
Minor Allele Frequencies and Molecular Pathways Differences for SNPs Associated with Amyotrophic Lateral Sclerosis in Subjects Participating in the UKBB and 1000 Genomes Project


Abstract
:1. Introduction
2. Materials and Methods
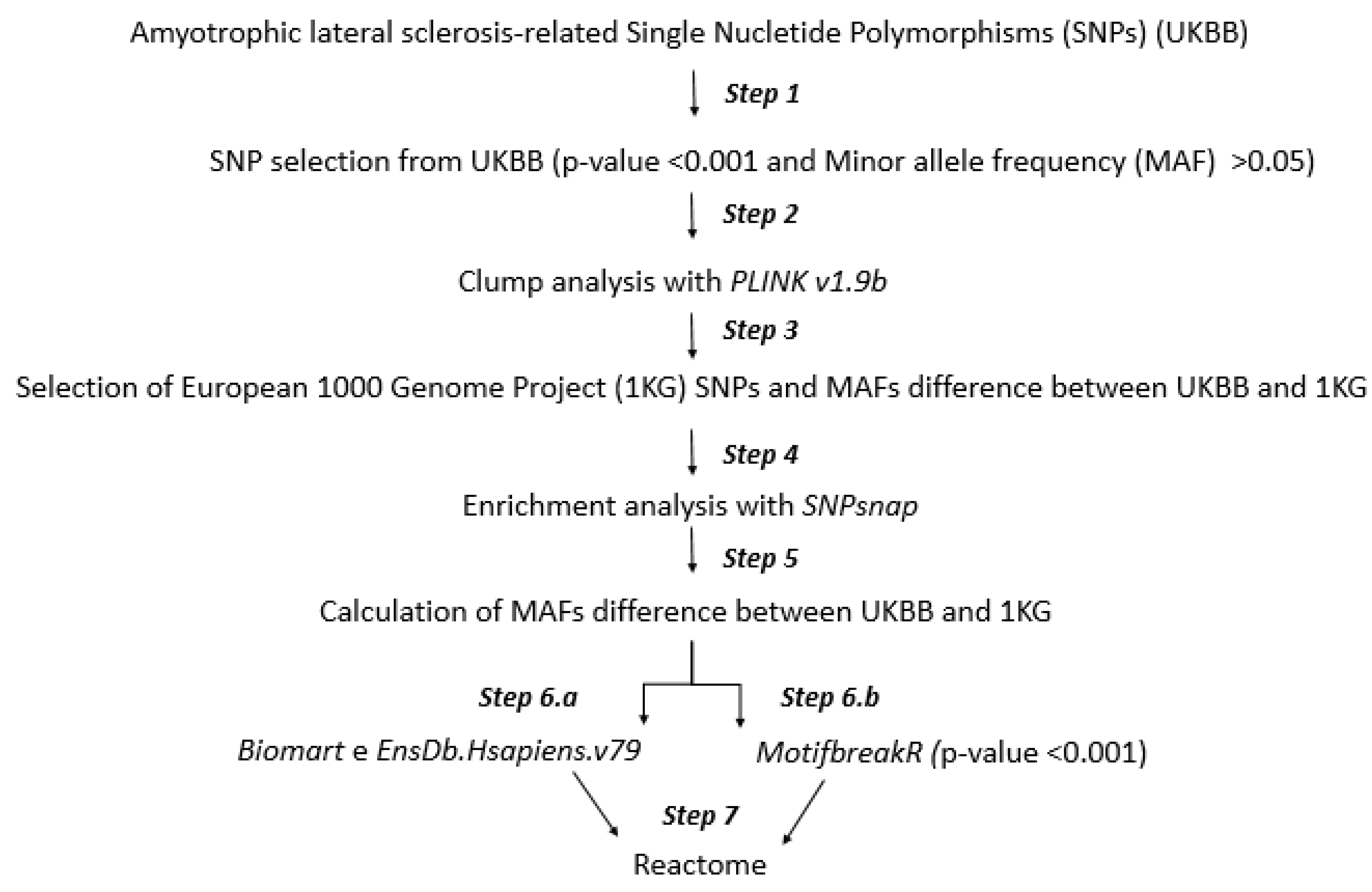
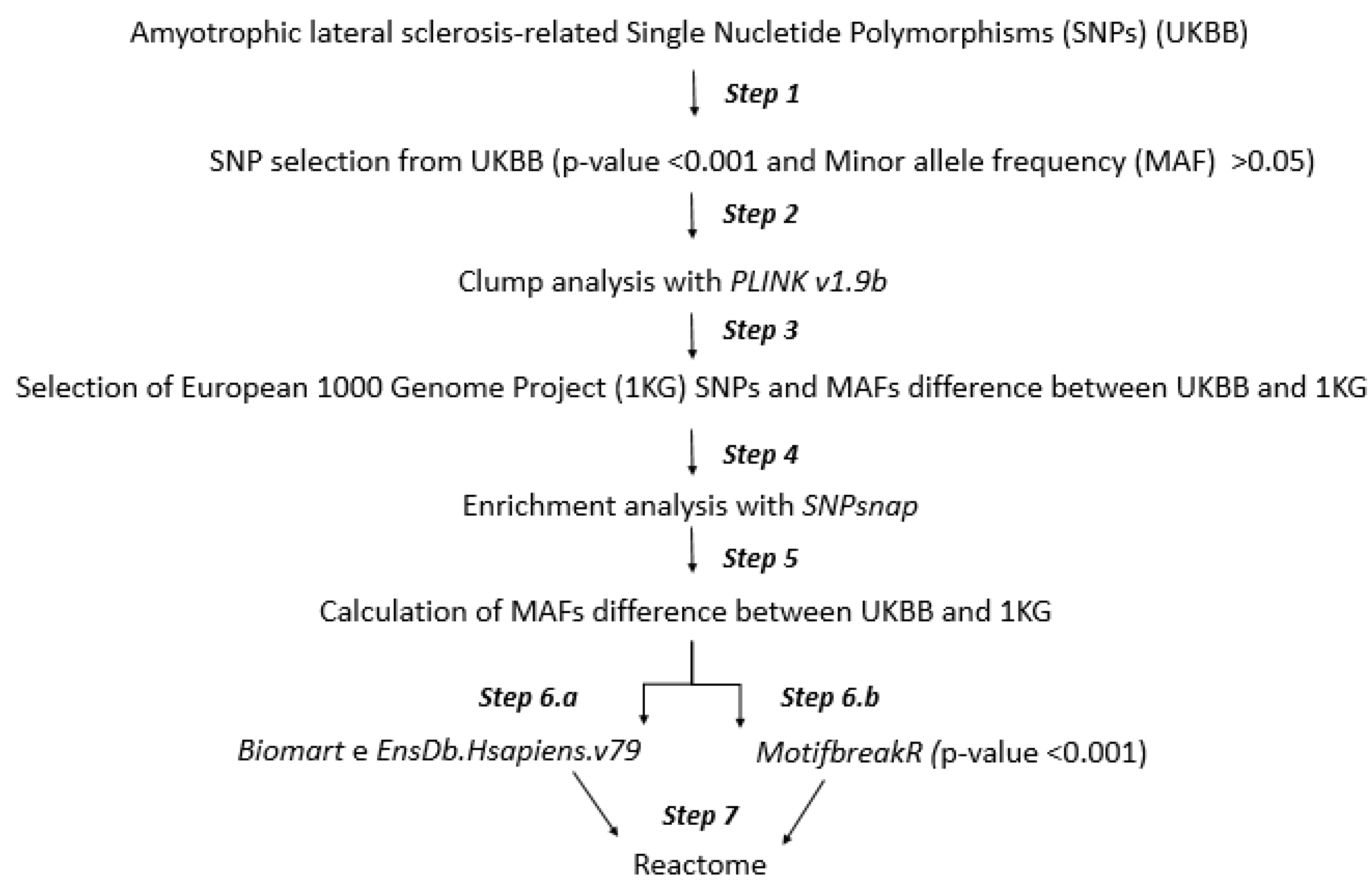
2.1. Workflow
2.2. Step 1: SNP Selection from UKBB
2.3. Step 2: Clump Analysis
2.4. Step 3: Minor Allele Frequency (MAF) Analysis
2.5. Step 4: SNP Enrichment Analysis
2.6. Step 5: Minor Allele Frequency Analysis of SNPs Enriched
2.7. Step 6.a and 6.b: Mapping to Gene Symbol and Altered Transcription Factor Binding
2.8. Step 7: Molecular Pathways
3. Results
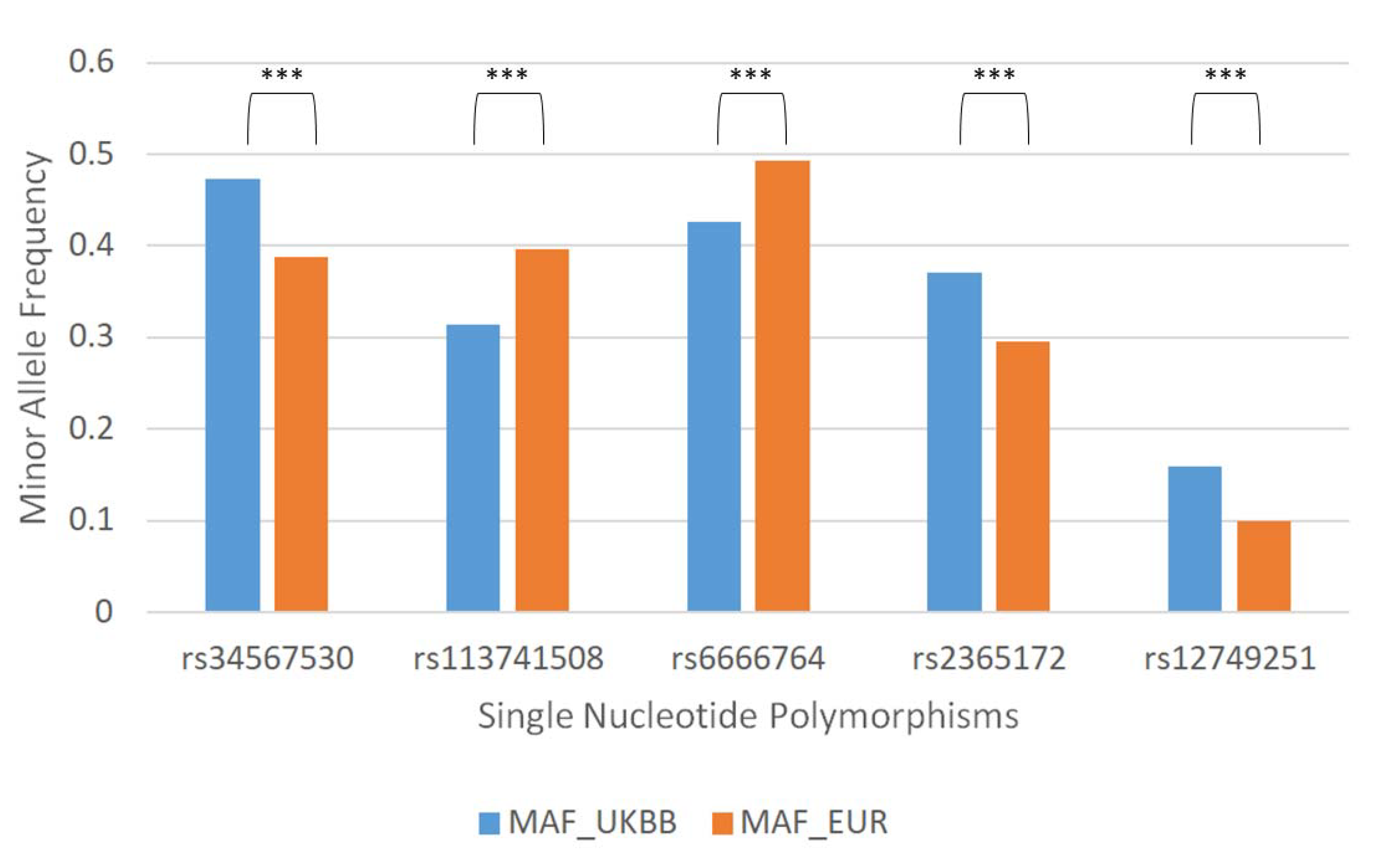
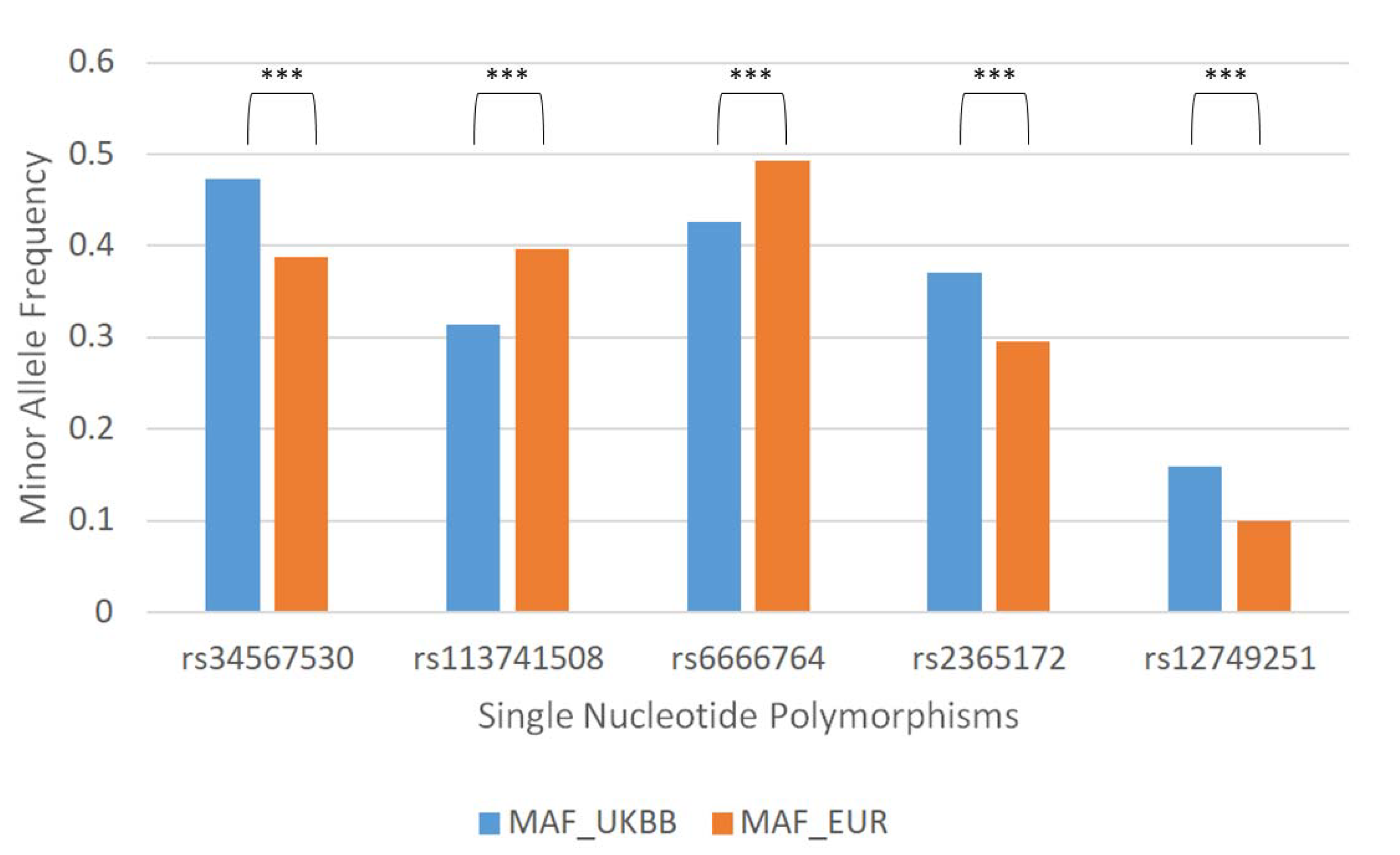
3.1. 84. SNPs Differ between United Kingdom Biobank (UKBB) and the European 1000 Genome Project (1KG)
3.2. Altered Transcription Factor Binding
3.3. Molecular Pathways
4. Discussion
4.1. 46 Genes Associated with 84 SNPs That Differ between UKBB and 1KG
4.1.1. KCNMB2 and Molecular Pathways
4.1.2. SLC15A2 and Molecular Pathway
4.1.3. DUSP10 and Molecular Pathway
4.2. Pathways Enriched by Genes Whose Motives of Binding Sites Were Altered by 70 of 84 SNPs
4.2.1. Regulation of Beta-Cell Development
4.2.2. Transcriptional Regulation by RUNX3
4.2.3. Transcriptional Regulation of Pluripotent Stem Cells
4.2.4. FOXO-Mediated Transcription of Cell Death Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Logroscino, G.; Traynor, B.J.; Hardiman, O.; Chio’, A.; Couratier, P.; Mitchell, J.D.; Swingler, R.J.; Beghi, E.; EURALS. Descriptive epidemiology of amyotrophic lateral sclerosis: New evidence and unsolved issues. J. Neurol. Neurosurg. Psychiatry 2008, 79, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Logroscino, G.; Traynor, B.J.; Hardiman, O.; Chiò, A.; Mitchell, D.; Swingler, R.J.; Millul, A.; Benn, E.; Beghi, E.; EURALS. Incidence of amyotrophic lateral sclerosis in Europe. J. Neurol. Neurosurg. Psychiatry 2010, 81, 385–390. [Google Scholar] [CrossRef]
- Mazzini, L.; Balzarini, C.; Colombo, R.; Mora, G.; Pastore, I.; De Ambrogio, R.; Caligari, M. Effects of creatine supplementation on exercise performance and muscular strength in amyotrophic lateral sclerosis: Preliminary results. J. Neurol. Sci. 2001, 191, 139–144. [Google Scholar] [CrossRef]
- Morgan, S.; Orrell, R.W. Pathogenesis of amyotrophic lateral sclerosis. Br. Med. Bull. 2016, 119, 87–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Toole, O.; Traynor, B.J.; Brennan, P.; Sheehan, C.; Frost, E.; Corr, B.; Hardiman, O. Epidemiology and clinical features of amyotrophic lateral sclerosis in Ireland between 1995 and 2004. J. Neurol Neurosurg. Psychiatry 2008, 79, 30–32. [Google Scholar] [CrossRef]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Polkey, M.I.; Lyall, R.A.; Moxham, J.; Leigh, P.N. Respiratory aspects of neurological disease. J. Neurol. Neurosurg. Psychiatry 1999, 66, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Abhinav, K.; Stanton, B.; Johnston, C.; Hardstaff, J.; Orrell, R.W.; Howard, R.; Clarke, J.; Sakel, M.; Ampong, M.A.; et al.; Amyotrophic lateral sclerosis in South-East England: A population-based study The South-East England register for amyotrophic lateral sclerosis (SEALS Registry). Neuroepidemiology 2007, 29, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Greenway, M.J.; Andersen, P.M.; Russ, C.; Ennis, S.; Cashman, S.; Donaghy, C.; Patterson, V.; Swingler, R.; Kieran, D.; Prehn, J.; et al. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat. Genet. 2006, 38, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Ticozzi, N.; Vance, C.; Leclerc, A.L.; Keagle, P.; Glass, J.D.; McKenna-Yasek, D.; Sapp, P.C.; Silani, V.; Bosco, D.A.; Shaw, C.E.; et al. Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Valdmanis, P.N.; Rouleau, G.A. Genetics of familial amyotrophic lateral sclerosis. Neurology 2008, 70, 144–152. [Google Scholar] [CrossRef]
