
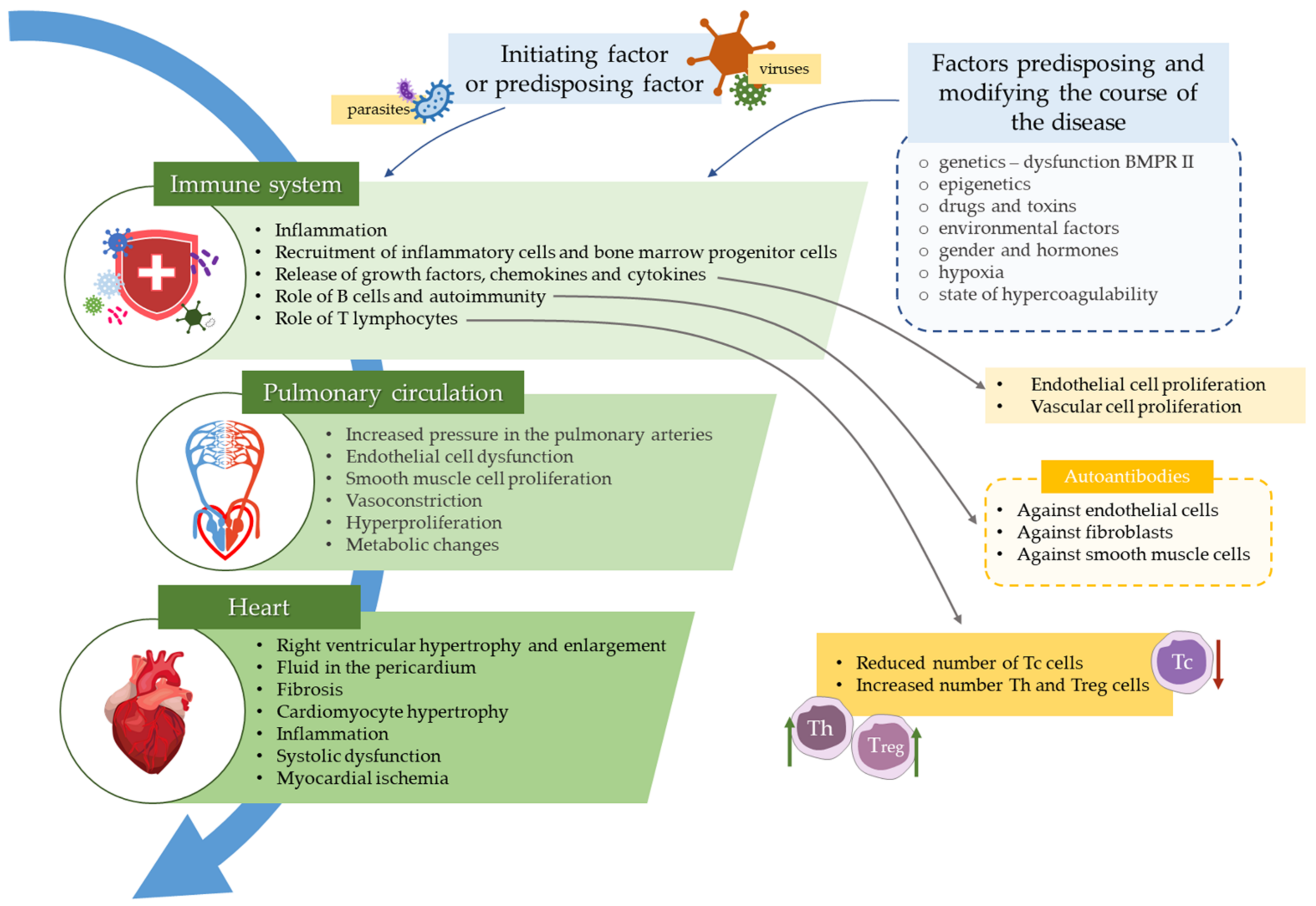
Role of the Immune System Elements in Pulmonary Arterial Hypertension


 ,
,  ,
,  ,
,  and
and 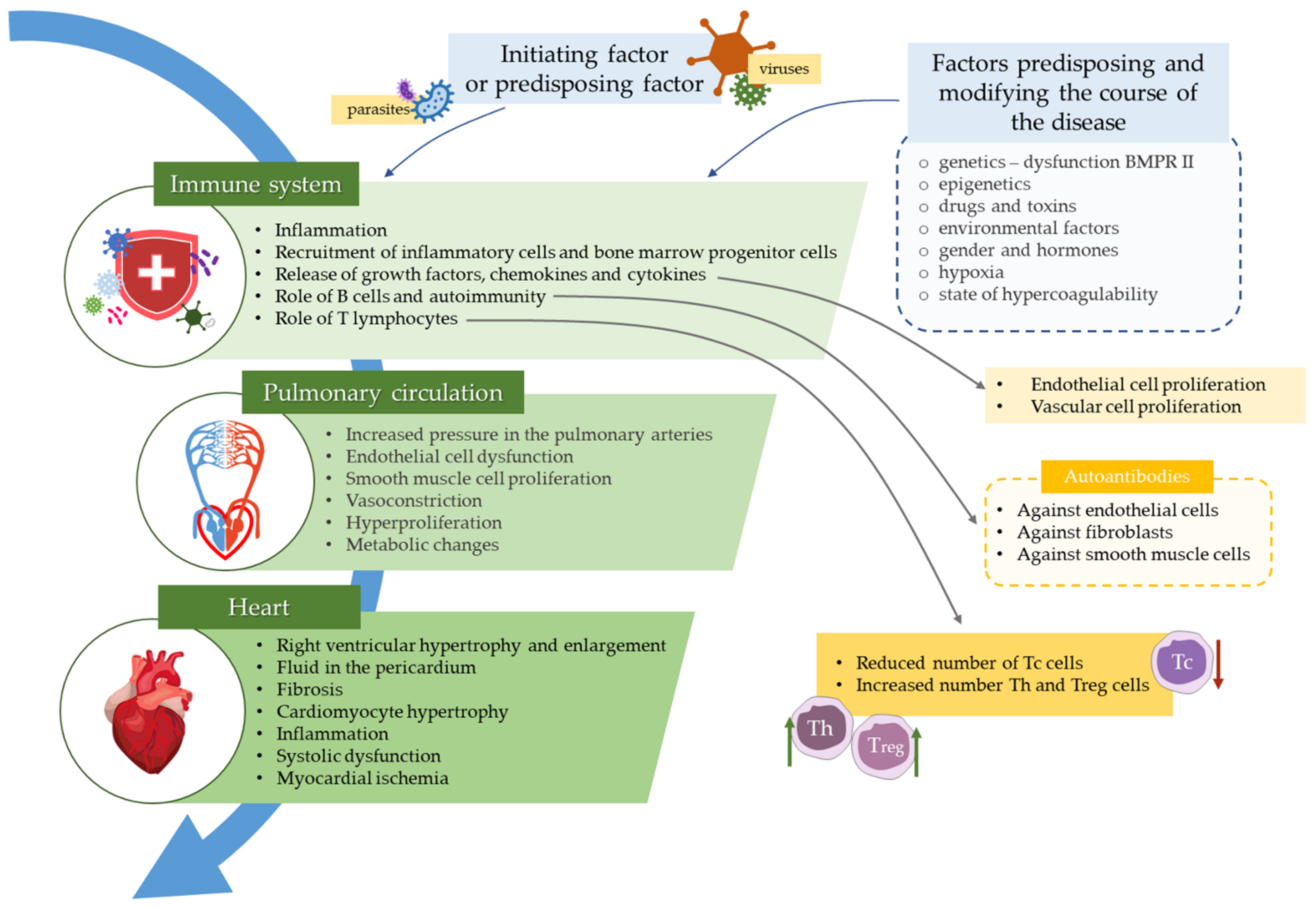{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Immune System Cells
2.1. NK Cells and T Cells in Pulmonary Arterial Hypertension
2.2. Regulatory T Cells in Pulmonary Arterial Hypertension
2.3. B Lymphocytes in Pulmonary Arterial Hypertension
2.4. Macrophages in Pulmonary Arterial Hypertension
3. Cytokines
3.1. IL-2
3.2. IL-6
3.3. IL-10
3.4. IL-4
3.5. Interferons
4. Immunoregulatory Molecules
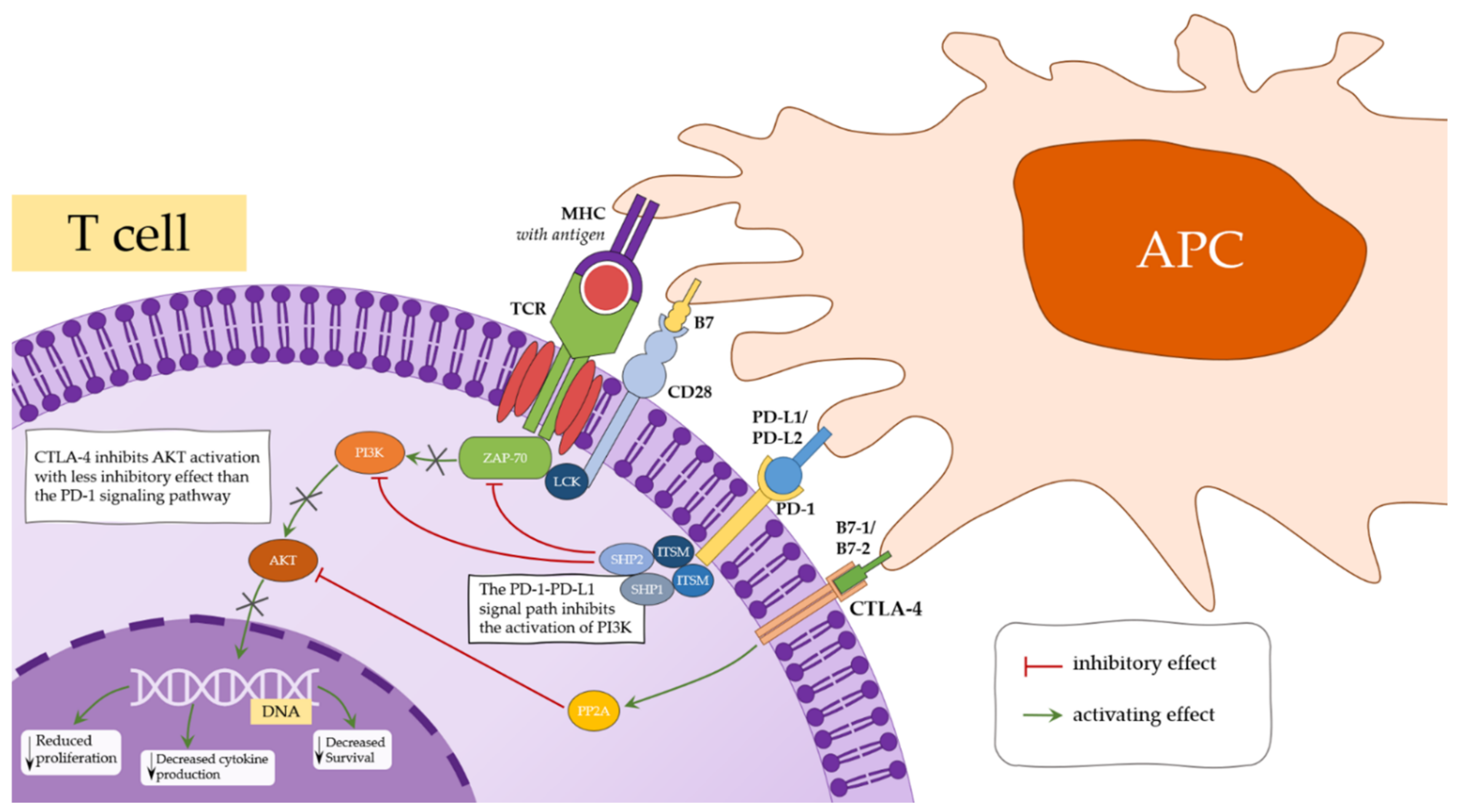
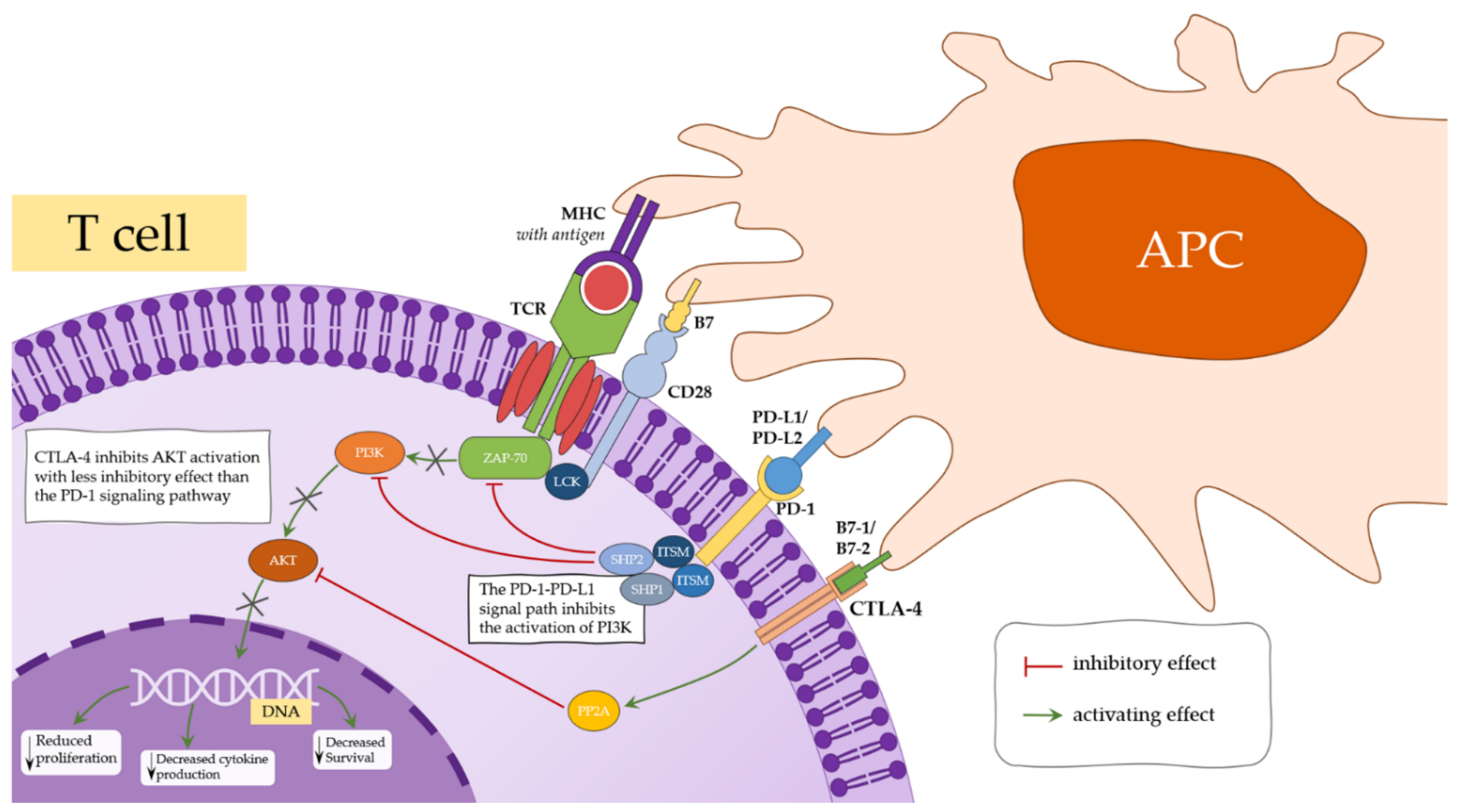
4.1. CTLA-4
4.2. CD200 and CD200R
4.3. PD1/PD-L1
5. Immunotherapeutic Approach
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Galiè, N.; Channick, R.N.; Frantz, R.; Grünig, E.; Jing, Z.-C.; Moiseeva, O.; Preston, I.R.; Pulido, T.; Safdar, Z.; Tamura, Y.; et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801889. [Google Scholar] [CrossRef]
- Thenappan, T.; Ormiston, M.L.; Ryan, J.J.; Archer, S.L. Pulmonary arterial hypertension: Pathogenesis and clinical manage-ment. BMJ 2018, 360, j5492. [Google Scholar] [CrossRef] [PubMed]
- Kopeć, G.; Kurzyna, M.; Mroczek, E.; Chrzanowski, L.; Mularek-Kubzdela, T.; Skoczylas, I.; Kuśmierczyk, B.; Pruszczyk, P.; Błaszczak, P.; Lewicka, E.; et al. Characterization of Patients with Pulmonary Arterial Hypertension: Data from the Polish Registry of Pulmonary Hypertension (BNP-PL). J. Clin. Med. 2020, 9, 173. [Google Scholar] [CrossRef] [Green Version]
- Constantinescu, T.; Magda, S.L.; Niculescu, R.; Mincu, R.I.; Zaharia, D.; Toma, C.L.; Cinteza, M.; Bogdan, M.A. New Echo-cardiographic Tehniques in Pulmonary Arterial Hypertension vs. Right Heart Catheterization—A Pilot Study. Maedica 2013, 8, 116–123. [Google Scholar] [PubMed]
- Jacobs, W.; van de Veerdonk, M.C.; Trip, P.; Man, F.H.-D.; Heymans, M.; Marcus, J.T.; Kawut, S.M.; Bogaard, H.-J.; Boonstra, A.; Noordegraaf, A.V. The Right Ventricle Explains Sex Differences in Survival in Idiopathic Pulmonary Arterial Hypertension. Chest 2014, 145, 1230–1236. [Google Scholar] [CrossRef] [Green Version]
- Hatton, N.; Ryan, J.J. Sex differences in response to pulmonary arterial hypertension therapy: Is what’s good for the goose, good for the gender? Chest 2014, 145, 1184–1186. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.D.J.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Noordegraaf, A.V.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 2015, 37, 67–119. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Newman, K.C.; Riley, E.M. Whatever turns you on: Accessory-cell-dependent activation of NK cells by pathogens. Nat. Rev. Immunol. 2007, 7, 279–291. [Google Scholar] [CrossRef]
- Sun, J.C.; Lanier, L.L. NK cell development, homeostasis and function: Parallels with CD8+ T cells. Nat. Rev. Immunol. 2011, 11, 645–657. [Google Scholar] [CrossRef]
- Ślebioda, T.J.; Kaszubowska, L.; Kmieć, Z. Nowe Mechanizmy aktywacji komórek NK w przebiegu infekcji wirusowych. Postępy Biol. Komórkowej 2012, 39, 61–83. [Google Scholar]
- Kopeć-Szlęzak, J.; Podstawka, U. Biologia komórek NK (Natural Killer). Onkol. Pol. 2007, 10, 115–119. [Google Scholar]
- Lodoen, M.B.; Lanier, L.L. Natural killer cells as an initial defense against pathogens. Curr. Opin. Immunol. 2006, 18, 391–398. [Google Scholar] [CrossRef]
- Cheent, K.; Khakoo, S.I. Natural killer cells: Integrating diversity with function. Immunology 2009, 126, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Babić, M.; Krmpotić, A.; Jonjić, S. All is fair in virus–host interactions: NK cells and cytomegalovirus. Trends Mol. Med. 2011, 17, 677–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Liu, P.; Song, R.; Zhang, Y.; Lei, S.; Wu, S. Immune cells and autoantibodies in pulmonary arterial hypertension. Acta Biochim. Biophys. Sin. 2017, 49, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Ormiston, M.; Chang, C.; Long, L.L.; Soon, E.; Jones, D.; Machado, R.D.; Treacy, C.; Toshner, M.R.; Campbell, K.; Riding, A.; et al. Impaired Natural Killer Cell Phenotype and Function in Idiopathic and Heritable Pulmonary Arterial Hypertension. Circulation 2012, 126, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Savai, R.; Pullamsetti, S.S.; Kolbe, J.; Bieniek, E.; Voswinckel, R.; Fink, L.; Scheed, A.; Ritter, C.; Dahal, B.K.; Vater, A.; et al. Immune and Inflammatory Cell Involvement in the Pathology of Idiopathic Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 897–908. [Google Scholar] [CrossRef]
- Edwards, A.L.; Gunningham, S.P.; Clare, G.C.; Hayman, M.W.; Smith, M.; Frampton, C.M.; Robinson, B.; Troughton, R.W.; El Beckert, L. Professional killer cell deficiencies and decreased survival in pulmonary arterial hypertension. Respirology 2013, 18, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, P.M.; Mouthon, L.; Barberà, J.A.; Eddahibi, S.; Flores, S.C.; Grimminger, F.; Jones, P.L.; Maitland, M.L.; Michelakis, E.D.; Morrell, N.; et al. Inflammation, Growth Factors, and Pulmonary Vascular Remodeling. J. Am. Coll. Cardiol. 2009, 54, S10–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zandman-Goddard, G.; Shoenfeld, Y. HIV and autoimmunity. Autoimmun. Rev. 2002, 1, 329–337. [Google Scholar] [CrossRef]
