
Understanding Chronic Venous Disease: A Critical Overview of Its Pathophysiology and Medical Management

 ,
,  , ,
, ,  ,
,  , ,
, ,  ,
, 

Abstract
1. Introduction
2. Epidemiology and Risk Factors
3. Clinical Manifestations
4. Diagnosis
5. Etiology and Pathogenesis of CVD
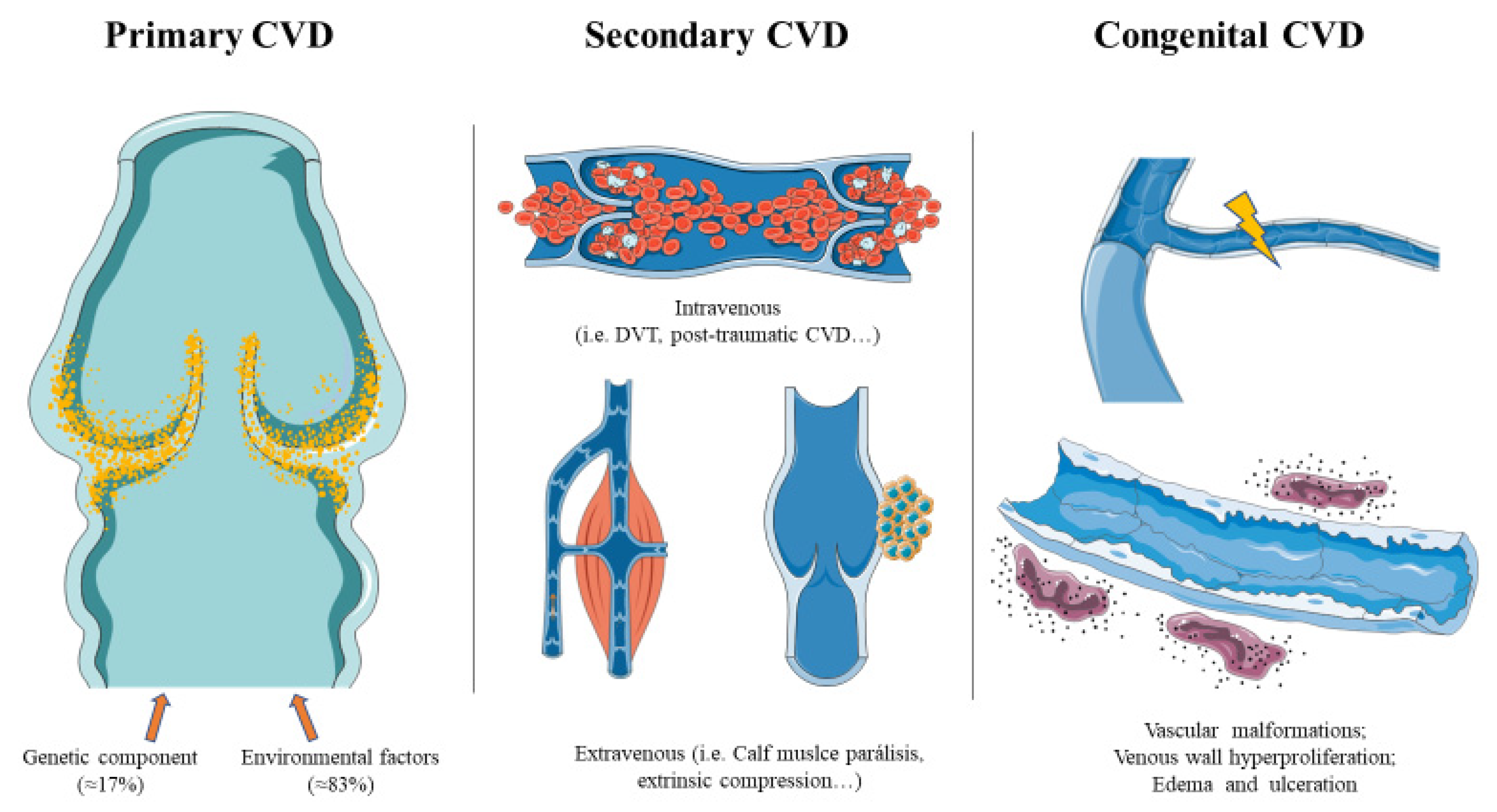
5.1. Primary CVD
5.2. Secondary CVD
5.3. Congenital CVD
6. Pathophysiology
6.1. Hemodynamic and Microcirculatory Alterations
6.2. Inflammation and the Role of Endothelial Dysfunction
6.3. The Hypoxic Environment
6.4. Molecular Basis of CVD: The Venous Wall Remodeling
6.5. Genetics and Epigenetics Mechanisms of CVD
6.5.1. Genetics
6.5.2. Epigenetics
6.6. Systemic Affections
7. Therapeutical Approaches in CVD
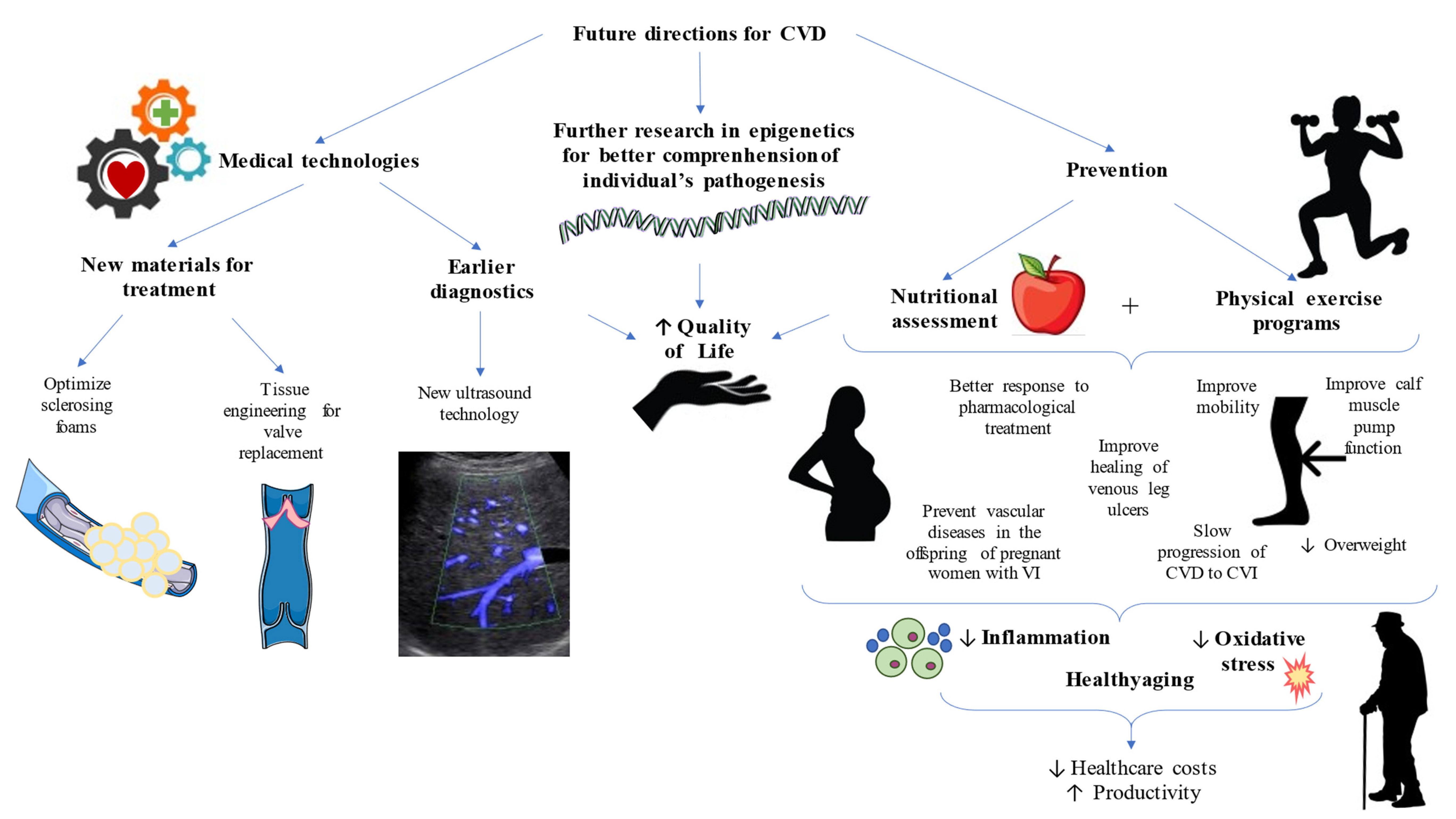
8. Future Directions in CVD—Towards Clinical and Translational Improvements
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nicolaides, A.N.; Labropoulos, N. Burden and Suffering in Chronic Venous Disease. Adv. Ther. 2019, 36. [Google Scholar] [CrossRef]
- Davies, A.H. The Seriousness of Chronic Venous Disease: A Review of Real-World Evidence. Adv. Ther. 2019, 36, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Ligi, D.; Croce, L.; Mannello, F. Chronic venous disorders: The dangerous, the good, and the diverse. Int. J. Mol. Sci. 2018, 19, 2544. [Google Scholar] [CrossRef]
- Eklof, B.; Perrin, M.; Delis, K.T.; Rutherford, R.B.; Gloviczki, P. Updated terminology of chronic venous disorders: The VEIN-TERM transatlantic interdisciplinary consensus document. J. Vasc. Surg. 2009, 49, 498–501. [Google Scholar] [CrossRef]
- Piazza, G. Varicose veins. Circulation 2014, 130, 582–587. [Google Scholar] [CrossRef]
- Youn, Y.J.; Lee, J. Chronic venous insufficiency and varicose veins of the lower extremities. Korean J. Intern. Med. 2019, 34, 269–283. [Google Scholar] [CrossRef]
- Tucker, W.D.; Mahajan, K. Anatomy, Blood Vessels; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Jacobs, B.N.; Andraska, E.A.; Obi, A.T.; Wakefield, T.W. Pathophysiology of varicose veins. J. Vasc. Surg. Venous Lymphat. Disord. 2017, 5, 460–467. [Google Scholar] [CrossRef]