- Zhang, K.; Liu, Q.; Shen, D.; Tai, H.; Liu, S.; Wang, Z.; Shi, J.; Fu, H.; Wu, S.; Ding, Q.; et al. Mutation analysis of KIF5A in Chinese amyotrophic lateral sclerosis patients. Neurobio. Aging 2019, 73, 229.e1–229.e4. [Google Scholar] [CrossRef]
- Bonafede, R.; Mariotti, R. ALS Pathogenesis and Therapeutic Approaches: The Role of Mesenchymal Stem Cells and Extracellular Vesicles. Front. Cell Neurosci. 2017, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K. Motor neuron disease: The bare essentials. Pr. Neurol. 2009, 9, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Benjaminsen, E.; Alstadhaug, K.B.; Gulsvik, M.; Baloch, F.K.; Odeh, F. Amyotrophic lateral sclerosis in Nordland county, Norway, 2000–2015: Prevalence, incidence, and clinical features. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 522–527. [Google Scholar] [CrossRef]
- Longinetti, E.; Regodón Wallin, A.; Samuelsson, K.; Press, R.; Zachau, A.; Ronnevi, L.O.; Kierkegaard, M.; Andersen, P.M.; Hillert, J.; Fang, F.; et al. The Swedish motor neuron disease quality registry. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 528–537. [Google Scholar] [CrossRef]
- Jun, K.Y.; Park, J.; Oh, K.W.; Kim, E.M.; Bae, J.S.; Kim, I.; Kim, S.H. Epidemiology of ALS in Korea using nationwide big data. J. Neurol. Neurosurg. Psychiatry 2019, 90, 395–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palese, F.; Sartori, A.; Verriello, L.; Ros, S.; Passadore, P.; Manganotti, P.; Barbone, F.; Pisa, F.E. Epidemiology of amyotrophic lateral sclerosis in Friuli-Venezia Giulia, North-Eastern Italy, 2002–2014: A retrospective population-based study. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Leighton, D.J.; Newton, J.; Stephenson, L.J.; Colville, S.; Davenport, R.; Gorrie, G.; Morrison, I.; Swingler, R.; Chandran, S.; Pal, S. CARE-MND Consortium. Changing epidemiology of motor neurone disease in Scotland. J. Neurol. 2019, 266, 817–825. [Google Scholar] [CrossRef] [Green Version]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International HapMap Consortium. The International HapMap Project. Nature 2003, 426, 789–796. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [Green Version]
- SNPsnap-Broad Institute. Available online: https://data.broadinstitute.org/mpg/snpsnap/documentation.html (accessed on 3 April 2021).
- Pers, T.H.; Timshel, P.; Hirschhorn, J.N. SNPsnap: A Web-based tool for identification and annotation of matched SNPs. Bioinformatics 2015, 31, 418–420. [Google Scholar] [CrossRef]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef] [Green Version]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Rainer, J.; Gatto, L.; Weichenberger, C.X. Ensembldb: An R package to create and use Ensembl-based annotation resources. Bioinformatics 2019, 35, 3151–3153. [Google Scholar] [CrossRef] [Green Version]
