
Arterial Stiffness in Type 1 Diabetes: The Case for the Arterial Wall Itself as a Target Organ

 , ,
, ,
Abstract
1. Introduction
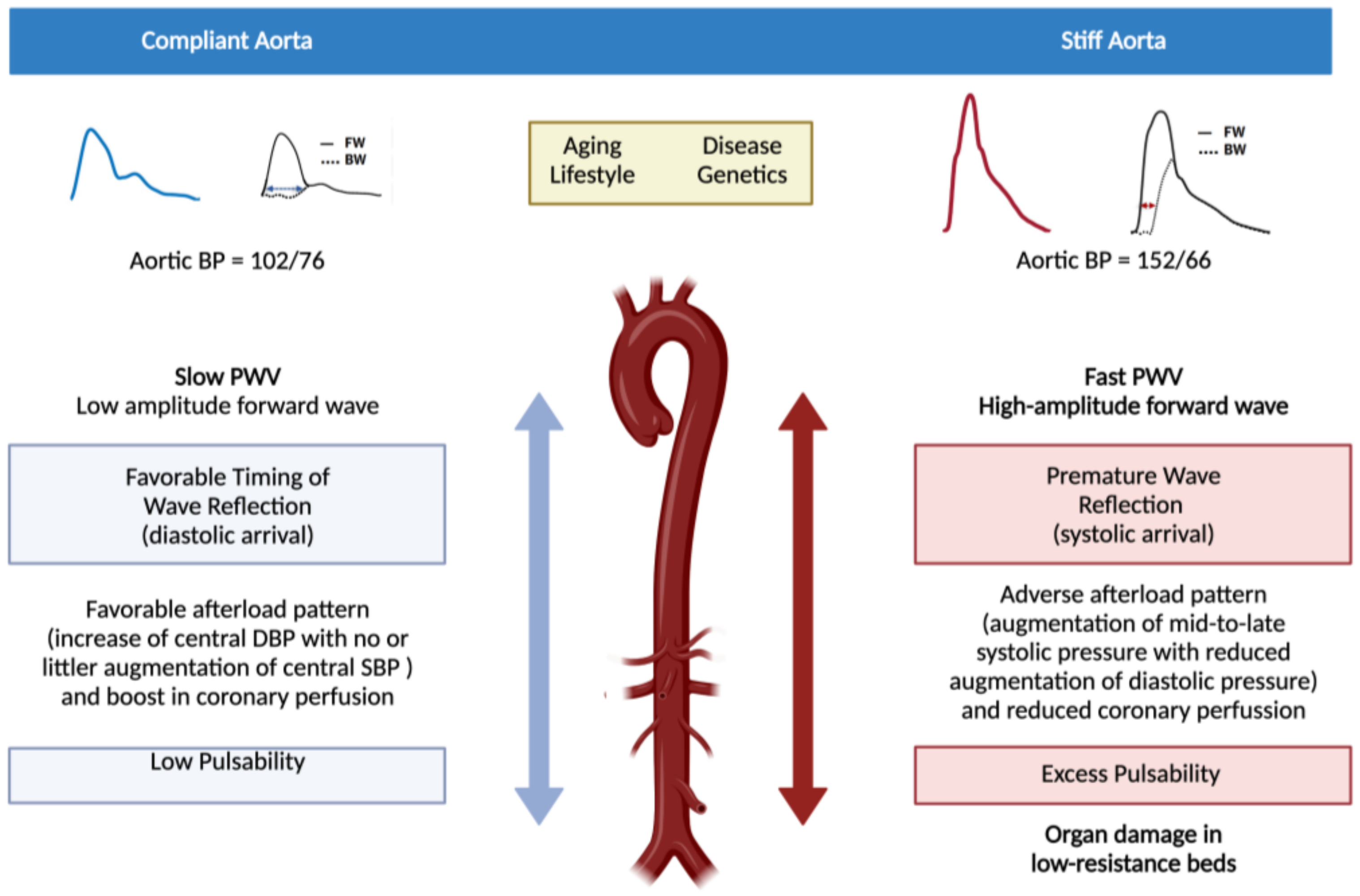
2. Pathophysiological and Clinical Consequences of Arterial Stiffness
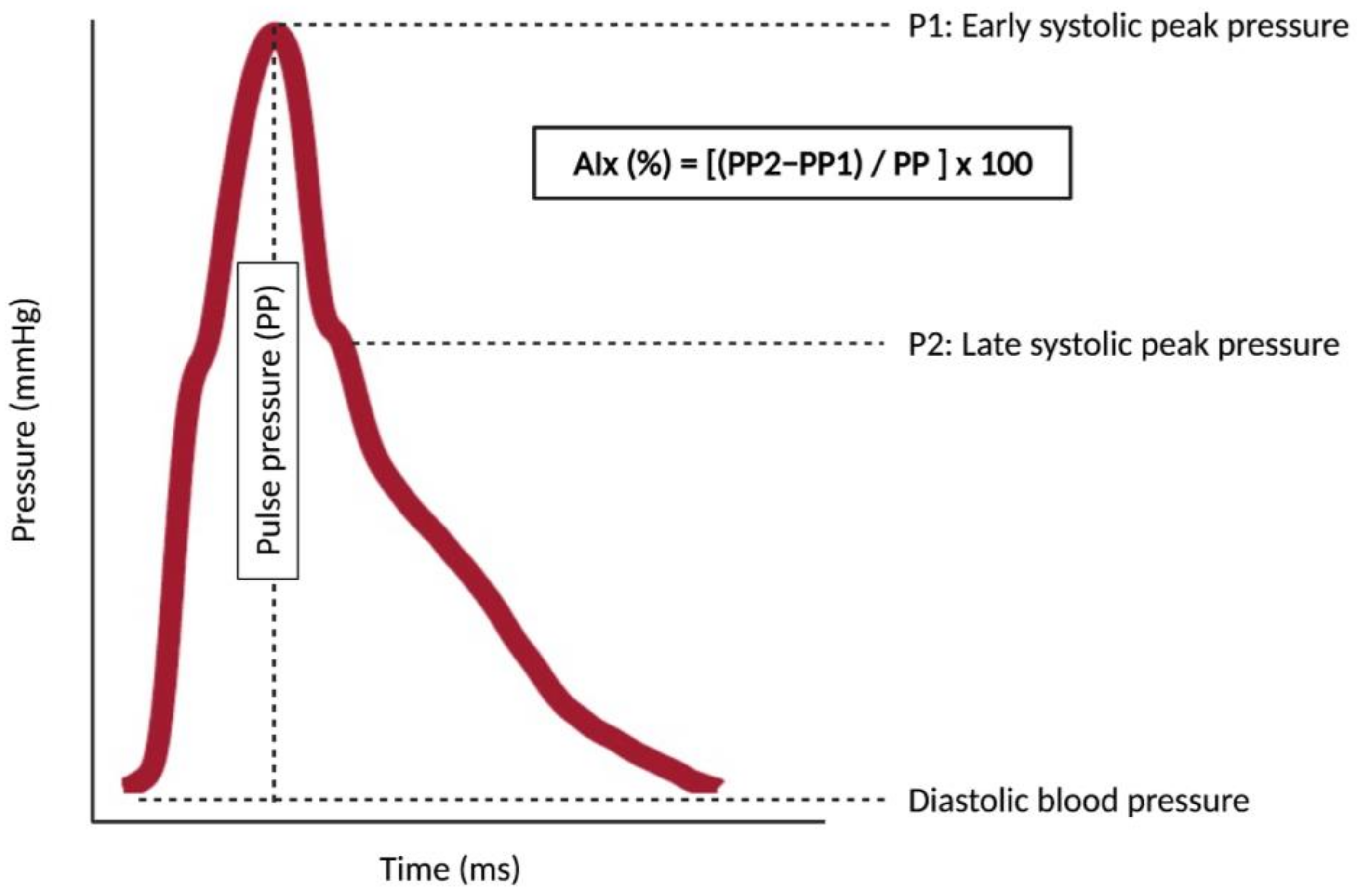
3. Measuring Aortic Stiffness in Daily Clinical Practice
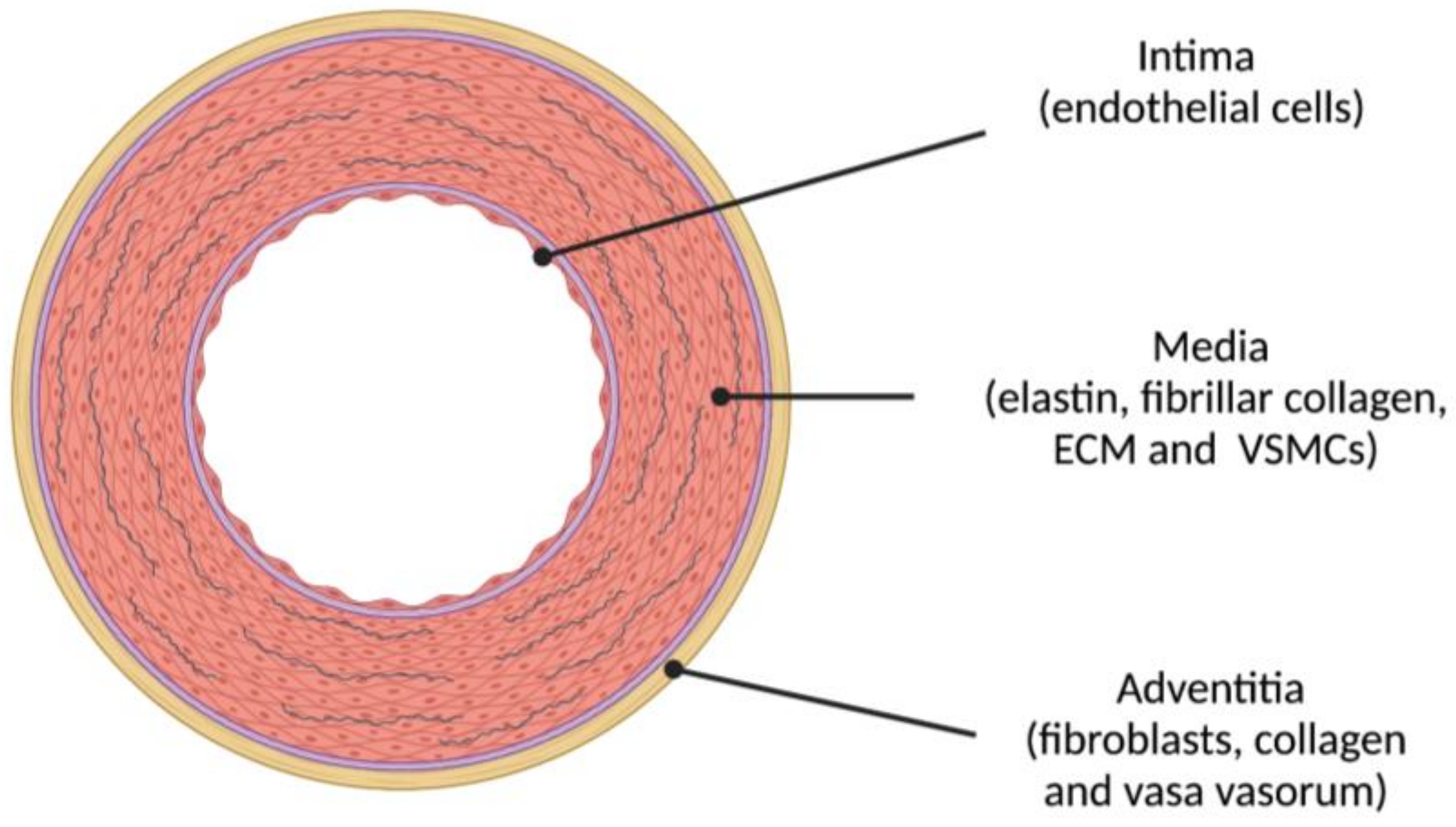
4. Pathophysiological Mechanisms Implicated in Stiffening of Large Elastic Arteries
5. Arterial Stiffness in Diabetes Mellitus. The Case of Type 1 Diabetes Mellitus in Adults
6. Arterial Stiffness in Diabetes: Future Directions
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- Lloyd-Jones, D.M.; Wilson, P.W.; Larson, M.; Beiser, A.; Leip, E.P.; D’Agostino, R.B.; Levy, D. Framingham risk score and prediction of lifetime risk for coronary heart disease. Am. J. Cardiol. 2004, 94, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Conroy, R.; Pyörälä, K.; Fitzgerald, A.; Sans, S.; Menotti, A.; De Backer, G.; De Bacquer, D.; Ducimetière, P.; Jousilahti, P.; Keil, U.; et al. Estimation of ten-year risk of fatal cardiovascular disease in Europe: The SCORE project. Eur. Heart J. 2003, 24, 987–1003. [Google Scholar] [CrossRef]
- Vistisen, D.; Andersen, G.S.; Hansen, C.S.; Hulman, A.; Henriksen, J.E.; Bech-Nielsen, H.; Jørgensen, M.E. Prediction of first cardiovascular disease event in type 1 diabetes mellitus: The steno type 1 risk engine. Circulation 2016, 133, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.J.; Kothari, V.; Adler, A.I.; Stratton, I.M. The UKPDS risk engine: A model for the risk of coronary heart disease in Type II diabetes (UKPDS 56). Clin. Sci. 2001, 101, 671–679. [Google Scholar] [CrossRef]
- Studziński, K.; Tomasik, T.; Krzysztoń, J.; Jóźwiak, J.; Windak, A. Effect of using cardiovascular risk scoring in routine risk assessment in primary prevention of cardiovascular disease: An overview of systematic reviews. BMC Cardiovasc. Disord. 2019, 19, 11. [Google Scholar] [CrossRef] [PubMed]
- Shirwany, A.N.; Zou, M.-H. Arterial stiffness: A brief review. Acta Pharmacol. Sin. 2010, 31, 1267–1276. [Google Scholar] [CrossRef]
- Mitchell, G.F.; Hwang, S.-J.; Vasan, R.S.; Larson, M.G.; Levy, D.; Benjamin, E.J.; Pencina, M.J.; Hamburg, N.M.; Vita, J.A. Response to letters regarding article, “Arterial stiffness and cardiovascular events: The Framingham Heart Study”. Circulation 2010, 122, e515. [Google Scholar] [CrossRef]
- Chirinos, J.A.; Segers, P.; Hughes, T.; Townsend, R. Large-artery stiffness in health and disease: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2019, 74, 1237–1263. [Google Scholar] [CrossRef]