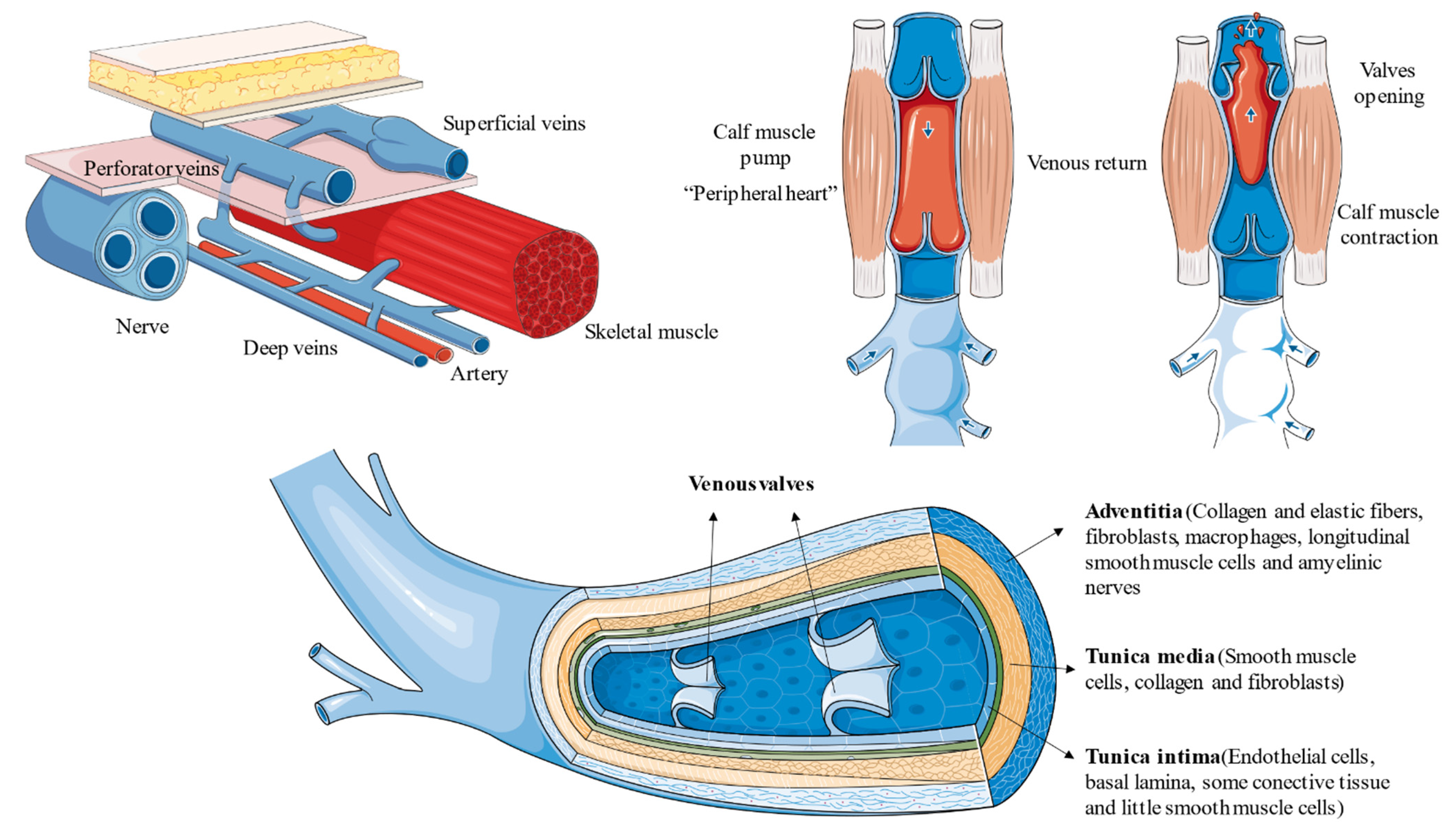
- Tansey, E.A.; Montgomery, L.E.A.; Quinn, J.G.; Roe, S.M.; Johnson, C.D. Understanding basic vein physiology and venous blood pressure through simple physical assessments. Adv. Physiol. Educ. 2019, 43, 423–429. [Google Scholar] [CrossRef]
- Recek, C. Calf pump activity influencing venous hemodynamics in the lower extremity. Int. J. Angiol. 2013, 22, 23–30. [Google Scholar] [CrossRef]
- Uhl, J.F.; Gillot, C. Anatomy of the veno-muscular pumps of the lower limb. Phlebology 2015, 30, 180–193. [Google Scholar] [CrossRef]
- Raetz, J.; Wilson, M.; Collins, K. Varicose Veins: Diagnosis and Treatment. Am. Fam. Physicians 2019, 99, 682–688. [Google Scholar]
- Sukhovatykh, B.S.; Sukhovatykh, M.B. Perforating veins insufficiency in patients with varicose disease. Khirurgiia Mosk 2015, 5, 14–18. [Google Scholar] [CrossRef]
- Nicolaides, A.; Kakkos, S.; Baekgaard, N.; Comerota, A.; de Maeseneer, M.; Eklof, B.; Giannoukas, A.D.; Lugli, M.; Maleti, O.; Myers, K.; et al. Management of chronic venous disorders of the lower limbs Guidelines According to Scientific Evidence PART I. Int. Angiol. 2018, 37, 181–259. [Google Scholar] [CrossRef] [PubMed]
- Hyder, O.N.; Soukas, P.A. Chronic Venous Insufficiency: Novel Management Strategies for an Under-diagnosed Disease Process. Rhode Isl. Med. J. 2017, 100, 37–39. [Google Scholar]
- Moura, R.M.F.; Gonçalves, G.S.; Navarro, T.P.; Britto, R.R.; Dias, R.C. Relationship between quality of life and the ceap clinical classification in chronic venous disease. Rev. Bras. Fisioter. 2010, 14, 99–105. [Google Scholar] [CrossRef]
- Carman, T.L.; Al-Omari, A. Evaluation and Management of Chronic Venous Disease Using the Foundation of CEAP. Curr. Cardiol. Rep. 2019, 21, 1–8. [Google Scholar] [CrossRef]
- Santler, B.; Goerge, T. Chronic venous insufficiency—A review of pathophysiology, diagnosis, and treatment. JDDG J. Dtsch. Soc. Dermatol. 2017, 15, 538–556. [Google Scholar] [CrossRef]