- Coetzee, S.G.; Coetzee, G.A.; Hazelett, D.J. motifbreakR: An R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics 2015, 31, 3847–3849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Forner, O.; Marin-Garcia, P.; Arnau, V.; D’Eustachio, P.; Stein, L.; Hermjakob, H. Reactome pathway analysis: A high-performance in-memory approach. BMC Bioinformatics 2017, 18, 142. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, R.; Misawa, K.; Tohnai, G.; Nakatochi, M.; Furuhashi, S.; Atsuta, N.; Hayashi, N.; Yokoi, D.; Watanabe, H.; Watanabe, H.; et al. A multi-ethnic meta-analysis identifies novel genes, including ACSL5, associated with amyotrophic lateral sclerosis. Commun. Biol. 2020, 3, 526. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saberi, S.; Stauffer, J.E.; Schulte, D.J.; Ravits, J. Neuropathology of Amyotrophic Lateral Sclerosis and Its Variants. Neurol. Clin. 2015, 33, 855–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Verbeeck, C.; Deng, Q.; Dejesus-Hernandez, M.; Taylor, G.; Ceballos-Diaz, C.; Kocerha, J.; Golde, T.; Das, P.; Rademakers, R.; Dickson, D.W.; et al. Expression of Fused in sarcoma mutations in mice recapitulates the neuropathology of FUS proteinopathies and provides insight into disease pathogenesis. Mol. Neurodegener. 2012, 7, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takanashi, K.; Yamaguchi, A. Aggregation of ALS-linked FUS mutant sequesters RNA binding proteins and impairs RNA granules formation. Biochem. Biophys. Res. Commun. 2014, 452, 600–607. [Google Scholar] [CrossRef]
- Ince, P.G.; Tomkins, J.; Slade, J.Y.; Thatcher, N.M.; Shaw, P.J. Amyotrophic lateral sclerosis associated with genetic abnormalities in the gene encoding Cu/Zn superoxide dismutase: Molecular pathology of five new cases, and comparison with previous reports and 73 sporadic cases of ALS. J. Neuropathol. Exp. Neurol. 1998, 57, 895–904. [Google Scholar] [CrossRef]
- Bosco, D.A.; Morfini, G.; Karabacak, N.M.; Song, Y.; Gros-Louis, F.; Pasinelli, P.; Goolsby, H.; Fontaine, B.A.; Lemay, N.; McKenna-Yasek, D.; et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 2010, 13, 1396–1403. [Google Scholar] [CrossRef] [Green Version]
- Forsberg, K.; Jonsson, P.A.; Andersen, P.M.; Bergemalm, D.; Graffmo, K.S.; Hultdin, M.; Jacobsson, J.; Rosquist, R.; Marklund, S.L.; Brännström, T. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS ONE 2010, 5, e11552. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, K.; Andersen, P.M.; Marklund, S.L.; Brännström, T. Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2011, 121, 623–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesiridis, G.S.; Lee, V.M.; Trojanowski, J.Q. Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum. Mol. Genet. 2009, 18, R156–R162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Deerlin, V.M.; Leverenz, J.B.; Bekris, L.M.; Bird, T.D.; Yuan, W.; Elman, L.B.; Clay, D.; Wood, E.M.; Chen-Plotkin, A.S.; Martinez-Lage, M.; et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: A genetic and histopathological analysis. Lancet Neurol. 2008, 7, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, A.; Kenna, K.P.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1268–1283.e6. [Google Scholar] [CrossRef] [Green Version]
- Miki, H.; Setou, M.; Kaneshiro, K.; Hirokawa, N. All kinesin superfamily protein, KIF, genes in mouse and human. Proc. Natl. Acad. Sci. USA 2001, 98, 7004–7011. [Google Scholar] [CrossRef] [Green Version]
- Kanai, Y.; Okada, Y.; Tanaka, Y.; Harada, A.; Terada, S.; Hirokawa, N. KIF5C, a novel neuronal kinesin enriched in motor neurons. J. Neurosci. 2000, 20, 6374–6384. [Google Scholar] [CrossRef] [Green Version]
- Chevalier-Larsen, E.; Holzbaur, E.L. Axonal transport and neurodegenerative disease. Biochim Biophys Acta. 2006, 1762, 1094–1108. [Google Scholar] [CrossRef] [Green Version]