- Townsend, R.R.; Wilkinson, I.B.; Schiffrin, E.L.; Avolio, A.P.; Chirinos, J.A.; Cockcroft, J.R.; Heffernan, K.S.; Lakatta, E.G.; McEniery, C.M.; Mitchell, G.F.; et al. Recommendations for improving and standardizing vascular research on arterial stiffness: A scientific statement from the American Heart Association. Hypertension 2015, 66, 698–722. [Google Scholar] [CrossRef]
- Aroor, A.R.; Jia, G.; Sowers, J.R. Cellular mechanisms underlying obesity-induced arterial stiffness. Am. J. Physiol. Integr. Comp. Physiol. 2018, 314, R387–R398. [Google Scholar] [CrossRef]
- Safar, M.E.; Asmar, R.; Benetos, A.; Blacher, J.; Boutouyrie, P.; Lacolley, P.; Laurent, S.; London, G.; Pannier, B.; Protogerou, A.; et al. Interaction between hypertension and arterial stiffness. Hypertension 2018, 72, 796–805. [Google Scholar] [CrossRef]
- Laurent, S.; Boutouyrie, P. Arterial stiffness and hypertension in the elderly. Front. Cardiovasc. Med. 2020, 7, 544302. [Google Scholar] [CrossRef] [PubMed]
- Boutouyrie, P.; Bruno, R.M. The clinical significance and application of vascular stiffness measurements. Am. J. Hypertens. 2019, 32, 4–11. [Google Scholar] [CrossRef]
- Saji, N.; Toba, K.; Sakurai, T. Cerebral small vessel disease and arterial stiffness: Tsunami effect in the brain? Pulse 2016, 3, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Tsiachris, D.; Tsioufis, C.; Syrseloudis, D.; Roussos, D.; Tatsis, I.; Dimitriadis, K.; Toutouzas, K.; Tsiamis, E.; Stefanadis, C. Subendocardial viability ratio as an index of impaired coronary flow reserve in hypertensives without significant coronary artery stenoses. J. Hum. Hypertens. 2011, 26, 64–70. [Google Scholar] [CrossRef]
- Zureik, M.; Bureau, J.-M.; Temmar, M.; Adamopoulos, C.; Courbon, D.; Bean, K.; Touboul, P.-J.T.; Benetos, A.; Ducimetière, P. Echogenic carotid plaques are associated with aortic arterial stiffness in subjects with subclinical carotid atherosclerosis. Hypertension 2003, 41, 519–527. [Google Scholar] [CrossRef]
- Boesen, M.E.; Singh, D.; Menon, B.K.; Frayne, R. A systematic literature review of the effect of carotid atherosclerosis on local vessel stiffness and elasticity. Atherosclerosis 2015, 243, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-L.; Kim, S.-H. Pulse wave velocity in atherosclerosis. Front. Cardiovasc. Med. 2019, 6, 41. [Google Scholar] [CrossRef]
- Liao, S.; McLachlan, C.S. Cholesterol efflux: Does it contribute to aortic stiffening? J. Cardiovasc. Dev. Dis. 2018, 5, 23. [Google Scholar] [CrossRef]
- Laurent, S.; Cockcroft, J.; Van Bortel, L.; Boutouyrie, P.; Giannattasio, C.; Hayoz, D.; Pannier, B.; Vlachopoulos, C.; Wilkinson, I.; Struijker-Boudier, H.; et al. Expert consensus document on arterial stiffness: Methodological issues and clinical applications. Eur. Heart J. 2006, 27, 2588–2605. [Google Scholar] [CrossRef] [PubMed]