- Kemény, L.; Kiss, M.; Gyulai, R.; Kenderessy, A.S.; Adám, E.; Nagy, F.; Dobozy, A. Human herpesvirus 8 in classic Kaposi sarcoma. Acta Microbiol. Immunol. Hung. 1996, 43, 391–395. [Google Scholar] [PubMed]
- Taraseviciene-Stewart, L.; Nicolls, M.R.; Kraskauskas, D.; Scerbavicius, R.; Burns, N.; Cool, C.; Wood, K.; Parr, J.E.; Boackle, S.A.; Voelkel, N.F. Absence of T Cells Confers Increased Pulmonary Arterial Hypertension and Vascular Remodeling. Am. J. Respir. Crit. Care Med. 2007, 175, 1280–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ormiston, M.; Deng, Y.; Stewart, D.J.; Courtman, D.W. Innate Immunity in the Therapeutic Actions of Endothelial Progenitor Cells in Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2010, 43, 546–554. [Google Scholar] [CrossRef]
- Ratsep, M.T.; Moore, S.D.; Jafri, S.; Mitchell, M.; Brady, H.; Mandelboim, O.; Southwood, M.; Morrell, N.W.; Colucci, F.; Ormiston, M.L. Spontaneous pulmonary hypertension in genetic mouse models of natural killer cell deficiency. Am. J. Physiol. Cell. Mol. Physiol. 2018, 315, L977–L990. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Romagnani, S. Heterogeneity of human effector CD4+ T cells. Arthritis Res. Ther. 2009, 11, 257. [Google Scholar] [CrossRef] [Green Version]
- Suvas, S.; Kumaraguru, U.; Pack, C.D.; Lee, S.; Rouse, B.T. CD4+CD25+ T Cells Regulate Virus-specific Primary and Memory CD8+ T Cell Responses. J. Exp. Med. 2003, 198, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, C.; Thornton, A.M. Cornerstone of peripheral tolerance: Naturally occurring CD4+CD25+ regulatory T cells. Trends Immunol. 2004, 25, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Malek, T.R.; Bayer, A. Tolerance, not immunity, crucially depends on IL-2. Nat. Rev. Immunol. 2004, 4, 665–674. [Google Scholar] [CrossRef]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; Van Der Veeken, J.; DeRoos, P.; Liu, H.; Cross, J.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Kopeć, G.; Dzikowska-Diduch, O.; Mroczek, E.; Mularek-Kubzdela, T.; Chrzanowski, L.; Skoczylas, I.; Tomaszewski, M.; Peregud-Pogorzelska, M.; Karasek, D.; Lewicka, E.; et al. Characteristics and outcomes of patients with chronic thromboembolic pulmonary hypertension in the era of modern therapeutic approaches: Data from the Polish multicenter registry (BNP-PL). Ther. Adv. Chronic Dis. 2021, 12. [Google Scholar] [CrossRef]
- Khanna, D.; Gladue, H.; Channick, R.; Chung, L.; Distler, O.; Furst, D.E.; Hachulla, E.; Humbert, M.; Langleben, D.; Mathai, S.C.; et al. Recommendations for screening and detection of connective tissue disease-associated pulmonary arterial hyper-tension. Arthritis Rheum. 2013, 65, 3194–3201. [Google Scholar] [CrossRef] [Green Version]
- Sada, Y.; Dohi, Y.; Uga, S.; Higashi, A.; Kinoshita, H.; Kihara, Y. Non-suppressive regulatory T cell subset expansion in pulmonary arterial hypertension. Heart Vessel. 2015, 31, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T Cells and Immune Tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S.; Miyara, M.; Costantino, C.M.; Ha, D.A. FOXP3+ regulatory T cells in the human immune system. Nat. Rev. Immunol. 2010, 10, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Tamosiuniene, R.; Tian, W.; Dhillon, G.; Wang, L.; Sung, Y.K.; Gera, L.; Patterson, A.J.; Agrawal, R.; Rabinovitch, M.; Ambler, K.; et al. Regulatory T Cells Limit Vascular Endothelial Injury and Prevent Pulmonary Hypertension. Circ. Res. 2011, 109, 867–879. [Google Scholar] [CrossRef]
- Tamosiuniene, R.; Manouvakhova, O.; Mesange, P.; Saito, T.; Qian, J.; Sanyal, M.; Lin, Y.-C.; Nguyen, L.P.; Luria, A.; Tu, A.B.; et al. Dominant Role for Regulatory T Cells in Protecting Females Against Pulmonary Hypertension. Circ. Res. 2018, 122, 1689–1702. [Google Scholar] [CrossRef]
- Rich, S.; Kieras, K.; Hart, K.; Groves, B.M.; Stobo, J.D.; Brundage, B.H. Antinuclear antibodies in primary pulmonary hyper-tension. J. Am. Coll. Cardiol. 1986, 8, 1307–1311. [Google Scholar] [CrossRef] [Green Version]
- Blum, L.K.; Cao, R.R.; Sweatt, A.J.; Bill, M.; Lahey, L.J.; Hsi, A.C.; Lee, C.S.; Kongpachith, S.; Ju, C.-H.; Mao, R.; et al. Circulating plasmablasts are elevated and produce pathogenic anti-endothelial cell autoantibodies in idiopathic pulmonary arterial hypertension. Eur. J. Immunol. 2018, 48, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, S.; Taraseviciene-Stewart, L.; Huber, L.C.; Speich, R.; Voelkel, N. Peripheral blood B lymphocytes derived from patients with idiopathic pulmonary arterial hypertension express a different RNA pattern compared with healthy controls: A cross sectional study. Respir. Res. 2008, 9, 20. [Google Scholar] [CrossRef] [Green Version]
- Perros, F.; Dorfmüller, P.; Montani, D.; Hammad, H.; Waelput, W.; Girerd, B.; Raymond, N.; Mercier, O.; Mussot, S.; Cohen-Kaminsky, S.; et al. Pulmonary Lymphoid Neogenesis in Idiopathic Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 311–321. [Google Scholar] [CrossRef]
- Breitling, S.; Hui, Z.; Zabini, D.; Hu, Y.; Hoffmann, J.; Goldenberg, N.M.; Tabuchi, A.; Buelow, R.; Dos Santos, C.; Kuebler, W.M. The mast cell–B cell axis in lung vascular remodeling and pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2017, 312, L710–L721. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Kempuraj, D.; Tagen, M.; Conti, P.; Kalogeromitros, D. Differential release of mast cell mediators and the pathogenesis of inflammation. Immunol. Rev. 2007, 217, 65–78. [Google Scholar] [CrossRef]
