. Trichol. 2016, 8, 186–187. [Google Scholar] [CrossRef]
- Hays, S.B.; Camisa, C. Acquired pili torti in two patients treated with synthetic retinoids. Cutis 1985, 35, 466–468. [Google Scholar]
- Caneppele, S.; Mazereeuw-Hautier, J.; Bonafé, J.L. Sodium valproate-induced kinky hair syndrome. Ann. Dermatol. Venereol. 2001, 128, 134–135. [Google Scholar]
- Bolck, F.; Ziegler, V.; Sieler, H. Bleaching of hair by carbamide perhydrate. Contact Dermat. 1977, 3, 214–215. [Google Scholar] [CrossRef]
- Kanti, V.; Röwert-Huber, J.; Vogt, A.; Blume-Peytavi, U. Cicatricial alopecia. Jddg J. Dtsch. Dermatol. Ges. 2018, 16, 435–461. [Google Scholar] [CrossRef] [PubMed]
- Errichetti, E.; Figini, M.; Croatto, M.; Stinco, G. Therapeutic management of classic lichen planopilaris: A systematic review. Clin. Cosmet. Investig. Dermatol. 2018, 11, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Eftekhari, H.; Azimi, S.Z.; Rafiei, R.; Darjani, A.; Alizadeh, N.; Rafiei, E.; Ghadarjani, R.; Gharaei Nejad, K. Dermoscopic features of lichen planopilaris in Northern Iran: A prospective observational study. Int. J. Dermatol. 2019, 58, 1406–1414. [Google Scholar] [CrossRef]
- Waśkiel, A.; Rakowska, A.; Sikora, M.; Olszewska, M.; Rudnicka, L. Obraz trichoskopowy liszaja płaskiego mieszkowego. Dermatol. Rev. 2018, 105, 63–75. [Google Scholar] [CrossRef]
- Kossard, S. Postmenopausal frontal fibrosing alopecia. Scarring alopecia in a pattern distribution. Arch. Dermatol. 1994, 130, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Fanti, P.A.; Baraldi, C.; Misciali, C.; Piraccini, B.M. Cicatricial alopecia. G Ital. Dermatol. Venereol 2018, 153, 230–242. [Google Scholar] [CrossRef]
- Ferrari, B.; Vincenzi, C.; Tosti, A. Pili Torti as a Sign of Eyebrow Involvement in Frontal Fibrosing Alopecia. Ski. Appendage Disord. 2019, 5, 393–395. [Google Scholar] [CrossRef]
- Inui, S.; Nakajima, T.; Shono, F.; Itami, S. Dermoscopic findings in frontal fibrosing alopecia: Report of four cases. Int. J. Dermatol. 2008, 47, 796–799. [Google Scholar] [CrossRef]
- Rudnicka, L.; Olszewska, M.; Rakowska, A.; Slowinska, M. Trichoscopy update 2011. J. Dermatol. Case Rep. 2011, 5, 82–88. [Google Scholar] [CrossRef]
- Qi, S.; Zhao, Y.; Zhang, X.; Li, S.; Cao, H.; Zhang, X. Clinical features of primary cicatricial alopecia in Chinese patients. Indian J. Dermatol. Venereol. Leprol. 2014, 80, 306–312. [Google Scholar] [CrossRef]
- Mathur, M.; Acharya, P. Trichoscopy of primary cicatricial alopecias: An updated review. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 473–484. [Google Scholar] [CrossRef]
- Saceda-Corralo, D.; Moreno-Arrones, O.M.; Rodrigues-Barata, R.; Rubio-Lombrana, M.; Mir-Bonafe, J.F.; Morales-Raya, C.; Miguel-Gomez, L.; Hermosa-Gelbard, A.; Jaen-Olasolo, P.; Vano-Galvan, S. Trichoscopy activity scale for folliculitis decalvans. J. Eur. Acad. Dermatol. Venereol. 2020, 34, e55–e57. [Google Scholar] [CrossRef]