- Hirokawa, N.; Niwa, S.; Tanaka, Y. Molecular motors in neurons: Transport mechanisms and roles in brain function, development, and disease. Neuron 2010, 68, 610–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millecamps, S.; Julien, J.P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, N.C.; Wang, Y.D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski TJJr Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; Valdmanis, P.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [Green Version]
- Kanai, Y.; Dohmae, N.; Hirokawa, N. Kinesin transports RNA: Isolation and characterization of an RNA-transporting granule. Neuron 2004, 43, 513–525. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, F.; Shirane, M.; Matsumoto, M.; Nakayama, K.I. Protrudin serves as an adaptor molecule that connects KIF5 and its cargoes in vesicular transport during process formation. Mol. Biol. Cell. 2011, 22, 4602–4620. [Google Scholar] [CrossRef]
- Nishimura, A.L.; Al-Chalabi, A.; Zatz, M. A common founder for amyotrophic lateral sclerosis type 8 (ALS8) in the Brazilian population. Hum. Genet. 2005, 118, 499–500. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brown, A. A hereditary spastic paraplegia mutation in kinesin-1A/KIF5A disrupts neurofilament transport. Mol. Neurodegener. 2010, 5, 52. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.H.; Roberts, E.A.; Her, L.S.; Liu, X.; Williams, D.S.; Cleveland, D.W.; Goldstein, L.S. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J. Cell Biol. 2003, 161, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Sausbier, U.; Sausbier, M.; Sailer, C.A.; Arntz, C.; Knaus, H.G.; Neuhuber, W.; Ruth, P. Ca2+ -activated K+ channels of the BK-type in the mouse brain. Histochem. Cell Biol. 2006, 125, 725–741. [Google Scholar] [CrossRef]
- Contet, C.; Goulding, S.P.; Kuljis, D.A.; Barth, A.L. BK Channels in the Central Nervous System. Int. Rev. Neurobiol. 2016, 128, 281–342. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Shao, L.R.; Chavoshy, S.; Gu, N.; Trieb, M.; Behrens, R.; Laake, P.; Pongs, O.; Knaus, H.G.; Ottersen, O.P.; et al. Presynaptic Ca2+-activated K+ channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release. J. Neurosci. 2001, 21, 9585–9597. [Google Scholar] [CrossRef] [Green Version]
- Faber, E.S.; Sah, P. Calcium-activated potassium channels: Multiple contributions to neuronal function. Neuroscientist 2003, 9, 181–194. [Google Scholar] [CrossRef]
- Raffaelli, G.; Saviane, C.; Mohajerani, M.H.; Pedarzani, P.; Cherubini, E. BK potassium channels control transmitter release at CA3-CA3 synapses in the rat hippocampus. J. Physiol. 2004, 557Pt 1, 147–157. [Google Scholar] [CrossRef]
- Griguoli, M.; Sgritta, M.; Cherubini, E. Presynaptic BK channels control transmitter release: Physiological relevance and potential therapeutic implications. J. Physiol. 2016, 594, 3489–3500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, E.A.; Han, J.; Jung, I.D.; Park, W.S. Physiological roles of K+ channels in vascular smooth muscle cells. J. Smooth Muscle Res. 2008, 44, 65–81. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.A.; Yang, Y.; Ella, S.R.; Davis, M.J.; Braun, A.P. Large conductance, Ca2+-activated K+ channels (BKCa) and arteriolar myogenic signaling. FEBS Lett. 2010, 584, 2033–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamoorthy-Natarajan, G.; Koide, M. BK Channels in the Vascular System. Int. Rev. Neurobiol. 2016, 128, 401–438. [Google Scholar] [CrossRef]