- The Reference Values for Arterial Stiffness’ Collaboration. Determinants of pulse wave velocity in healthy people and in the presence of cardiovascular risk factors: ‘Establishing normal and reference values’. Eur Heart J. 2010, 31, 2338–2350. [Google Scholar] [CrossRef]
- Van Bortel, L.M.; Laurent, S.; Boutouyrie, P.; Chowienczyk, P.; Cruickshank, J.K.; De Backer, T.; Filipovsky, J.; Huybrechts, S.; Mattace-Raso, F.U.S.; Protogerou, A.G. Expert consensus document on the measurement of aortic stiffness in daily practice using carotid-femoral pulse wave velocity. J. Hypertens. 2012, 30, 445–448. [Google Scholar] [CrossRef]
- Segers, P.; Rietzschel, E.R.; Chirinos, J.A. How to measure arterial stiffness in humans. Arter. Thromb. Vasc. Biol. 2020, 40, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Boutouyrie, P.; Fliser, D.; Goldsmith, D.; Covic, A.; Wiecek, A.; Ortiz, A.; Martinez-Castelao, A.; Lindholm, B.; Massy, Z.A.; Suleymanlar, G.; et al. Assessment of arterial stiffness for clinical and epidemiological studies: Methodological considerations for validation and entry into the European Renal and Cardiovascular Medicine registry. Nephrol. Dial. Transplant. 2014, 29, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, J.L.; Lima, J.A.; Redheuil, A.; Al-Mallah, M.H. Aortic stiffness: Current understanding and future directions. J. Am. Coll. Cardiol. 2011, 57, 1511–1522. [Google Scholar] [CrossRef]
- Laurent, S.; Marais, L.; Boutouyrie, P. The noninvasive assessment of vascular aging. Can. J. Cardiol. 2016, 32, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Battistoni, A.; Michielon, A.; Marino, G.; Savoia, C. Vascular Aging and central aortic blood pressure: From pathophysiology to treatment. High Blood Press. Cardiovasc. Prev. 2020, 27, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Townsend, R.R. Arterial stiffness: Recommendations and standardization. Pulse 2016, 4, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Gomez, D. Smooth muscle cell phenotypic diversity. Arter. Thromb. Vasc. Biol. 2019, 39, 1715–1723. [Google Scholar] [CrossRef]
- Lacolley, P.; Regnault, V.; Laurent, S. Mechanisms of arterial stiffening: From mechanotransduction to epigenetics. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Maegdefessel, L.; Rayner, K.J.; Leeper, N.J. MicroRNA regulation of vascular smooth muscle function and phenotype: Early career committee contribution. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2–6. [Google Scholar] [CrossRef]
- Rossman, M.J.; Gioscia-Ryan, R.A.; Clayton, Z.S.; Murphy, M.P.; Seals, D.R. Targeting mitochondrial fitness as a strategy for healthy vascular aging. Clin. Sci. 2020, 134, 1491–1519. [Google Scholar] [CrossRef]