- Segurado-Miravalles, G.; Camacho-Martinez, F.M.; Arias-Santiago, S.; Serrano-Falcon, C.; Serrano-Ortega, S.; Rodrigues-Barata, R.; Jaen Olasolo, P.; Vano-Galvan, S. Epidemiology, clinical presentation and therapeutic approach in a multicentre series of dissecting cellulitis of the scalp. J. Eur. Acad. Dermatol. Venereol. 2017, 31, e199–e200. [Google Scholar] [CrossRef]
- Abedini, R.; Kamyab Hesari, K.; Daneshpazhooh, M.; Ansari, M.S.; Tohidinik, H.R.; Ansari, M. Validity of trichoscopy in the diagnosis of primary cicatricial alopecias. Int. J. Dermatol. 2016, 55, 1106–1114. [Google Scholar] [CrossRef]
- Ogunleye, T.A.; McMichael, A.; Olsen, E.A. Central centrifugal cicatricial alopecia: What has been achieved, current clues for future research. Dermatol. Clin. 2014, 32, 173–181. [Google Scholar] [CrossRef]
- Herskovitz, I.; Miteva, M. Central centrifugal cicatricial alopecia:Challenges and solutions. Clin. Cosmet. Investig. Dermatol. 2016, 9, 175–181. [Google Scholar] [CrossRef]
- Munoz Moreno-Arrones, O.; Vano-Galvan, S. Bitemporal hair loss related to traction alopecia. Dermatol. Online J. 2016, 22, 16. [Google Scholar] [CrossRef]
- Simakou, T.; Butcher, J.P.; Reid, S.; Henriquez, F.L. Alopecia areata: A multifactorial autoimmune condition. J. Autoimmun. 2019, 98, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Amberg, N.; Sotiropoulou, P.A.; Heller, G.; Lichtenberger, B.M.; Holcmann, M.; Camurdanoglu, B.; Baykuscheva-Gentscheva, T.; Blanpain, C.; Sibilia, M. EGFR Controls Hair Shaft Differentiation in a p53-Independent Manner. iScience 2019, 15, 243–256. [Google Scholar] [CrossRef]
- Duverger, O.; Morasso, M.I. Role of homeobox genes in the patterning, specification, and differentiation of ectodermal appendages in mammals. J. Cell. Physiol. 2008, 216, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Alting, K.; van Hunsel, F. Curling of Hair in Two Female Patients Taking Alitretinoin. Drug Saf. Case Rep. 2018, 5, 26. [Google Scholar] [CrossRef]
- Yasemin, G. Curly Hair Induced by Valproate in Bipolar Disorder. Clin. Psychopharmacol. Neurosci. 2016, 14, 114. [Google Scholar] [CrossRef][Green Version]
- Rakowska, A.; Górska, R.; Rudnicka, L.; Zadurska, M. Trichoscopic Hair Evaluation in Patients with Ectodermal Dysplasia. J. Pediatric 2015, 167, 193–195. [Google Scholar] [CrossRef]
- Waśkiel, A.; Rakowska, A.; Sikora, M.; Olszewska, M.; Rudnicka, L. Trichoscopy of alopecia areata: An update. J. Dermatol. 2018, 45, 692–700. [Google Scholar] [CrossRef]
- Gelles, L.N. Picture of the month. Pili torti. Arch. Pediatric Adolesc. Med. 1999, 153, 647–648. [Google Scholar] [CrossRef][Green Version]



{kind=link}