- Jin, W.; Sugaya, A.; Tsuda, T.; Ohguchi, H.; Sugaya, E. Relationship between large conductance calcium-activated potassium channel and bursting activity. Brain Res. 2000, 860, 21–28. [Google Scholar] [CrossRef]
- Gu, N.; Vervaeke, K.; Storm, J.F. BK potassium channels facilitate high-frequency firing and cause early spike frequency adaptation in rat CA1 hippocampal pyramidal cells. J. Physiol. 2007, 580 Pt 3, 859–882. [Google Scholar] [CrossRef]
- Beecham, G.W.; Hamilton, K.; Naj, A.C.; Martin, E.R.; Huentelman, M.; Myers, A.J.; Corneveaux, J.J.; Hardy, J.; Vonsattel, J.P.; Younkin, S.G.; et al. Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet. 2014, 10, e1004606. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.S.; Marx, S.O. The BK potassium channel in the vascular smooth muscle and kidney: α- and β-subunits. Kidney Int. 2010, 78, 963–974. [Google Scholar] [CrossRef] [Green Version]
- Wallner, M.; Meera, P.; Toro, L. Molecular basis of fast inactivation in voltage and Ca2+-activated K+ channels: A transmembrane beta-subunit homolog. Proc. Natl. Acad. Sci. USA 1999, 96, 4137–4142. [Google Scholar] [CrossRef] [PubMed]
- Weaver, A.K.; Bomben, V.C.; Sontheimer, H. Expression and function of calcium-activated potassium channels in human glioma cells. Glia 2006, 54, 223–233. [Google Scholar] [CrossRef] [Green Version]
- Charles, A. Intercellular calcium waves in glia. Glia 1998, 24, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Guo, L.; Li, N.; Henzel, K.S.; Gu, H.; Ran, X.; Sun, W.; Liu, S.; Lu, Y.; Ehninger, D.; et al. Overexpression of Kcnmb2 in Dorsal CA1 of Offspring Mice Rescues Hippocampal Dysfunction Caused by a Methyl Donor-Rich Paternal Diet. Front. Cell Neurosci. 2018, 12, 360. [Google Scholar] [CrossRef]
- Francis, S.H.; Corbin, J.D. Cyclic nucleotide-dependent protein kinases: Intracellular receptors for cAMP and cGMP action. Crit. Rev. Clin. Lab. Sci. 1999, 36, 275–328. [Google Scholar] [CrossRef] [PubMed]
- Moro, M.A.; Fernández-Tomé, P.; Leza, J.C.; Lorenzo, P.; Lizasoain, I. Neuronal death induced by SIN-1 in the presence of superoxide dismutase: Protection by cyclic GMP. Neuropharmacology 1998, 37, 1071–1079. [Google Scholar] [CrossRef]
- Nakamizo, T.; Kawamata, J.; Yoshida, K.; Kawai, Y.; Kanki, R.; Sawada, H.; Kihara, T.; Yamashita, H.; Shibasaki, H.; Akaike, A.; et al. Phosphodiesterase inhibitors are neuroprotective to cultured spinal motor neurons. J. Neurosci. Res. 2003, 71, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Wen, J.; Liu, J.; Li, L. The roles of free radicals in amyotrophic lateral sclerosis: Reactive oxygen species and elevated oxidation of protein, DNA, and membrane phospholipids. FASEB J. 1999, 13, 2318–2328. [Google Scholar] [CrossRef]
- Wareham, L.K.; Buys, E.S.; Sappington, R.M. The nitric oxide-guanylate cyclase pathway and glaucoma. Nitric Oxide 2018, 77, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Berka, V.; Tsai, A.L.; Murad, F. Soluble guanylyl cyclase: The nitric oxide receptor. Methods Enzymol. 2005, 396, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, B.H.; Kim, B.C.; Shin, A.; Kim, J.S.; Hong, S.H.; Hwang, J.A.; Lee, J.A.; Nam, S.; Lee, S.H.; et al. SLC15A2 genomic variation is associated with the extraordinary response of sorafenib treatment: Whole-genome analysis in patients with hepatocellular carcinoma. Oncotarget 2015, 6, 16449–16460. [Google Scholar] [CrossRef] [PubMed]
- Berger, U.V.; Hediger, M.A. Distribution of peptide transporter PEPT2 mRNA in the rat nervous system. Anat. Embryol. 1999, 199, 439–449. [Google Scholar] [CrossRef]