- Rubin, J.; Nambi, V.; Chambless, L.E.; Steffes, M.W.; Juraschek, S.; Coresh, J.; Sharrett, A.R.; Selvin, E. Hyperglycemia and arterial stiffness: The Atherosclerosis Risk in the Communities study. Atherosclerosis 2012, 225, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Lovshin, J.A.; Lytvyn, Y.; Lovblom, L.E.; Katz, A.; Boulet, G.; Bjornstad, P.; Lai, V.; Cham, L.; Tse, J.; Orszag, A.; et al. Retinopathy and RAAS activation: Results from the Canadian Study of Longevity in type 1 diabetes. Diabetes Care 2018, 42, 273–280. [Google Scholar] [CrossRef]
- Kim, M.; Kim, M.; Yoo, H.J.; Bang, Y.J.; Lee, S.H.; Lee, J.H. Apolipoprotein A5 gene variants are associated with decreased adiponectin levels and increased arterial stiffness in subjects with low high-density lipoprotein-cholesterol levels. Clin. Genet. 2018, 94, 438–444. [Google Scholar] [CrossRef]
- Nanoudis, S.; Pikilidou, M.; Yavropoulou, M.; Zebekakis, P. The Role of MicroRNAs in Arterial Stiffness and Arterial Calci-fication. An Update and Review of the Literature. Front. Genet. 2017, 8, 209. [Google Scholar] [CrossRef]
- Parthenakis, F.; Marketou, M.; Kontaraki, J.; Patrianakos, A.; Nakou, H.; Touloupaki, M.; Vernardos, M.; Kochiadakis, G.; Chlouverakis, G.; Vardas, P. Low levels of MicroRNA-21 are a marker of reduced arterial stiffness in well-controlled hypertension. J. Clin. Hypertens. 2016, 19, 235–240. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.S.; El Ghormli, L.; Gidding, S.; Bacha, F.; Nadeau, K.J.; Katz, L.E.L.; Tryggestad, J.B.; Leibel, N.; Hale, D.E.; Urbina, E.M. Prevalence of arterial stiffness in adolescents with type 2 diabetes in the TODAY cohort: Relationships to glycemic control and other risk factors. J. Diabetes Complicat. 2018, 32, 740–745. [Google Scholar] [CrossRef]
- Dabelea, D.; Stafford, J.M.; Mayer-Davis, E.J.; D’Agostino, R.; Dolan, L.; Imperatore, G.; Linder, B.; Lawrence, J.M.; Marcovina, S.M.; Mottl, A.K.; et al. Association of type 1 diabetes vs type 2 diabetes diagnosed during childhood and adolescence with complications during teenage years and young adulthood. JAMA 2017, 317, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, S.J.; Levin, D.; Looker, H.C.; Lindsay, R.; Wild, S.H.; Joss, N.; Leese, G.; Leslie, P.; McCrimmon, R.; Metcalfe, W.; et al. Estimated life expectancy in a scottish cohort with type 1 diabetes, 2008–2010. JAMA 2015, 313, 37–44. [Google Scholar] [CrossRef]
- Soedamah-Muthu, S.S.; Fuller, J.H.; Mulnier, H.E.; Raleigh, V.S.; Lawrenson, R.A.; Colhoun, H. High Risk of Cardiovascular Disease in Patients With Type 1 Diabetes in the U.K.: A cohort study using the General Practice Research Database. Diabetes Care 2006, 29, 798–804. [Google Scholar] [CrossRef]