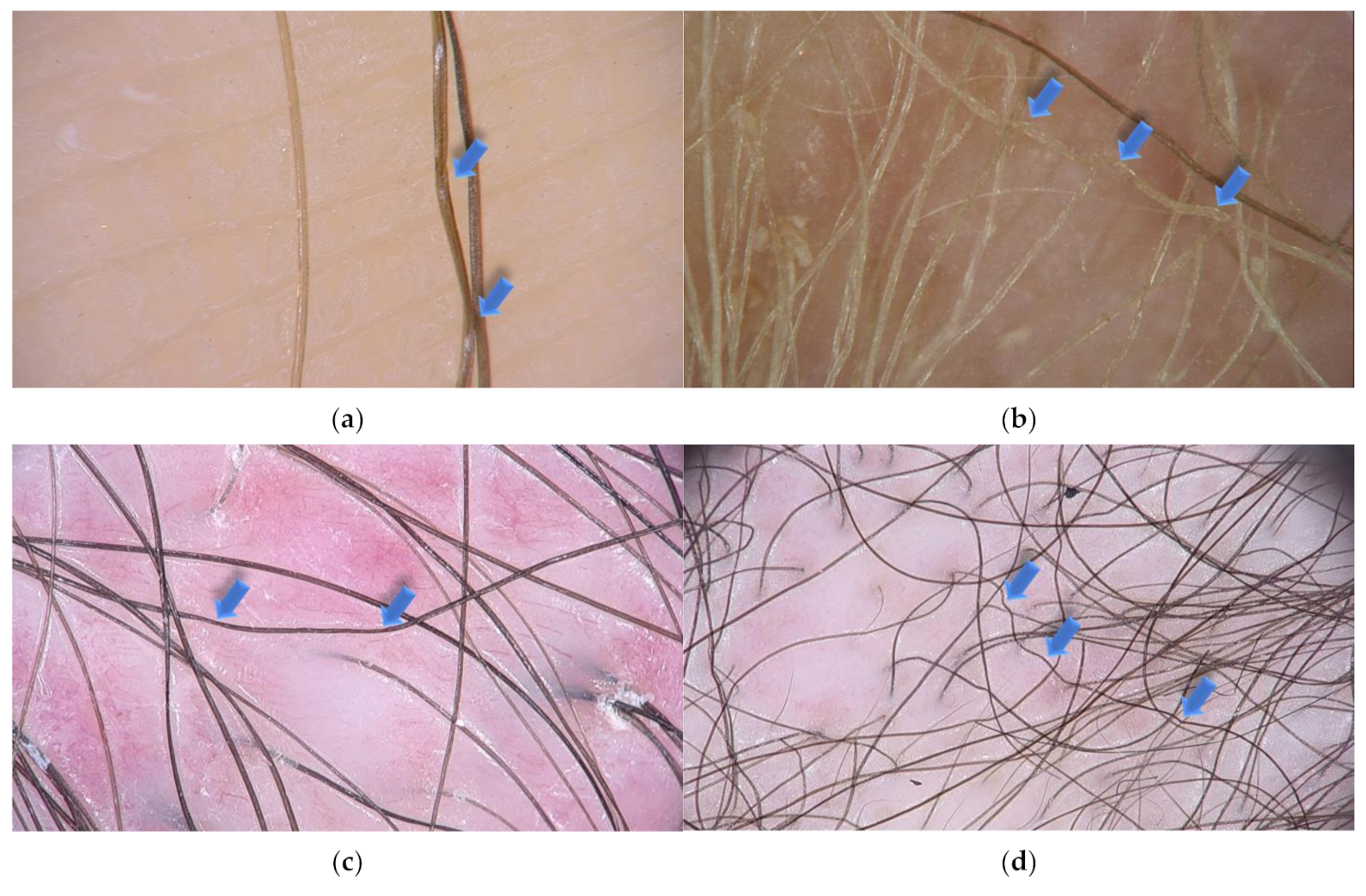{kind=link}
| Disease/Syndrome | Genetic Defect, Inheritance | Other Clinical Findings | Other Hair Shaft Abnormalities |
|---|---|---|---|
| Abnormal hair, joint laxity, and developmental delay [14,15] | HEPHL1 gene (AR) | growth and developmental delay, joint laxity, neurologic abnormalities | trichorrhexis nodosa |
| Acrofacial dysostosis, Palagonia type [16] | NK (AD) | short stature, vertebral anomalies, syndactyly, oligodontia, cleft lip | - |
| Arginosuccinic aciduria [17] | ASL gene (AR) | lethargy, vomiting, seizures, cerebral edema, hepatomegaly | trichorrhexis nodosa |
| Autosomal recessive ichthyosis with hypotrichosis [18] | ST14 gene (AR) | lamellar ichthyosis, follicular atrophoderma, hypohidrosis | dysplastic hair, pili bifurcati |
| Bazex-Dupre-Christol syndrome [19] | ACTRT1 gene (XD) | follicular atrophoderma, multiple basal cell carcinomas, milia, hypohidrosis | trichorrhexis nodosa |
| Björnstad syndrome [20,21] | BCS1L gene (AR, AD) | sensorineural hearing loss | - |
| Citrullinemia [22] | ASS1 gene (AR) | hyperammonemia, lethargy, poor feeding, vomiting, high intracranial pressure, scaly skin eruption | trichorrhexis nodosa |
| Congenital disorder of glycosylation, type Ia [23] | PMM2 gene (AR) | hypotonia, strabismus, cerebellar hypoplasia, seizures, mental and physical retardation, hepatomegaly, liver fibrosis, fat pads, ‘orange peel’ skin | trichorrhexis nodosa |
| Congenital erythropoietic porphyria [24] | UROS gene (AR) | severe skin photosensitivity (scarring, blistering), erythrodontia, reddish-colored urine, anemia | - |
| Congenital hypotrichosis with juvenile macular dystrophy [25] | CDH3 gene (AR) | retinal degeneration | - |
| Conradi-Hünermann syndrome [26] | EBP gene (XD) | short stature, asymmetric short limbs, vertebral malformations, hip dysplasia, chondrodysplasia punctata, follicular atrophoderma, abnormal nails, craniofacial anomalies, cataracts | - |
| Crandall syndrome [27] | NK (AR) | neurosensory deafness, hypogonadism with decreased levels of luteinizing hormone and growth hormone | - |
| Giant axonal neuropathy [28] | GAN gene (AR) | progressive sensorimotor peripheral neuropathy, axonal loss, optic atrophy, ophthalmoplegia, skeletal deformations | trichorrhexis nodosa |
| Hypotrichosis 6 [29,30] | DSG4 gene (AR) | hyperkeratotic follicular papules, erythema, scaling, dry skin | monilethrix-like hair, trichoschisis, trichorrhexis nodosa-like defects, tapered hair |
| Laron syndrome [31] | GHR gene (AR) | short stature, obesity, facial dysmorphism, hypogenitalism, elevated serum growth hormone, undetectable or low serum insulin-like growth factor 1 | - |
| Marie Unna hypotrichosis [32] | U2HR gene (AD) | - | - |