- Sayed, B.A.; Christy, A.; Quirion, M.R.; Brown, M.A. The Master Switch: The Role of Mast Cells in Autoimmunity and Tolerance. Annu. Rev. Immunol. 2008, 26, 705–739. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Monti, G.; Brenot, F.; Sitbon, O.; Portier, A.; Grangeot-Keros, L.; Duroux, P.; Galanaud, P.; Simonneau, G.; Emilie, D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am. J. Respir. Crit. Care. Med. 1995, 151, 1628–1631. [Google Scholar] [CrossRef] [PubMed]
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated Levels of Inflammatory Cytokines Predict Survival in Idiopathic and Familial Pulmonary Arterial Hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef] [Green Version]
- Steiner, M.K.; Syrkina, O.L.; Kolliputi, N.; Mark, E.J.; Hales, C.A.; Waxman, A.B. Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 2009, 104, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Farkas, L.; Al Husseini, A.; Farkas, D.; Gomez-Arroyo, J.; Kraskauskas, D.; Nicolls, M.R.; Cool, C.D.; Bogaard, H.J.; Voelkel, N.F. Severe Pulmonary Arterial Hypertension Induced by SU5416 and Ovalbumin Immunization. Am. J. Respir. Cell Mol. Biol. 2012, 47, 679–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, S.; Fujimoto, M.; Hasegawa, M.; Takehara, K.; Tedder, T.F. Altered B lymphocyte function induces systemic autoimmunity in systemic sclerosis. Mol. Immunol. 2004, 41, 1123–1133. [Google Scholar] [CrossRef]
- Fujimoto, M.; Sato, S. B lymphocytes and systemic sclerosis. Curr. Opin. Rheumatol. 2005, 17, 746–751. [Google Scholar] [CrossRef] [PubMed]
- Kuebler, W.M.; Bonnet, S.; Tabuchi, A. Inflammation and autoimmunity in pulmonary hypertension: Is there a role for endothelial adhesion molecules? Pulm. Circ. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergadi, E.; Chang, M.S.; Lee, C.; Liang, O.D.; Liu, X.; Fernandez-Gonzalez, A.; Mitsialis, S.A.; Kourembanas, S. Early Macrophage Recruitment and Alternative Activation Are Critical for the Later Development of Hypoxia-Induced Pulmonary Hypertension. Circulation 2011, 123, 1986–1995. [Google Scholar] [CrossRef] [Green Version]
- Zawia, A.; Arnold, N.D.; West, L.; Pickworth, J.A.; Turton, H.; Iremonger, J.; Braithwaite, A.T.; Cañedo, J.; Johnston, S.A.; Thompson, A.R.; et al. Altered Macrophage Polarization Induces Experimental Pulmonary Hypertension and Is Observed in Patients with Pulmonary Arterial Hypertension. Arter. Thromb. Vasc. Biol. 2020, 41, 430–445. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Hao, Y.; Gao, D.; Gao, L.; Li, G.; Zhang, Z. Phenotype and function of macrophage polarization in monocrotaline-induced pulmonary arterial hypertension rat model. Physiol. Res. 2021, 70, 213–226. [Google Scholar] [CrossRef]
- Itoh, T.; Nagaya, N.; Ishibashi-Ueda, H.; Kyotani, S.; Oya, H.; Sakamaki, F.; Kimura, H.; Nakanishi, N. Increased plasma monocyte chemoattractant protein-1 level in idiopathic pulmonary arterial hypertension. Respirology 2006, 11, 158–163. [Google Scholar] [CrossRef]
- Cracowski, J.-L.; Chabot, F.; Labarere, J.; Faure, P.; Degano, B.; Schwebel, C.; Chaouat, A.; Reynaud-Gaubert, M.; Cracowski, C.; Sitbon, O.; et al. Proinflammatory cytokine levels are linked to death in pulmonary arterial hypertension. Eur. Respir. J. 2013, 43, 915–917. [Google Scholar] [CrossRef] [Green Version]
- Harris, D.P.; Haynes, L.; Sayles, P.C.; Duso, D.K.; Eaton, S.M.; Lepak, N.M.; Johnson, L.L.; Swain, S.L.; Lund, F.E. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat. Immunol. 2000, 1, 475–482. [Google Scholar] [CrossRef]
- Minami, Y.; Kono, T.; Miyazaki, T.; Taniguchi, T. The IL-2 Receptor Complex: Its Structure, Function, and Target Genes. Annu. Rev. Immunol. 1993, 11, 245–268. [Google Scholar] [CrossRef] [PubMed]
- Olejniczak, K.; Kasprzak, A. Biological properties of interleukin 2 and its role in pathogenesis of selected diseases—A review. Med. Sci. Monit. 2008, 14, 179–189. [Google Scholar]
- Malek, T.R. The main function of IL-2 is to promote the development of T regulatory cells. J. Leukoc. Biol. 2003, 74, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L.; Liu, K. Overview of interleukin-2 function, production and clinical applications. Cytokine 2004, 28, 109–123. [Google Scholar] [CrossRef]
- Parijs, L.V.; Kinzler, K.W.; Vogelstein, B. Homeostasis and Self-Tolerance in the Immune System: Turning Lymphocytes off. Science 1998, 280, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Chen, L.; Xiong, Y.; Wang, N.; Xie, X.; Hong, Y.; Meng, Z. An upregulation of CD8+CD25+Foxp3+ T cells with suppressive function through interleukin 2 pathway in pulmonary arterial hypertension. Exp. Cell Res. 2017, 358, 182–187. [Google Scholar] [CrossRef]
- Glauser, F.L.; Deblois, G.G.; Bechard, D.E.; Merchant, R.E.; Grant, A.J.; Fowler, A.A.; Fairman, R.P. Cardiopulmonary effects of recombinant interleukin-2 infusion in sheep. J. Appl. Physiol. 1988, 64, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, T.; Miura, S.; Hirokawa, M.; Hokari, R.; Higuchi, H.; Watanabe, N.; Tsuzuki, Y.; Kimura, H.; Tada, S.; Nakatsumi, R.C.; et al. Induction of endothelin-1 synthesis by IL-2 and its modulation of rat intestinal epithelial cell growth. Am. J. Physiol. Content 1998, 275, G556–G563. [Google Scholar] [CrossRef] [PubMed]