- Rönnback, M.; Fagerudd, J.; Forsblom, C.; Pettersson-Fernholm, K.; Reunanen, A.; Groop, P.-H. Altered age-related blood pressure pattern in type 1 diabetes. Circulation 2004, 110, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Llaurado, G.; Mallafré, V.C.; Vilardell, C.; Simo, R.; Freixenet, N.; Vendrell, J.; Gonzalez-Clemente, J.M. Arterial stiffness is increased in patients with type 1 diabetes without cardiovascular disease: A potential role of low-grade inflammation. Diabetes Care 2012, 35, 1083–1089. [Google Scholar] [CrossRef]
- Prince, C.T.; Secrest, A.M.; Mackey, R.H.; Arena, V.C.; Kingsley, L.A.; Orchard, T. Cardiovascular autonomic neuropathy, HDL cholesterol, and smoking correlate with arterial stiffness markers determined 18 years later in type 1 diabetes. Diabetes Care 2009, 33, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Prince, C.T.; Secrest, A.M.; Mackey, R.H.; Arena, V.C.; Kingsley, L.A.; Orchard, T.J. Pulse wave analysis and prevalent cardiovascular disease in type 1 diabetes. Atherosclerosis 2010, 213, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Gordin, D.; Wadén, J.; Forsblom, C.; Thorn, L.; Rosengård-Bärlund, M.; Tolonen, N.; Saraheimo, M.; Harjutsalo, H.; Groop, P.-H. Pulse pressure predicts incident cardio-vascular disease but not diabetic nephropathy in patients with type 1 diabetes (The FinnDiane Study). Diabetes Care 2011, 34, 886–891. [Google Scholar] [CrossRef][Green Version]
- Theilade, S.; Lajer, M.; Jorsal, A.; Tarnow, L.; Parving, H.H.; Rossing, P. Arterial stiffness and endothelial dysfunction independently and synergistically predict cardiovascular and renal outcome in patients with type 1 diabetes. Diabet. Med. 2012, 29, 990–994. [Google Scholar] [CrossRef]
- Gordin, D.; Wadén, J.; Forsblom, C.; Thorn, L.M.; Rosengård-Bärlund, M.; Heikkilä, O.; Saraheimo, M.; Tolonel, N.; Hietala, K.; Soro-Paavonen, A.; et al. Arterial stiffness and vascular compli-cations in patients with type 1 diabetes: The Finnish Diabetic Nephropathy (FinnDiane) Study. Ann. Med. 2012, 44, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Theilade, S.; Lajer, M.; Persson, F.; Joergensen, C.; Rossing, P. Arterial stiffness is associated with cardiovascular, renal, retinal, and autonomic disease in type 1 diabetes. Diabetes Care 2013, 36, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Theilade, S.; Lajer, M.; Hansen, T.W.; Rossing, P. Pulse wave reflection is associated with diabetes duration, albuminuria and cardiovascular disease in type 1 diabetes. Acta Diabetol. 2014, 51, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Theilade, S.; Hansen, T.W.; Rossing, P. Central hemodynamics are associated with cardiovascular disease and albuminuria in type 1 diabetes. Am. J. Hypertens. 2014, 27, 1152–1159. [Google Scholar] [CrossRef]