- Limbad, C.; Oron, T.R.; Alimirah, F.; Davalos, A.R.; Tracy, T.E.; Gan, L.; Desprez, P.Y.; Campisi, J. Astrocyte senescence promotes glutamate toxicity in cortical neurons. PLoS ONE 2020, 15, e0227887. [Google Scholar] [CrossRef]
- Kakaroubas, N.; Brennan, S.; Keon, M.; Saksena, N.K. Pathomechanisms of Blood-Brain Barrier Disruption in ALS. Neurosci. J. 2019, 2019, 2537698. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, I.T.; Skurray, R.A. The POT family of transport proteins. Trends Biochem. Sci. 1994, 19, 404. [Google Scholar] [CrossRef]
- Ayka, A.; Şehirli, A.Ö. The Role of the SLC Transporters Protein in the Neurodegenerative Disorders. Clin. Psychopharmacol. Neurosci. 2020, 18, 174–187. [Google Scholar] [CrossRef] [Green Version]
- Seifert, G.; Schilling, K.; Steinhäuser, C. Astrocyte dysfunction in neurological disorders: A molecular perspective. Nat. Rev. Neurosci. 2006, 7, 194–206. [Google Scholar] [CrossRef]
- Mishra, A.; Oulès, B.; Pisco, A.O.; Ly, T.; Liakath-Ali, K.; Walko, G.; Viswanathan, P.; Tihy, M.; Nijjher, J.; Dunn, S.J.; et al. A protein phosphatase network controls the temporal and spatial dynamics of differentiation commitment in human epidermis. Elife 2017, 6, e27356. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Martínez, M.; Stamatakis, K.; Fresno, M. The Dual-Specificity Phosphatase 10 (DUSP10): Its Role in Cancer, Inflammation, and Immunity. Int. J. Mol. Sci. 2019, 20, 1626. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.M.; Grum-Tokars, V.L.; Schavocky, J.P.; Saeed, F.; Staniszewski, A.; Teich, A.F.; Arancio, O.; Bachstetter, A.D.; Webster, S.J.; Van Eldik, L.J.; et al. Targeting human central nervous system protein kinases: An isoform selective p38αMAPK inhibitor that attenuates disease progression in Alzheimer’s disease mouse models. ACS Chem. Neurosci. 2015, 6, 666–680. [Google Scholar] [CrossRef] [Green Version]
- Sama, R.R.; Fallini, C.; Gatto, R.; McKeon, J.E.; Song, Y.; Rotunno, M.S.; Penaranda, S.; Abdurakhmanov, I.; Landers, J.E.; Morfini, G.; et al. ALS-linked FUS exerts a gain of toxic function involving aberrant p38 MAPK activation. Sci. Rep. 2017, 7, 115. [Google Scholar] [CrossRef]
- Dolenšek, J.; Rupnik, M.S.; Stožer, A. Structural similarities and differences between the human and the mouse pancreas. Islets 2015, 7, e1024405. [Google Scholar] [CrossRef] [Green Version]
- Perurena, O.H.; Festoff, B.W. Reduction in insulin receptors in amyotrophic lateral sclerosis correlates with reduced insulin sensitivity. Neurology 1987, 37, 1375–1379. [Google Scholar] [CrossRef]
- Muddapu, V.R.; Dharshini, S.A.P.; Chakravarthy, V.S.; Gromiha, M.M. Neurodegenerative Diseases-Is Metabolic Deficiency the Root Cause? Front. Neurosci. 2020, 14, 213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, H.; Zelenin, P.; Fontanet, P.; Wanderoy, S.; Petitpré, C.; Comai, G.; Bellardita, C.; Xue-Franzén, Y.; Huettl, R.E.; et al. Muscle-selective RUNX3 dependence of sensorimotor circuit development. Development 2019, 146, dev181750. [Google Scholar] [CrossRef] [Green Version]
- Lallemend, F.; Sterzenbach, U.; Hadjab-Lallemend, S.; Aquino, J.B.; Castelo-Branco, G.; Sinha, I.; Villaescusa, J.C.; Levanon, D.; Wang, Y.; Franck, M.C.; et al. Positional differences of axon growth rates between sensory neurons encoded by Runx3. EMBO J. 2012, 31, 3718–3729. [Google Scholar] [CrossRef] [Green Version]
- Levanon, D.; Bettoun, D.; Harris-Cerruti, C.; Woolf, E.; Negreanu, V.; Eilam, R.; Bernstein, Y.; Goldenberg, D.; Xiao, C.; Fliegauf, M.; et al. The Runx3 transcription factor regulates development and survival of TrkC dorsal root ganglia neurons. EMBO J. 2002, 21, 3454–3463. [Google Scholar] [CrossRef]