- Diehl, S.; Rincón, M. The two faces of IL-6 on Th1/Th2 differentiation. Mol. Immunol. 2002, 39, 531–536. [Google Scholar] [CrossRef]
- Croker, B.; Krebs, D.L.; Zhang, J.-G.; Wormald, S.; Willson, T.A.; Stanley, E.G.; Robb, L.; Greenhalgh, C.J.; Förster, I.; Clausen, B.; et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat. Immunol. 2003, 4, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Ferro, T.J.; Johnson, A.; Everitt, J.; Malik, A.B. IL-2 induces pulmonary edemaand vasoconstriction independent of circulating lymphocytes. J. Immunol. 1989, 142, 1916–1921. [Google Scholar] [PubMed]
- Rincon, M. Interleukin-6: From an inflammatory marker to a target for inflammatory diseases. Trends Immunol. 2012, 33, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Kaminsky, S.; Hautefort, A.; Price, L.; Humbert, M.; Perros, F. Inflammation in pulmonary hypertension: What we know and what we could logically and safely target first. Drug Discov. Today 2014, 19, 1251–1256. [Google Scholar] [CrossRef]
- Haberka, M.; Machnik, G.; Kowalówka, A.; Biedroń, M.; Skudrzyk, E.; Regulska-Ilow, B.; Gajos, G.; Manka, R.; Deja, M.; Okopień, B.; et al. Epicardial, paracardial and perivascular fat quantity, genes expression and serum cytokines in coronary artery disease and diabetes. Pol. Arch. Intern. Med. 2019, 129, 738–746. [Google Scholar] [CrossRef] [Green Version]
- Heresi, G.A.; Aytekin, M.; Hammel, J.P.; Wang, S.; Chatterjee, S.; Dweik, R.A. Plasma interleukin-6 adds prognostic information in pulmonary arterial hypertension. Eur. Respir. J. 2013, 43, 912–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhodes, C.J.; Wharton, J.; Howard, L.; Gibbs, J.S.R.; Wilkins, M.R. Red cell distribution width outperforms other potential circulating biomarkers in predicting survival in idiopathic pulmonary arterial hypertension. Heart 2011, 97, 1054–1060. [Google Scholar] [CrossRef]
- Prins, K.; Archer, S.L.; Pritzker, M.; Rose, L.; Weir, E.K.; Sharma, A.; Thenappan, T. Interleukin-6 is independently associated with right ventricular function in pulmonary arterial hypertension. J. Hear. Lung Transplant. 2018, 37, 376–384. [Google Scholar] [CrossRef]
- Jasiewicz, M.; Knapp, M.; Waszkiewicz, E.; Ptaszynska-Kopczynska, K.; Szpakowicz, A.; Sobkowicz, B.; Musial, W.J.; Kaminski, K.A. Enhanced IL-6 trans-signaling in pulmonary arterial hypertension and its potential role in disease-related systemic damage. Cytokine 2015, 76, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Shimazaki, C.; Fujimoto, Y.; Shimura, K.; Uchiyama, H.; Matsumoto, Y.; Kuroda, J.; Horiike, S.; Taniwaki, M. Tocilizumab is effective for pulmonary hypertension associated with multicentric Castleman’s disease. Int. J. Hematol. 2009, 90, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Arita, Y.; Sakata, Y.; Sudo, T.; Maeda, T.; Matsuoka, K.; Tamai, K.; Higuchi, K.; Shioyama, W.; Nakaoka, Y.; Kanakura, Y.; et al. The efficacy of tocilizumab in a patient with pulmonary arterial hypertension associated with Castleman’s disease. Heart Vessel. 2010, 25, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Furuya, Y.; Satoh, T.; Kuwana, M. Interleukin-6 as a Potential Therapeutic Target for Pulmonary Arterial Hypertension. Int. J. Rheumatol. 2010, 2010, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Karavolias, G.K.; Georgiadou, P.; Gkouziouta, A.; Kariofillis, P.; Karabela, G.; Tsiapras, D.; Sbarouni, E.; Chaidaroglou, A.; Degiannis, D.; Adamopoulos, S.; et al. Short and long term anti-inflammatory effects of bosentan therapy in patients with pulmonary arterial hypertension: Relation to clinical and hemodynamic responses. Expert Opin. Ther. Targets 2010, 14, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ikeda, U. Inflammatory cytokines and cardiovascular disease. Curr. Drug Targets-Inflamm. Allergy 2003, 2, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Okada, T.; Miyashita, H.; Nomoto, T.; Nonaka-Sarukawa, M.; Uchibori, R.; Maeda, Y.; Urabe, M.; Mizukami, H.; Kume, A.; et al. Interleukin-10 Expression Mediated by an Adeno-Associated Virus Vector Prevents Monocrotaline-Induced Pulmonary Arterial Hypertension in Rats. Circ. Res. 2007, 101, 734–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Kapturczak, M.H.; Wasserfall, C.; Glushakova, O.Y.; Campbell-Thompson, M.; Deshane, J.S.; Joseph, R.; Cruz, P.E.; Hauswirth, W.W.; Madsen, K.M.; et al. Interleukin 10 attenuates neointimal proliferation and inflammation in aortic allografts by a heme oxygenase-dependent pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 7251–7256. [Google Scholar] [CrossRef] [Green Version]
- Mazighi, M.; Pellé, A.; Gonzalez, W.; Mtairag, E.M.; Philippe, M.; Hénin, D.; Michel, J.-B.; Feldman, L.J. IL-10 inhibits vascular smooth muscle cell activation in vitro and in vivo. Am. J. Physiol. Circ. Physiol. 2004, 287, H866–H871. [Google Scholar] [CrossRef]