| McCune-Albright syndrome [33] | GNAS1 gene (not inherited) | polyostotic fibrous dysplasia, cafe-au-lait skin pigmentation, multiple endocrine dysfunction | - |
| Menkes disease [5] | ATP7A gene (XR) | growth retardation, vascular, neurological, and skeletal abnormalities, pale skin | trichoclasis, trichorrhexis nodosa, trichoptilosis |
| Mitochondrial diseases [34] | all modes of inheritance can be expected | mental retardation, failure to thrive, hypotonia, hypoparathyroidism | longitudinal grooving with cuticle loss |
| Netherton syndrome [35,36] | SPINK5 gene (AR) | congenital erythroderma, atopic manifestations, increased IgE level | trichorrhexis I vaginata, trichorrhexis nodosa, trichoschisis, trichoptilosis |
| Occipital horn syndrome [37] | ATP7A gene (XR) | tallness, pectus excavatum, dorsal kyphosis, occipital horn exostoses, joint laxity, loose skin, decreased serum copper and ceruloplasmin | - |
| Olmsted syndrome [38] | TRPV3 gene (AD) | constriction of digits (‘pseudoainhum’), mutilating palmoplantar keratoderma, onychodystrophy, periorificial keratotic plaques | trichorrhexis nodosa |
| Peeling skin syndrome [39] | CDSN gene (AR) | superficial patchy peeling of the entire skin, erythroderma, atopy, nail anomalies | trichorrhexis invaginata-like changes, monili-form hair shaft diameter reductions, irregular hair shaft torsions |
| Salti-Salem syndrome [40] | NK (AD) | hypogonadotropic hypogonadism | - |
| Steatocystoma multiplex [41] | KRT17 gene (AD) | subcutaneous cysts | pili canaliculi |
| Tricho-hepato-enteric syndrome [42] | TTC37, SKIV2L genes (AR) | low birth weight, failure to thrive, facial dysmorphism, diarrhoea, liver disease | trichorrhexis nodosa, aniso- and poilkilotrichosis |
| Trichothiodystrophy, photosensitive [43,44] | ERCC2, XPD genes (AR) | mental and physical retardation, short stature, facial dysmorphism, ichthyosis, photosensitivity, ocular abnormalities | Pili annulati (‘tiger-tail’ hair), trichoschisis, trichorrhexis nodosa |
| Disease/Syndrome | Genetic Defect, Inheritance | Other Clinical Findings | Other Hair Shaft Abnormalities |
|---|---|---|---|
| Ankyloblepharon-ectodermal defects-cleft lip and palate syndrome [60] | TP63 gene (AD) | hypoplastic maxilla, palmoplantar hyperkeratosis, dystrophic nails, cleft lip/palate, dental anomalies, ankyloblepharon, lacrimal duct atresia, auricular abnormalities | pili canaliculi, trichoclasis, trichorrhexis nodosa, pili annulati, pili triangulati |
| Basan syndrome [61] | SMARCAD1 gene (AD) | neonatal blisters and milia, adermatoglyphia, traumatic blistering and fissuring, hypohidrosis | - |
| Cleft lip/palate-ectodermal dysplasia syndrome [25] | PVRL1 gene (AR) | mental retardation, facial dysmorphism (protruding and malformed ears, micrognathia, bilateral cleft lip/palate), syndactyly, palmoplantar keratoderma, hypohidrosis, teeth, and nail anomalies | - |