- de Nooij, J.C.; Doobar, S.; Jessell, T.M. Etv1 inactivation reveals proprioceptor subclasses that reflect the level of NT3 expression in muscle targets. Neuron 2013, 77, 1055–1068. [Google Scholar] [CrossRef] [Green Version]
- Patel, T.D.; Kramer, I.; Kucera, J.; Niederkofler, V.; Jessell, T.M.; Arber, S.; Snider, W.D. Peripheral NT3 signaling is required for ETS protein expression and central patterning of proprioceptive sensory afferents. Neuron 2003, 38, 403–416. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, S.; Senzaki, K.; Yoshikawa, M.; Nishimura, M.; Inoue, K.; Ito, Y.; Ozaki, S.; Shiga, T. Dynamic regulation of the expression of neurotrophin receptors by Runx3. Development 2008, 135, 1703–1711. [Google Scholar] [CrossRef] [Green Version]
- Assou, S.; Le Carrour, T.; Tondeur, S.; Ström, S.; Gabelle, A.; Marty, S.; Nadal, L.; Pantesco, V.; Réme, T.; Hugnot, J.P.; et al. A meta-analysis of human embryonic stem cells transcriptome integrated into a web-based expression atlas. Stem Cells 2007, 25, 961–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assou, S.; Cerecedo, D.; Tondeur, S.; Pantesco, V.; Hovatta, O.; Klein, B.; Hamamah, S.; De Vos, J. A gene expression signature shared by human mature oocytes and embryonic stem cells. BMC Genom. 2009, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Xu, H.; Faiola, F.; Ma’ayan, A.; Wang, J. Oct4 links multiple epigenetic pathways to the pluripotency network. Cell Res. 2012, 22, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Martí, M.; Mulero, L.; Pardo, C.; Morera, C.; Carrió, M.; Laricchia-Robbio, L.; Esteban, C.R.; Izpisua Belmonte, J.C. Characterization of pluripotent stem cells. Nat. Protoc. 2013, 8, 223–253. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.G.; Lee, J.J.; Jung, D.Y.; Jeon, J.; Heo, H.S.; Kang, H.C.; Shin, J.H.; Cho, Y.S.; Cha, K.J.; Kim, C.G.; et al. Profiling of differentially expressed genes in human stem cells by cDNA microarray. Mol. Cells 2006, 21, 343–355. [Google Scholar] [PubMed]
- Player, A.; Wang, Y.; Bhattacharya, B.; Rao, M.; Puri, R.K.; Kawasaki, E.S. Comparisons between transcriptional regulation and RNA expression in human embryonic stem cell lines. Stem Cells Dev. 2006, 15, 315–323. [Google Scholar] [CrossRef]
- Rao, R.R.; Calhoun, J.D.; Qin, X.; Rekaya, R.; Clark, J.K.; Stice, S.L. Comparative transcriptional profiling of two human embryonic stem cell lines. Biotechnol. Bioeng. 2004, 88, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Rao, S.; Chu, J.; Shen, X.; Levasseur, D.N.; Theunissen, T.W.; Orkin, S.H. A protein interaction network for pluripotency of embryonic stem cells. Nature 2006, 444, 364–368. [Google Scholar] [CrossRef]
- Clarke, D.D.; Sokoloff, L. Circulation and energy metabolism of the brain. In Basic Neurochemistry: Molecular, Cellular, and Medical Aspects; Siegel, G.J., Agranoff, B.W., Albers, R.W., Molinoff, P.B., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 1998; pp. 637–669. [Google Scholar]
- Link, W. Introduction to FOXO Biology. Methods Mol. Biol. 2019, 1890, 1–9. [Google Scholar] [CrossRef]
- Paik, J.H.; Ding, Z.; Narurkar, R.; Ramkissoon, S.; Muller, F.; Kamoun, W.S.; Chae, S.S.; Zheng, H.; Ying, H.; Mahoney, J.; et al. FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell 2009, 5, 540–553. [Google Scholar] [CrossRef] [Green Version]