- Yoshioka, T.; Okada, T.; Maeda, Y.; Ikeda, U.; Shimpo, M.; Nomoto, T.; Takeuchi, K.; Nonaka-Sarukawa, M.; Ito, T.; Takahashi, M.; et al. Adeno-associated virus vector-mediated interleukin-10 gene transfer inhibits atherosclerosis in apolipoprotein E-deficient mice. Gene Ther. 2004, 11, 1772–1779. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Khosraviani, S.; Noel, S.; Mohan, D.; Donner, T.; Hamad, A.R.A. Interleukin-10 paradox: A potent immunoregulatory cytokine that has been difficult to harness for immunotherapy. Cytokine 2014, 74, 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadani, S.P.; Cronk, J.C.; Norris, G.T.; Kipnis, J. IL-4 in the Brain: A Cytokine to Remember. J. Immunol. 2012, 189, 4213–4219. [Google Scholar] [CrossRef]
- Yamaji-Kegan, K.; Su, Q.; Angelini, D.J.; Champion, H.C.; Johns, R.A. Hypoxia-induced mitogenic factor has proangiogenic and proinflammatory effects in the lung via VEGF and VEGF receptor-2. Am. J. Physiol. Cell. Mol. Physiol. 2006, 291, 1159–1168. [Google Scholar] [CrossRef]
- Yamaji-Kegan, K.; Su, Q.; Angelini, D.J.; Myers, A.C.; Cheadle, C.; Johns, R.A. Hypoxia-Induced Mitogenic Factor (HIMF/FIZZ1/RELMα) Increases Lung Inflammation and Activates Pulmonary Microvascular Endothelial Cells via an IL-4–Dependent Mechanism. J. Immunol. 2010, 185, 5539–5548. [Google Scholar] [CrossRef] [Green Version]
- Yamaji-Kegan, K.; Takimoto, E.; Zhang, A.; Weiner, N.C.; Meuchel, L.W.; Berger, A.E.; Cheadle, C.; Johns, R.A. Hypoxia-induced mitogenic factor (FIZZ1/RELMα) induces endothelial cell apoptosis and subsequent interleukin-4-dependent pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2014, 306, 1090–1103. [Google Scholar] [CrossRef] [Green Version]
- Katze, M.G.; He, Y.; Gale, M. Viruses and interferon: A fight for supremacy. Nat. Rev. Immunol. 2002, 2, 675–687. [Google Scholar] [CrossRef]
- Pollard, K.M.; Cauvi, D.M.; Toomey, C.B.; Morris, K.V.; Kono, D.H. Interferon-γ and systemic autoimmunity. Discov. Med. 2013, 16, 123–131. [Google Scholar]
- Al-Zahrani, H.; Gupta, V.; Minden, M.; Messner, H.; Lipton, J. Vascular Events Associated with Alpha Interferon Therapy. Leuk. Lymphoma 2003, 44, 471–475. [Google Scholar] [CrossRef]
- Ledinek, A.; Jazbec, S.; Drinovec, I.; Rot, U. Pulmonary arterial hypertension associated with interferon beta treatment for multiple sclerosis: A case report. Mult. Scler. J. 2009, 15, 885–886. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S.; Kaker, A.; Dosanjh, S.D.K.; Japra, D.; VanThiel, D.H. Irreversible Pulmonary Hypertension Associated with the use of Interferon Alpha for Chronic Hepatitis C. Dig. Dis. Sci. 2010, 55, 1785–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, G.R.; Zeuzem, S.; Pianko, S.; Sarin, S.K.; Piratvisuth, T.; Shah, S.; Andreone, P.; Sood, A.; Chuang, W.-L.; Lee, C.-M.; et al. Decline in pulmonary function during chronic hepatitis C virus therapy with modified interferon alfa and ribavirin. J. Viral Hepat. 2013, 20, e115–e123. [Google Scholar] [CrossRef] [PubMed]
- Pentcheva-Hoang, T.; Corse, E.; Allison, J.P. Negative regulators of T-cell activation: Potential targets for therapeutic intervention in cancer, autoimmune disease, and persistent infections. Immunol. Rev. 2009, 229, 67–87. [Google Scholar] [CrossRef] [PubMed]
- Kong, K.-F.; Fu, G.; Zhang, Y.; Yokosuka, T.; Casas, J.; Canonigo-Balancio, A.J.; Bécart, S.; Kim, G.; Yates, J.R.; Kronenberg, M.; et al. Protein kinase C-η controls CTLA-4–mediated regulatory T cell function. Nat. Immunol. 2014, 15, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Han, X. Anti–PD-1/PD-L1 therapy of human cancer: Past, present, and future. J. Clin. Investig. 2015, 125, 3384–3391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmberg, D.; Cilio, C.M.; Lundholm, M.; Motta, V. CTLA-4 (CD152) and its involvement in autoimmune disease. Autoimmunity 2005, 38, 225–233. [Google Scholar] [CrossRef]
- Reddy, M.; Eirikis, E.; Davis, C.; Davis, H.M.; Prabhakar, U. Comparative analysis of lymphocyte activation marker expression and cytokine secretion profile in stimulated human peripheral blood mononuclear cell cultures: An in vitro model to monitor cellular immune function. J. Immunol. Methods 2004, 293, 127–142. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinzerling, L.; Ott, P.A.; Hodi, F.S.; Husain, A.N.; Tajmir-Riahi, A.; Tawbi, H.; Pauschinger, M.; Gajewski, T.F.; Lipson, E.J.; Luke, J.J. Cardiotoxicity associated with CTLA4 and PD1 blocking immunotherapy. J. Immunother. Cancer 2016, 4, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomaszewski, M.; Grywalska, E.; Tomaszewski, A.; Błaszczak, P.; Kurzyna, M.; Roliński, J.; Kopeć, G. Overexpression of PD-1 on Peripheral Blood Lymphocytes in Patients with Idiopathic Pulmonary Arterial Hypertension and Its Association with High Viral Loads of Epstein-Barr Virus and Poor Clinical Parameters. J. Clin. Med. 2020, 9, 1966. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.; Rock, M.; Mosse, C.; Vnencak-Jones, C.; Yoder, S.; Robbins, I.; Loyd, J.; Meyrick, B. T lymphocyte subset abnormalities in the blood and lung in pulmonary arterial hypertension. Respir. Med. 2010, 104, 454–462. [Google Scholar] [CrossRef] [Green Version]
- Wright, G.; Jones, M.; Puklavec, M.J.; Brown, M.H.; Barclay, A.N. The unusual distribution of the neuronal/lymphoid cell surface CD200 (OX2) glycoprotein is conserved in humans. Immunology 2001, 102, 173–179. [Google Scholar] [CrossRef]