| Ectodermal dysplasia 4, hair/nail type [62] | KRT85, KRT74, HOXC13 genes (AR) | congenital nail dystrophy | - |
| Ectodermal dysplasia with corkscrew hairs [26,63] | NK (AR) | facial dysmorphism, cleft lip/palate, scalp keloids, follicular plugging, keratosis pilaris, xerosis, eczema, palmoplantar keratodermia, cutaneous syndactyly, onychodysplasia, teeth abnormalities | - |
| Ectodermal dysplasia with syndactyly [25] | PVRL-4 gene (AR) | highly arched palate, teeth abnormalities, syndactyly, hypoplastic nails, dry skin with hyperkeratosis | - |
| Ectrodactyly, ectodermal dysplasia, and cleft lip/palate syndrome 3 [64] | EEC3 gene (AD) | hearing loss, cleft lip/palate, dysplastic teeth, ectrodactyly, syndactyly, nail dystrophy, hypopigmentated skin, hyperkeratosis, skin atrophy, genitourinary anomalies | pseudomoniletrix, pili canaliculi, longitudinal grooving, trichothiodystrophy |
| Goltz syndrome [65] | PORCN gene (XD) | cleft palate, syndactyly, polydactyly, skin atrophy, telangiectasia, herniation of fat, papillomas, nail and teeth anomalies, ocular anomalies (coloboma of iris and choroid, strabismus, microphthalmia) | atrophic hair with reduced diameters, flattened hair shafts, trichorrhexis nodosa, pili trianguli et canaliculi |
| Hidrotic ectodermal dysplasia [66,67,68] | GJB6 gene (AD) | short stature, clubbed digits, palmoplantar hyperkeratosis, hyperpigmentation, nail dystrophy, cataract, photophobia, strabismus | trichorrhexis nodosa, trichoptilosis, pili bifurcati, variable diameter, damaged cuticles, irregular helical twists, pili canaliculi |
| Hypohidrotic Ectodermal Dysplasia [12] | EDA1/EDAR, EDARADD, WNT10A genes (XR, AR, AD) | facial dysmorphism (prominent forehead, thick lips, flattened nasal bridge), teeth abnormalities, hypohidrosis | trichorrhexis nodosa, pili bifurcati, variable shaft thickness |
| Hypotrichosis-osteolysis-periodontitis-palmoplantar keratoderma syndrome [25] | NK gene (AD) | onychogryphosis, acroosteolysis, linear or reticular palmoplantar keratoderma and erythematous, psoriasis-like skin lesions, periodontitis, premature teeth loss, lingua plicata, ventricular tachycardia | pili annulati |
| Oculo-dento-digital syndrome [69,70] | GJA1 gene (AD, AR) | facial dysmorphism (narrow, pinched nose, hypoplastic alae nasi, prominent columella, narrow nasal bridge), microphthalmia, microdontia, syndactyly, camptodactyly, clinodactyly, brittle nails | “tiger tail” aspect, monilethrix, pili annulati |
| Pachyonychia congenita-2 [71,72,73] | KRT17 gene (AD) | palmoplantar hyperkeratosis, nail dystrophy, hyperhidrosis, cystic lesions (steatocystoma multiplex, pilosebaceous cysts), folliculitis, natal teeth | - |