- Llauradó, G.; Canonge, R.S.; Albert, L.; Ballesta, S.; Mazarico, I.; Luchtenberg, M.-F.; González-Sastre, M.; Megia, A.; Simó, R.; Vendrell, J.; et al. Arterial stiffness is highly correlated with the scores obtained from the steno type 1 risk engine in subjects with T1DM. PLoS ONE 2019, 14, e0220206. [Google Scholar] [CrossRef]
- Tynjälä, A.; Forsblom, C.; Harjutsalo, V.; Groop, P.-H.; Gordin, D. Arterial stiffness predicts mortality in individuals with type 1 diabetes. Diabetes Care 2020, 43, 2266–2271. [Google Scholar] [CrossRef]
- González-Clemente, J.M.; Gimenez-Perez, G.; Richart, C.; Broch, M.; Caixàs, A.; Megia, A.; Giménez-Palop, O.; Simón, I.; Mauricio, D.; Vendrell, J. The tumour necrosis factor (TNF)-α system is activated in accordance with pulse pressure in normotensive subjects with type 1 diabetes mellitus. Eur. J. Endocrinol. 2005, 153, 687–691. [Google Scholar] [CrossRef]
- Llauradó, G.; Mallafré, V.C.; Vilardell, C.; Simó, R.; Gil, P.; Canonge, R.S.; Vendrell, J.; González-Clemente, J.-M. Advanced glycation end products are associated with arterial stiffness in type 1 diabetes. J. Endocrinol. 2014, 221, 405–413. [Google Scholar] [CrossRef]
- Llauradó, G.; Megía, A.; Cano, A.; Giménez-Palop, O.; Simón, I.; González-Sastre, M.; Berlanga, E.; Fernández-Veledo, S.; Vendrell, J.; González-Clemente, J.-M. FGF-23/Vitamin D axis in type 1 diabetes: The potential role of mineral metabolism in arterial stiffness. PLoS ONE 2015, 10, e0140222. [Google Scholar] [CrossRef]
- Zobel, E.H.; Theilade, S.; von Scholten, B.J.; Persson, F.; Tarnow, L.; Lajer, M.; Hansen, T.W.; Rossing, P. Higher parathyroid hormone level is associated with increased arterial stiffness in type 1 diabetes. Diabetes Care 2017, 40, e32–e33. [Google Scholar] [CrossRef]
- Llauradó, G.; Amigó, N.; Cano, A.; Ballesta, S.; Albert, L.; Mazarico, I.; Fernandez-Veledo, S.; Pedro-Botet, J.; Vandrell, J.; Gonzalez-Clament, J.-M. Specific nuclear magnetic resonance lipoprotein sub-class profiles and central arterial stiffness in type 1 diabetes mellitus: A case control study. J. Clin. Med. 2019, 8, 1875. [Google Scholar] [CrossRef] [PubMed]
- Kietsiriroje, N.; Pearson, S.; Campbell, M.; Ariëns, R.A.S.; Ajjan, R.A. Double diabetes: A distinct high-risk group? Diabetes Obes. Metab. 2019, 21, 2609–2618. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti, R.E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, I.B.; Mäki-Petäjä, K.M.; Mitchell, G.F. Uses of arterial stiffness in clinical practice. Arter. Thromb. Vasc. Biol. 2020, 40, 1063–1067. [Google Scholar] [CrossRef]