- Rijkers, E.S.; de Ruiter, T.; Baridi, A.; Veninga, H.; Hoek, R.M.; Meyaard, L. The inhibitory CD200R is differentially expressed on human and mouse T and B lymphocytes. Mol. Immunol. 2008, 45, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Akkaya, M.; Barclay, A.N. Heterogeneity in the CD200R paired receptor family. Immunogenetics 2009, 62, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chen, D.-X.; Kai, Y.; Khatri, I.; Lamptey, B.; Gorczynski, R.M. Identification of an Expressed Truncated Form of CD200, CD200tr, which is a Physiologic Antagonist of CD200-Induced Suppression. Transplantation 2008, 86, 1116–1124. [Google Scholar] [CrossRef]
- Walker, D.G.; Lue, L.-F. Understanding the neurobiology of CD200 and the CD200 receptor: A therapeutic target for controlling inflammation in human brains? Futur. Neurol. 2013, 8, 321–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenmalm, M.; Cherwinski, H.; Bowman, E.P.; Phillips, J.H.; Sedgwick, J.D. Regulation of Myeloid Cell Function through the CD200 Receptor. J. Immunol. 2005, 176, 191–199. [Google Scholar] [CrossRef]
- Fallarino, F.; Orabona, C.; Vacca, C.; Bianchi, R.; Gizzi, S.; Asselin-Paturel, C.; Fioretti, M.C.; Trinchieri, G.; Grohmann, U.; Puccetti, P. Ligand and cytokine dependence of the immunosuppressive pathway of tryptophan catabolism in plasmacytoid dendritic cells. Int. Immunol. 2005, 17, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.-H.; Zhang, E.; Kang, J.W.; Na Shin, Y.; Byun, J.Y.; Oh, S.-H.; Seo, J.H.; Lee, Y.H.; Kim, D.W. Expression of CD200 in alternative activation of microglia following an excitotoxic lesion in the mouse hippocampus. Brain Res. 2012, 1481, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Tsuchiya, K.; Ikemizu, S.; Yoshikawa, S.; Yamanishi, Y.; Watanabe, M.; Karasuyama, H. Novel CD200 homologues iSEC1 and iSEC2 are gastrointestinal secretory cell-specific ligands of inhibitory receptor CD200R. Sci. Rep. 2016, 6, 36457. [Google Scholar] [CrossRef]
- Wright, G.; Cherwinski, H.; Foster-Cuevas, M.; Brooke, G.; Puklavec, M.J.; Bigler, M.; Song, Y.; Jenmalm, M.; Gorman, D.; McClanahan, T.; et al. Characterization of the CD200 Receptor Family in Mice and Humans and Their Interactions with CD200. J. Immunol. 2003, 171, 3034–3046. [Google Scholar] [CrossRef] [Green Version]
- Najar, M.; Raicevic, G.; Jebbawi, F.; De Bruyn, C.; Meuleman, N.; Bron, D.; Toungouz, M.; Lagneaux, L. Characterization and functionality of the CD200–CD200R system during mesenchymal stromal cell interactions with T-lymphocytes. Immunol. Lett. 2012, 146, 50–56. [Google Scholar] [CrossRef]
- Soberman, R.J.; Mackay, C.R.; Vaine, C.A.; Ryan, G.B.; Cerny, A.M.; Thompson, M.R.; Nikolic, B.; Primo, V.; Christmas, P.; Sheiffele, P.; et al. CD200R1 Supports HSV-1 Viral Replication and Licenses Pro-Inflammatory Signaling Functions of TLR2. PLoS ONE 2012, 7, e47740. [Google Scholar] [CrossRef]
- Gao, S.; Hao, B.; Yang, X.F.; Chen, W.Q. Decreased CD200R expression on monocyte-derived macrophages correlates with Th17/Treg imbalance and disease activity in rheumatoid arthritis patients. Inflamm. Res. 2014, 63, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Koning, N.; Swaab, D.; Hoek, R.M.; Huitinga, I. Distribution of the Immune Inhibitory Molecules CD200 and CD200R in the Normal Central Nervous System and Multiple Sclerosis Lesions Suggests Neuron-Glia and Glia-Glia Interactions. J. Neuropathol. Exp. Neurol. 2009, 68, 159–167. [Google Scholar] [CrossRef]
- Snelgrove, R.J.; Goulding, J.; Didierlaurent, A.M.; Lyonga, D.; Vekaria, S.; Edwards, L.; Gwyer, E.; Sedgwick, J.D.; Barclay, A.N.; Hussell, T. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat. Immunol. 2008, 9, 1074–1083. [Google Scholar] [CrossRef]
- Fraser, S.D.; Sadofsky, L.R.; Kaye, P.; Hart, S.P. Reduced expression of monocyte CD200R is associated with enhanced proinflammatory cytokine production in sarcoidosis. Sci. Rep. 2016, 6, 38689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ElShal, M.F.; AlDahlawi, A.M.; Saadah, O.I.; McCoy, J.P. Reduced Dendritic Cells Expressing CD200R1 in Children with Inflammatory Bowel Disease: Correlation with Th17 and Regulatory T Cells. Int. J. Mol. Sci. 2015, 16, 28998–29010. [Google Scholar] [CrossRef] [Green Version]
- Boudakov, I.; Liu, J.; Fan, N.; Gulay, S.; Wong, K.; Gorczynski, R.M. Mice Lacking CD200R1 Show Absence of Suppression of Lipopolysaccharide-Induced Tumor Necrosis Factor-α and Mixed Leukocyte Culture Responses by CD200. Transplantation 2007, 84, 251–257. [Google Scholar] [CrossRef]
- Walker, D.G.; Dalsing-Hernandez, J.E.; Campbell, N.A.; Lue, L.-F. Decreased expression of CD200 and CD200 receptor in Alzheimer’s disease: A potential mechanism leading to chronic inflammation. Exp. Neurol. 2009, 215, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Sakthivel, P.; Breithaupt, A.; Gereke, M.; Copland, D.A.; Schulz, C.; Gruber, A.D.; Dick, A.D.; Schreiber, J.; Bruder, D. Soluble CD200 Correlates with Interleukin-6 Levels in Sera of COPD Patients: Potential Implication of the CD200/CD200R Axis in the Disease Course. Lung 2016, 195, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Pallasch, C.P.; Ulbrich, S.; Brinker, R.; Hallek, M.; Uger, R.A.; Wendtner, C.-M. Disruption of T cell suppression in chronic lymphocytic leukemia by CD200 blockade. Leuk. Res. 2009, 33, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Broderick, C.; Hoek, R.M.; Forrester, J.V.; Liversidge, J.; Sedgwick, J.D.; Dick, A.D. Constitutive Retinal CD200 Expression Regulates Resident Microglia and Activation State of Inflammatory Cells during Experimental Autoimmune Uveoretinitis. Am. J. Pathol. 2002, 161, 1669–1677. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, D.; Dick, A. Blocking CD200-CD200 receptor axis augments NOS-2 expression and aggravates experimental autoimmune uveoretinitis in Lewis rats. Ocul. Immunol. Inflamm. 2004, 12, 115–125. [Google Scholar] [CrossRef]