- Yeo, H.; Lyssiotis, C.A.; Zhang, Y.; Ying, H.; Asara, J.M.; Cantley, L.C.; Paik, J.H. FoxO3 coordinates metabolic pathways to maintain redox balance in neural stem cells. EMBO J. 2013, 32, 2589–2602. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Single Nucleotide Polymorphism | Gene Related |
|---|---|
| rs34567530 | SLC15A2 |
| rs113741508 | LOC107985998 |
| rs6666764 | - |
| rs2365172 | - |
| rs12749251 | HECTD3 |
| Single Nucleotide Polymorphisms | Minor Allele Frequency | Gene Related |
|---|---|---|
| rs11546322 | 0.352,024 | RASGEF1C |
| rs4575343 | 0.30,842 | MGAT4C |
| Single Nucleotide Polymorphisms | Minor Allele Frequency | Gene Related |
|---|---|---|
| rs34567530 | 0.3882 | SLC5A2 |
| rs76402 | 0.3626 | ICA |
| Pathway Name | p-Value | Submitted Entities Found |
|---|---|---|
| Ca2+ activated K+ channels | 6.923 × 10−4 | KCNMB2 |
| cGMP effects | 2.147 × 10−3 | KCNMB2 |
| Nitric oxide stimulates guanylate cyclase | 4.354 × 10−3 | KCNMB2 |
| Proton/oligopeptide cotransporters | 1.677 × 10−2 | SLC15A2 |
| Signaling by MAPK mutants | 2.917 × 10−2 | DUSP10 |
| Pathway Name | p-Value | Submitted Entities Found |
|---|---|---|
| Regulation of beta-celldevelopment | 6.661 × 10−16 | NR5A2, HNF4A, HNF6, HES1, HNF1B, STF1, HNF1A, FOXA3, FOXO1, FOXA2 |
| Generic Transcription Pathway | 9.880 × 10−15 | SPI1, RORA, NR2E3, BACH1, GLI2, MECP2, RORG, HNF4A, SOX9, TEAD1, TEAD3, TEAD4, MSX2, PAX5, ERR1, P73, RUNX3, FOXP3, RUNX2, FOXP2, RUNX1, ELF1, NR5A2, TAL1, DDIT3, PPARG, PPARD, NR1I3, TCF7, LEF1, FOXO4, FOXO3, NR2C2, FOXO1, RXRA, HES1, RXRG, E2F6, SMAD2, NFE2, SMAD1, SMAD4, ZFHX3, SMAD3, NFYA, NFYB, VDR, NFYC, NR1H2, NR1H4, NR0B1, NR1D1, TBX5, NR2F6, NR4A3, SP1 |
| Transcriptional regulation by RUNX3 | 7.648 × 10−10 | SPI1, RORA, NR2E3, BACH1, GLI2, MECP2, RORG, HNF4A, SOX9, TEAD1, TEAD3, TEAD4, MSX2, PAX5, ERR1, P73, RUNX3, FOXP3, RUNX2, FOXP2, RUNX1, ELF1, NR5A2, TAL1, DDIT3, PPARG, PPARD, NR1I3, TCF7, LEF1, FOXO4, FOXO3, NR2C2, FOXO1, RXRA, HES1, RXRG, E2F6, SMAD2, NFE2, SMAD1, SMAD4, ZFHX3, SMAD3, NFYA, NFYB, VDR, NFYC, NR1H2, NR1H4, NR0B1, NR1D1, TBX5, NR2F6, NR4A3, SP1 |
| Transcriptional regulation of pluripotent stem cells | 5.635 × 10−7 | SMAD2, FOXD3, SMAD4, EPAS1, STAT3, PBX1 |
| FOXO-mediated transcription of cell death genes | 6.742 × 10−7 | NFYA, NFYB, DDIT3, NFYC, FOXO4, FOXO3, FOXO1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Antona, S.; Bertoli, G.; Castiglioni, I.; Cava, C. Minor Allele Frequencies and Molecular Pathways Differences for SNPs Associated with Amyotrophic Lateral Sclerosis in Subjects Participating in the UKBB and 1000 Genomes Project. J. Clin. Med. 2021, 10, 3394. https://doi.org/10.3390/jcm10153394
D’Antona S, Bertoli G, Castiglioni I, Cava C. Minor Allele Frequencies and Molecular Pathways Differences for SNPs Associated with Amyotrophic Lateral Sclerosis in Subjects Participating in the UKBB and 1000 Genomes Project. Journal of Clinical Medicine. 2021; 10(15):3394. https://doi.org/10.3390/jcm10153394
Chicago/Turabian StyleD’Antona, Salvatore, Gloria Bertoli, Isabella Castiglioni, and Claudia Cava. 2021. "Minor Allele Frequencies and Molecular Pathways Differences for SNPs Associated with Amyotrophic Lateral Sclerosis in Subjects Participating in the UKBB and 1000 Genomes Project" Journal of Clinical Medicine 10, no. 15: 3394. https://doi.org/10.3390/jcm10153394
APA StyleD’Antona, S., Bertoli, G., Castiglioni, I., & Cava, C. (2021). Minor Allele Frequencies and Molecular Pathways Differences for SNPs Associated with Amyotrophic Lateral Sclerosis in Subjects Participating in the UKBB and 1000 Genomes Project. Journal of Clinical Medicine, 10(15), 3394. https://doi.org/10.3390/jcm10153394