- Vlachopoulos, C.; Xaplanteris, P.; Aboyans, V.; Brodmann, M.; Cífková, R.; Cosentino, F.; De Carlo, M.; Gallino, A.; Landmesser, U.; Laurent, S.; et al. The role of vascular biomarkers for primary and secondary prevention. A position paper from the European Society of Cardiology Working Group on peripheral circulation: Endorsed by the Association for Research into Arterial Structure and Physiology (ARTERY) Society. Atherosclerosis 2015, 241, 507–532. [Google Scholar] [CrossRef]
- Mandraffino, G.; Scicali, R.; Rodríguez-Carrio, J.; Savarino, F.; Mamone, F.; Scuruchi, M.; Cinquegrani, M.; Imbalzano, E.; Di Pino, A.; Piro, S.; et al. Arterial stiffness improvement after adding on PCSK9 inhibitors or ezetimibe to high-intensity statins in patients with familial hypercholesterolemia: A two–lipid center real-world experience. J. Clin. Lipidol. 2020, 14, 231–240. [Google Scholar] [CrossRef]
- Ikonomidis, I.; Pavlidis, G.; Thymis, J.; Birba, D.; Kalogeris, A.; Kousathana, F.; Kountouri, A.; Balampanis, K.; Parissis, J.; Andreadou, I.; et al. Effects of glucagon-like peptide-1 receptor agonists, sodium-glucose cotransporter-2 inhibitors, and their combination on endothelial glycocalyx, arterial function, and myocardial work index in patients with type 2 diabetes mellitus after 12-month treatment. J. Am. Heart Assoc. 2020, 9, e015716. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Recommendations for Measuring cfPWV |
|---|
| 1. Place: a quiet room with a stable room temperature (e.g., 21–23 °C) |
| 2. Position: supine after at least 10 min rest |
| 3. Measurements preferably done at the right common carotid and common femoral arteries |
| 4. Repeated measures should be done at the same time of the day |
| 5. No food, caffeine or smoking within 3 h before measurements |
| 6. No speaking or sleeping during measurements |
| 7. Data should be mean of registrations during one respiratory cycle (about 5–6 s) |
| 8. Be aware of possible white-coat effects |
| 9. Measure distance in a straight line (carotid-femoral). If not possible with a tape meter (e.g., obesity) an infantometer may be helpful used upside-down |
| 10. Take mean of at least two measurements; if difference between them is >0.5 m/s perform a third measurement and take the median (not mean) of the three measurements |
| 11. Circumstances in which measurements of cfPWV should not be performed: arrhythmia, unstable clinical situation, high-grade stenosis of carotid artery, carotid sinus syndrome. |
| Author, Year, Design | Surrogate of AS | Results |
|---|---|---|
| Prince, 2010, cross-sectional [47] | AIx, AP, SEVR | Cardiac autonomic neuropathy associates with higher AIx and AP and lower SEVR |
| Prince, 2010, cross-sectional [48] | AIx, AP, SEVR | Higher AP associates with prevalent coronary artery disease. Lower SEVR associates with lower ankle-brachial index. |