| Rapp-Hodgkin syndrome [74,75] | TP63 gene (AD) | short stature, hypohidrosis, facial dysmorphism (narrow nose, small mouth, cleft lip, hypoplastic maxilla, prominent, malformed auricles), dysplastic nails, teeth abnormalities, chronic epiphora | pili canaliculi |
| Reeds syndrome [76] | NK (AD) | lobster claw deformity, nasolacrimal obstruction, cleft lip/palate, teeth abnormalities | - |
| Salamon syndrome [77] | NK (AR) | everted lower lip, teeth abnormalities, protruding ears | - |
| Schöpf-Schulz-Passarge syndrome [78] | WNT10A gene (AR) | Palmoplantar keratoderma, nail dystrophy, hypodontia, eyelid cysts | - |
| Trichodysplasia-xeroderma [79] | NK (AD) | dry skin | trichorrhexis nodosa |
| Conditions Associated with Acquired Pili Torti |
|---|
| lichen planopilaris [89]; |
| frontal fibrosing alopecia [89]; |
| alopecia areata [90]; |
| central centrifugal cicatricial alopecia [91]; |
| discoid lupus erythematosus [89,92]; |
| dissecting cellulitis [89]; |
| folliculitis decalvans [89]; |
| pseudopelade of Brocq [89]; |
| traction alopecia [93]; |
| linear scleroderma en coup de sabre [94,95,96]; |
| repetitive trauma [9]; |
| scalp metastasis of breast cancer [97]; |
| cutaneous T-cell lymphoma [98]; |
| acne conglobate [99]; |
| anorexia nervosa [100,101]; |
| graft-vs.-host disease [102]; |
| hair transplantation [9]; |
| malnutrition [103]; |
| systemic sclerosis [24]; |
| cataracts [99]. |
| Drugs Associated with Pili Torti |
|---|
| epidermal growth factor receptor inhibitors [104,105]; |
| oral retinoids [106]; |
| sodium valproate [107]; |
| carbamide perhydrate [108]. |
| Disease | Epidemiology | Clinical Features | Trichoscopy |
|---|---|---|---|
| Lichen Planopilaris | women 40–60 years of age | multifocal, confluent areas of hair loss with perifollicular hyperkeratosis and erythema at the periphery; the vertex and the parietal area are most commonly affected | perifollicular scaling, hair casts, perifollicular erythema, white dots, white and milky red areas, loss of follicular openings |
| Frontal Fibrosing Alopecia | post-menopausal women | recession of the frontotemporal hairline, eyebrow loss | perifollicular erythema, perifollicular scaling, hair casts, white areas, loss of follicular openings |
| Discoid Lupus Erythematosus | women 20–40 years of age | well-demarcated annular or oval plaques with follicular plugging, erythema, telangiectasia, scaling, dyspigmentation | follicular red dots, large yellow or yellow-brown dots, “red spiders on yellow dots”, scattered brown discoloration, white and milky red areas, loss of follicular openings |
| Pseudopelade of Brocq | middle-aged white women | asymptomatic, asymmetrical, white, or porcelain-white patches involving the vertex or parietal area | white dots, white and milky-red areas, loss of follicular openings, variations in hair diameter |