- Zolotukhin, I.A.; Seliverstov, E.I.; Shevtsov, Y.N.; Avakiants, I.P.; Nikishkov, A.S.; Tatarintsev, A.M.; Kirienko, A.I. Prevalence and Risk Factors for Chronic Venous Disease in the General Russian Population. Eur. J. Vasc. Endovasc. Surg. 2017, 54, 752–758. [Google Scholar] [CrossRef]
- Beebe-Dimmer, J.L.; Pfeifer, J.R.; Engle, J.S.; Schottenfeld, D. The epidemiology of chronic venous insufficiency and varicose veins. Ann. Epidemiol. 2005, 15, 175–184. [Google Scholar] [CrossRef]
- Rabe, E.; Guex, J.J.; Puskas, A.; Scuderi, A.; Fernandez Quesada, F.; Coordinators, V. Epidemiology of chronic venous disorders in geographically diverse populations: Results from the Vein Consult Program. Int. Angiol. J. Int. Union Angiol. 2012, 31, 105–115. [Google Scholar]
- Salim, S.; Machin, M.; Patterson, B.O.; Onida, S.; Davies, A.H. Global Epidemiology of Chronic Venous Disease: A Systematic Review with Pooled Prevalence Analysis. Ann. Surg. 2020. [Google Scholar] [CrossRef]
- Rabe, E.; Berboth, G.; Pannier, F. Epidemiology of chronic venous diseases. Wien. Med. Wochenschr. 2016, 166, 260–263. [Google Scholar] [CrossRef]
- Brand, F.N.; Dannenberg, A.L.; Abbott, R.D.; Kannel, W.B. The epidemiology of varicose veins: The Framingham Study. Am. J. Prev. Med. 1988, 4, 96–101. [Google Scholar] [CrossRef]
- Vuylsteke, M.E.; Thomis, S.; Guillaume, G.; Modliszewski, M.L.; Weides, N.; Staelens, I. Epidemiological study on chronic venous disease in Belgium and Luxembourg: Prevalence, risk factors, and symptomatology. Eur. J. Vasc. Endovasc. Surg. 2015, 49, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Pannier, F.; Rabe, E. The relevance of the natural history of varicose veins and refunded care. Phlebology 2012, 27, 23–26. [Google Scholar] [CrossRef]
- Kim, Y.; Png, C.Y.M.; Sumpio, B.J.; DeCarlo, C.S.; Dua, A. Defining the human and health care costs of chronic venous insufficiency. Semin. Vasc. Surg. 2021, 34, 59–64. [Google Scholar] [CrossRef]