- Copland, D.A.; Calder, C.J.; Raveney, B.J.; Nicholson, L.B.; Phillips, J.; Cherwinski, H.; Jenmalm, M.; Sedgwick, J.D.; Dick, A.D. Monoclonal Antibody-Mediated CD200 Receptor Signaling Suppresses Macrophage Activation and Tissue Damage in Experimental Autoimmune Uveoretinitis. Am. J. Pathol. 2007, 171, 580–588. [Google Scholar] [CrossRef] [Green Version]
- Lyons, A.; Downer, E.; Crotty, S.; Nolan, Y.; Mills, K.; Lynch, M.A. CD200 Ligand Receptor Interaction Modulates Microglial Activation In Vivo and In Vitro: A Role for IL-4. J. Neurosci. 2007, 27, 8309–8313. [Google Scholar] [CrossRef]
- Sun, F.-J.; Zhang, C.-Q.; Chen, X.; Wei, Y.-J.; Li, S.; Liu, S.-Y.; Zang, Z.-L.; He, J.-J.; Guo, W.; Yang, H. Downregulation of CD47 and CD200 in patients with focal cortical dysplasia type IIb and tuberous sclerosis complex. J. Neuroinflamm. 2016, 13, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Tomaszewski, M.; Grywalska, E.; Topyła-Putowska, W.; Błaszczak, P.; Kurzyna, M.; Roliński, J.; Kopeć, G. High CD200 Expression on T CD4+ and T CD8+ Lymphocytes as a Non-Invasive Marker of Idiopathic Pulmonary Hypertension–Preliminary Study. J. Clin. Med. 2021, 10, 950. [Google Scholar] [CrossRef]
- Tang, P.A.; Heng, D.Y.C. Programmed Death 1 Pathway inhibition in Metastatic Renal Cell Cancer and Prostate Cancer. Curr. Oncol. Rep. 2012, 15, 98–104. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.F.; Atkins, M.B. Immune Therapy for Kidney Cancer: A Second Dawn? Semin. Oncol. 2013, 40, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Irving, B.A.; Hodi, F.S. Molecular Pathways: Next-Generation Immunotherapy—Inhibiting Programmed Death-Ligand 1 and Programmed Death-1. Clin. Cancer Res. 2012, 18, 6580–6587. [Google Scholar] [CrossRef] [Green Version]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Iwai, Y.; Terawaki, S.; Honjo, T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int. Immunol. 2004, 17, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Gianchecchi, E.; Delfino, D.V.; Fierabracci, A. Recent insights into the role of the PD-1/PD-L1 pathway in immunological tolerance and autoimmunity. Autoimmun. Rev. 2013, 12, 1091–1100. [Google Scholar] [CrossRef]
- Sanjo, N.; Nose, Y.; Shishido-Hara, Y.; Mizutani, S.; Sekijima, Y.; Aizawa, H.; Tanizawa, T.; Yokota, T. A controlled inflammation and a regulatory immune system are associated with more favorable prognosis of progressive multifocal leukoencephalopathy. J. Neurol. 2018, 266, 369–377. [Google Scholar] [CrossRef]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keir, M.E.; Liang, S.C.; Guleria, I.; Latchman, Y.E.; Qipo, A.; Albacker, L.; Koulmanda, M.; Freeman, G.J.; Sayegh, M.H.; Sharpe, A.H. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 2006, 203, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Latchman, Y.E.; Liang, S.C.; Wu, Y.; Chernova, T.; Sobel, R.A.; Klemm, M.; Kuchroo, V.K.; Freeman, G.J.; Sharpe, A.H. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc. Natl. Acad. Sci. USA 2004, 101, 10691–10696. [Google Scholar] [CrossRef] [Green Version]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2005, 439, 682–687. [Google Scholar] [CrossRef]
- Okazaki, T.; Tanaka, Y.; Nishio, R.; Mitsuiye, T.; Mizoguchi, A.; Wang, J.; Ishida, M.; Hiai, H.; Matsumori, A.; Minato, N.; et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat. Med. 2003, 9, 1477–1483. [Google Scholar] [CrossRef]
- Petrelli, A.; Mijnheer, G.; van Konijnenburg, D.H.; Van Der Wal, M.M.; Giovannone, B.; Mocholi, E.; Vazirpanah, N.; Broen, J.C.; Hijnen, D.; Oldenburg, B.; et al. PD-1+CD8+ T cells are clonally expanding effectors in human chronic inflammation. J. Clin. Investig. 2018, 128, 4669–4681. [Google Scholar] [CrossRef]
- Riley, J.L. PD-1 signaling in primary T cells. Immunol. Rev. 2009, 229, 114–125. [Google Scholar] [CrossRef]
- Bommarito, D.; Hall, C.; Taams, L.S.; Corrigall, V.M. Inflammatory cytokines compromise programmed cell death-1 (PD-1)-mediated T cell suppression in inflammatory arthritis through up-regulation of soluble PD-1. Clin. Exp. Immunol. 2017, 188, 455–466. [Google Scholar] [CrossRef] [Green Version]
- Speiser, D.; Utzschneider, D.; Oberle, S.G.; Münz, C.; Romero, P.; Zehn, D. T cell differentiation in chronic infection and cancer: Functional adaptation or exhaustion? Nat. Rev. Immunol. 2014, 14, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Utzschneider, D.T.; Alfei, F.; Roelli, P.; Barras, D.; Chennupati, V.; Darbre, S.; Delorenzi, M.; Pinschewer, D.D.; Zehn, D. High antigen levels induce an exhausted phenotype in a chronic infection without impairing T cell expansion and survival. J. Exp. Med. 2016, 213, 1819–1834. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Wherry, E.J.; Ahmed, R.; Freeman, G.J. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 2007, 8, 239–245. [Google Scholar] [CrossRef]