| Gordin, 2011, prospective [49] | Brachial PP | Higher brachial PP predicts CV events but not nephropathy. |
| Llauradó, 2012, cross-sectional [46] | cfPWV | cfPWV is increased in T1D and associates with low-grade inflammation. |
| Theilade, 2012, prospective [50] | Brachial PP | Higher brachial PP predicts all-cause and CV deaths and progression to end-stage renal disease. |
| Gordin, 2012, cross-sectional [51] | AIx | AIx is increased in T1D and is higher in the presence of nephropathy, retinopathy and CV disease. |
| Theilade, 2013, cross-sectional [52] | cfPWV | Higher cfPWV associates with higher prevalence of CV, retinal, renal and cardiac autonomic diseases. |
| Theilade, 2014, cross-sectional [53] | AIx, AP | Higher AIx and AP associates with albuminuria and CV disease but not with retinopathy or cardiac autonomic neuropathy. |
| Theilade, 2014, cross-sectional/longitudinal [54] | Aortic systolic and diastolic pressures, aortic PP, SEVR | Higher aortic systolic pressure and PP, and lower aortic diastolic pressure and SEVR associates with CV disease and albuminuria. Lower SEVR predicts morality or end-stage renal disease. |
| Llauradó, 2019, cross-sectional [55] | cfPWV | Higher cfPWV are highly positively associated with higher CV risk according to the ST1RE. |
| Tynjälä, 2020, prospective [56] | AIx | Higher AIx predicts total mortality and a combination of CV and/or diabetes-related death. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Clemente, J.-M.; Cano, A.; Albert, L.; Giménez-Palop, O.; Romero, A.; Berlanga, E.; Vendrell, J.; Llauradó, G. Arterial Stiffness in Type 1 Diabetes: The Case for the Arterial Wall Itself as a Target Organ. J. Clin. Med. 2021, 10, 3616. https://doi.org/10.3390/jcm10163616
González-Clemente J-M, Cano A, Albert L, Giménez-Palop O, Romero A, Berlanga E, Vendrell J, Llauradó G. Arterial Stiffness in Type 1 Diabetes: The Case for the Arterial Wall Itself as a Target Organ. Journal of Clinical Medicine. 2021; 10(16):3616. https://doi.org/10.3390/jcm10163616
Chicago/Turabian StyleGonzález-Clemente, José-Miguel, Albert Cano, Lara Albert, Olga Giménez-Palop, Ana Romero, Eugenio Berlanga, Joan Vendrell, and Gemma Llauradó. 2021. "Arterial Stiffness in Type 1 Diabetes: The Case for the Arterial Wall Itself as a Target Organ" Journal of Clinical Medicine 10, no. 16: 3616. https://doi.org/10.3390/jcm10163616
APA StyleGonzález-Clemente, J.-M., Cano, A., Albert, L., Giménez-Palop, O., Romero, A., Berlanga, E., Vendrell, J., & Llauradó, G. (2021). Arterial Stiffness in Type 1 Diabetes: The Case for the Arterial Wall Itself as a Target Organ. Journal of Clinical Medicine, 10(16), 3616. https://doi.org/10.3390/jcm10163616