- Lee, A.J.; Robertson, L.A.; Boghossian, S.M.; Allan, P.L.; Ruckley, C.V.; Fowkes, F.G.; Evans, C.J. Progression of varicose veins and chronic venous insufficiency in the general population in the Edinburgh Vein Study. J. Vasc. Surg. Venous Lymphat. Disord. 2015, 3, 18–26. [Google Scholar] [CrossRef]
- Vuylsteke, M.E.; Colman, R.; Thomis, S.; Guillaume, G.; Van Quickenborne, D.; Staelens, I. An Epidemiological Survey of Venous Disease Among General Practitioner Attendees in Different Geographical Regions on the Globe: The Final Results of the Vein Consult Program. Angiology 2018, 69, 779–785. [Google Scholar] [CrossRef]
- Musil, D.; Kaletova, M.; Herman, J. Age, body mass index and severity of primary chronic venous disease. Biomed. Pap. Med. Fac. Univ. Palacky. Olomouc. Czech Repub. 2011, 155, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Vuylsteke, M.E.; Colman, R.; Thomis, S.; Guillaume, G.; Degrande, E.; Staelens, I. The influence of age and gender on venous symptomatology. An epidemiological survey in Belgium and Luxembourg. Phlebology 2016, 31, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.M.; Bush, R.L. Venous disease in women: Epidemiology, manifestations, and treatment. J. Vasc. Surg. 2013, 57, 37S–45S. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.A.; Sánchez-Trujillo, L.; Bravo, C.; Fraile-Martinez, O.; García-Montero, C.; Saez, M.A.; Alvarez-Mon, M.A.; Sainz, F.; Alvarez-Mon, M.; Bujan, J.; et al. Newborns of Mothers with Venous Disease during Pregnancy Show Increased Levels of Lipid Peroxidation and Markers of Oxidative Stress and Hypoxia in the Umbilical Cord. Antioxidants 2021, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- Cornu-Thenard, A.; Boivin, P. Chronic venous disease during pregnancy—Servier—PhlebolymphologyServier—Phlebolymphology. Phlebolymphology 2014, 21, 138–145. [Google Scholar]