- Karachaliou, N.; Gonzalez-Cao, M.; Crespo, G.; Drozdowskyj, A.; Aldeguer, E.; Gimenez-Capitan, A.; Teixido, C.; Molina-Vila, M.A.; Viteri, S.; Gil, M.D.L.L.; et al. Interferon gamma, an important marker of response to immune checkpoint blockade in non-small cell lung cancer and melanoma patients. Ther. Adv. Med Oncol. 2018, 10. [Google Scholar] [CrossRef]
- Bryant, A.J.; Fu, C.; Lu, Y.; Brantly, M.L.; Mehrad, B.; Moldawer, L.L.; Brusko, T.M.; Brittain, E.L.; West, J.D.; Austin, E.D.; et al. A checkpoint on innate myeloid cells in pulmonary arterial hypertension. Pulm. Circ. 2018, 9. [Google Scholar] [CrossRef]
- Price, L.C.; Wort, S.J.; Perros, F.; Dorfmüller, P.; Huertas, A.; Montani, D.; Cohen-Kaminsky, S.; Humbert, M. Inflammation in Pulmonary Arterial Hypertension. Chest 2012, 141, 210–221. [Google Scholar] [CrossRef] [Green Version]
- Hoeper, M.M.; Barst, R.J.; Bourge, R.C.; Feldman, J.; Frost, A.E.; Galié, N.; Sánchez, M.A.G.; Grimminger, F.; Grünig, E.; Hassoun, P.M.; et al. Ghofrani. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: Results of the randomized IMPRES study. Circulation 2013, 127, 1128–1138. [Google Scholar] [CrossRef] [Green Version]
- Hurst, L.A.; Dunmore, B.J.; Long, L.; Crosby, A.; Al-Lamki, R.; Deighton, J.; Southwood, M.; Yang, X.; Nikolic, M.; Herrera, B.; et al. TNFα drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat. Commun. 2017, 8, 14079. [Google Scholar] [CrossRef]
- Tian, W.; Jiang, X.; Sung, Y.K.; Shuffle, E.; Wu, T.H.; Kao, P.N.; Tu, A.B.; Dorfmüller, P.; Cao, A.; Wang, L.; et al. Phenotypically silent bone morphogenetic protein receptor 2 mutations predispose rats to inflammation-induced pulmonary arterial hypertension by enhancing the risk for neointimal transformation. Circulation 2019, 140, 1409–1425. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and Immunity in the Pathogenesis of Pulmonary Arterial Hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, E.; Tian, X.; Cai, J.; Hopper, P.K.; Sudheendra, D.; Li, C.G.; Bizri, N.; Sawada, H.; Haghighat, R.; Chan, R.; et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J. Clin. Investig. 2013, 123, 3600–3613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiekerkoetter, E.; Sung, Y.K.; Sudheendra, D.; Scott, V.; del Rosario, P.; Bill, M.; Haddad, F.; Long-Boyle, J.; Hedlin, H.; Za-manian, R.T. Randomised placebo-controlled safety and tolerability trial of FK506 (tacrolimus) for pulmonary arterial hypertension. Eur. Respir. J. 2017, 50, 1602449. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Lin, F.; Xiao, Z.; Sun, B.; Wei, Z.; Liu, B.; Xue, L.; Xiong, C. Investigational pharmacotherapy and immunotherapy of pulmonary arterial hypertension: An update. Biomed. Pharmacother. 2020, 129, 110355. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Wang, A.; Cao, J.; Chen, L. Apelin/APJ system: An emerging therapeutic target for respiratory diseases. Cell. Mol. Life Sci. 2020, 77, 2919–2930. [Google Scholar] [CrossRef] [PubMed]
- Pankey, E.A.; Byun, R.J.; Smith, W.B.; Bhartiya, M.; Bueno, F.R.; Badejo, A.M.; Stasch, J.-P.; Murthy, S.N.; Nossaman, B.D.; Kadowitz, P.J. The Rho kinase inhibitor azaindole-1 has long-acting vasodilator activity in the pulmonary vascular bed of the intact chest rat. Can. J. Physiol. Pharmacol. 2012, 90, 825–835. [Google Scholar] [CrossRef]
- Zhang, M.; Chang, Z.; Zhang, P.; Jing, Z.; Yan, L.; Feng, J.; Hu, Z.; Xu, Q.; Zhou, W.; Ma, P.; et al. Protective effects of 18β-glycyrrhetinic acid on pulmonary arterial hypertension via regulation of Rho A/Rho kinsase pathway. Chem. Biol. Interact. 2019, 311, 108749. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Zhang, Y.; Liu, R.; Yang, X. The acute effects of 30 mg vs 60 mg of intravenous Fasudil on patients with congenital heart defects and severe pulmonary arterial hypertension. Congen. Heart Dis. 2019, 14, 645–650. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomaszewski, M.; Bębnowska, D.; Hrynkiewicz, R.; Dworzyński, J.; Niedźwiedzka-Rystwej, P.; Kopeć, G.; Grywalska, E. Role of the Immune System Elements in Pulmonary Arterial Hypertension. J. Clin. Med. 2021, 10, 3757. https://doi.org/10.3390/jcm10163757
Tomaszewski M, Bębnowska D, Hrynkiewicz R, Dworzyński J, Niedźwiedzka-Rystwej P, Kopeć G, Grywalska E. Role of the Immune System Elements in Pulmonary Arterial Hypertension. Journal of Clinical Medicine. 2021; 10(16):3757. https://doi.org/10.3390/jcm10163757
Chicago/Turabian StyleTomaszewski, Michał, Dominika Bębnowska, Rafał Hrynkiewicz, Jakub Dworzyński, Paulina Niedźwiedzka-Rystwej, Grzegorz Kopeć, and Ewelina Grywalska. 2021. "Role of the Immune System Elements in Pulmonary Arterial Hypertension" Journal of Clinical Medicine 10, no. 16: 3757. https://doi.org/10.3390/jcm10163757
APA StyleTomaszewski, M., Bębnowska, D., Hrynkiewicz, R., Dworzyński, J., Niedźwiedzka-Rystwej, P., Kopeć, G., & Grywalska, E. (2021). Role of the Immune System Elements in Pulmonary Arterial Hypertension. Journal of Clinical Medicine, 10(16), 3757. https://doi.org/10.3390/jcm10163757