| Folliculitis Decalvans | young to middle-aged adult men of African descent | tender, recurrent papulo-pustular lesions on the vertex and occipital area | hair tufts consisting of 5–20 hair surrounded by yellowish tubular scaling, starburst sign, coiled capillary loops, white and milky-red areas, loss of follicular openings |
| Dissecting Cellulitis | young men of African descent | perifollicular pustules, painful nodules, abscesses with sinus tracts involving the vertex and occipital area | 3D yellow dots, yellow structureless areas, black dots, pinpoint-like vessels with whitish halo, white areas, loss of follicular openings |
| Central Centrifugal Cicatricial Alopecia | middle-aged women of African descent | scarring hair loss initially involving the vertex or crown of the scalp and slowly progressing peripherally | peripilar gray/white halo, perifollicular scaling, loss of follicular openings |
| Traction Alopecia | women and children of African descent | hair loss and thinning, pustules, inflammatory papules; may progress to scarring alopecia | perifollicular erythema, hair thinning, focal decrease in hair density, honeycomb pattern, pinpoint white dots, irregular white patches |
| Linear Scleroderma en Coup de Sabre | Children and women within the first two decades of life | single erythematous or violaceous linear indurated plaque, progressing to hyperpigmented or hypopigmented streak on the forehead | scattered black dots, broken hairs, short thick linear and branching tortuous vessels on the periphery of the lesion, white areas, loss of follicular openings |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, A.; Waśkiel-Burnat, A.; Żółkiewicz, J.; Blicharz, L.; Rakowska, A.; Goldust, M.; Olszewska, M.; Rudnicka, L. Pili Torti: A Feature of Numerous Congenital and Acquired Conditions. J. Clin. Med. 2021, 10, 3901. https://doi.org/10.3390/jcm10173901
Hoffmann A, Waśkiel-Burnat A, Żółkiewicz J, Blicharz L, Rakowska A, Goldust M, Olszewska M, Rudnicka L. Pili Torti: A Feature of Numerous Congenital and Acquired Conditions. Journal of Clinical Medicine. 2021; 10(17):3901. https://doi.org/10.3390/jcm10173901
Chicago/Turabian StyleHoffmann, Aleksandra, Anna Waśkiel-Burnat, Jakub Żółkiewicz, Leszek Blicharz, Adriana Rakowska, Mohamad Goldust, Małgorzata Olszewska, and Lidia Rudnicka. 2021. "Pili Torti: A Feature of Numerous Congenital and Acquired Conditions" Journal of Clinical Medicine 10, no. 17: 3901. https://doi.org/10.3390/jcm10173901
APA StyleHoffmann, A., Waśkiel-Burnat, A., Żółkiewicz, J., Blicharz, L., Rakowska, A., Goldust, M., Olszewska, M., & Rudnicka, L. (2021). Pili Torti: A Feature of Numerous Congenital and Acquired Conditions. Journal of Clinical Medicine, 10(17), 3901. https://doi.org/10.3390/jcm10173901