- Vlajinac, H.D.; Marinkovic, J.M.; Maksimovic, M.Z.; Matic, P.A.; Radak, D.J. Body mass index and primary chronic venous disease—A cross-sectional study. Eur. J. Vasc. Endovasc. Surg. 2013, 45, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.; Shah, P.; Gandhi, F. A study of chronic venous insufficiency in relation with body mass index and diameter of saphenofemoral junction and great saphenous vein. Indian J. Vasc. Endovasc. Surg. 2021, 8, 58. [Google Scholar] [CrossRef]
- Cavezzi, A. Medicine and Phlebolymphology: Time to Change? J. Clin. Med. 2020, 9, 4091. [Google Scholar] [CrossRef]
- Jones, W.S.; Vemulapalli, S.; Parikh, K.S.; Coeytaux, R.R.; Crowley, M.J.; Raitz, G.; Johnston, A.L.; Hasselblad, V.; McBroom, A.J.; Lallinger, K.R.; et al. Treatment Strategies for Patients with Lower Extremity Chronic Venous Disease (LECVD); Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2017. [Google Scholar]
- Sudoł-Szopińska, I.; Bogdan, A.; Szopiński, T.; Panorska, A.K.; Kołodziejczak, M. Prevalence of chronic venous disorders among employees working in prolonged sitting and standing postures. Int. J. Occup. Saf. Ergon. 2011, 17, 165–173. [Google Scholar] [CrossRef]
- Sharma, S.; Vashist, M.; Vashist, M.G. EJBPS|Family History as Major Predisposing Factor in Varicose Veins Disorder. Eur. J. Biomed. Pharm. Sci. 2015, 73, 392–396. [Google Scholar]
- Vlajinac, H.D.; Radak, D.J.; Marinković, J.M.; Maksimović, M.Ž. Risk factors for chronic venous disease. Phlebology 2012, 27, 416–422. [Google Scholar] [CrossRef]
- Kondo, T.; Nakano, Y.; Adachi, S.; Murohara, T. Effects of Tobacco Smoking on Cardiovascular Disease. Circ. J. 2019, 83, 1980–1985. [Google Scholar] [CrossRef]
- Fukaya, E.; Flores, A.M.; Lindholm, D.; Gustafsson, S.; Zanetti, D.; Ingelsson, E.; Leeper, N.J. Clinical and genetic determinants of varicose veins: Prospective, community-based study of ≈500,000 individuals. Circulation 2018, 138, 2869–2880. [Google Scholar] [CrossRef]
- Spácil, J. Does body height affect the severity of chronic venous disease in lower extremities? Vnitr. Lek. 2015, 61, 202–206. [Google Scholar]
- Rabe, E.; Pannier, F. Clinical, aetiological, anatomical and pathological classification (CEAP): Gold standard and limits. Phlebology 2012, 27, 114–118. [Google Scholar] [CrossRef]
- Sinabulya, H.; Holmberg, A.; Blomgren, L. Interobserver variability in the assessment of the clinical severity of superficial venous insufficiency. Phlebology 2015, 30, 61–65. [Google Scholar] [CrossRef]
- Castro-Ferreira, R.; Cardoso, R.; Leite-Moreira, A.; Mansilha, A. The Role of Endothelial Dysfunction and Inflammation in Chronic Venous Disease. Ann. Vasc. Surg. 2018, 46, 380–393. [Google Scholar] [CrossRef]
- Nakano, L.C.U.; Cacione, D.G.; Baptista-Silva, J.C.C.; Flumignan, R.L.G. Treatment for telangiectasias and reticular veins. Cochrane Database Syst. Rev. 2017, 2017. [Google Scholar] [CrossRef]
- Imbernón-Moya, A.; Ortiz-de Frutos, F.J.; Sanjuan-Alvarez, M.; Portero-Sanchez, I. Chronic venous disease of legs. Med. Clin. 2017, 148, 371–376. [Google Scholar] [CrossRef]
- Trayes, K.P.; Studdiford, J.S.; Pickle, S.; Tully, A.S. Edema: Diagnosis and management. Am. Fam. Physician 2013, 88, 102–110. [Google Scholar]
- Shaydakov, M.E.; Comerota, A.J.; Lurie, F. Primary venous insufficiency increases risk of deep vein thrombosis. J. Vasc. Surg. Venous Lymphat. Disord. 2016, 4, 161–166. [Google Scholar] [CrossRef]
- Clement, D.L. Superficial vein thrombosis: More dangerous than anticipated. Phlebolymphology 2013, 20, 188–192. [Google Scholar]
- Gilbert, J.D.; Byard, R.W. Ruptured varicose veins and fatal hemorrhage. Forensic Sci. Med. Pathol. 2018, 14, 244–247. [Google Scholar] [CrossRef]
- Nicolaides, A.N. The Most Severe Stage of Chronic Venous Disease: An Update on the Management of Patients with Venous Leg Ulcers. Adv. Ther. 2020, 37, 19–24. [Google Scholar] [CrossRef]
- Middleton, H. Exploring the aetiology and management of venous eczema. Br. J. Community Nurs. 2007, 12, S16–S23. [Google Scholar] [CrossRef]
- Caggiati, A.; Rosi, C.; Franceschini, M.; Innocenzi, D. The nature of skin pigmentations in chronic venous insufficiency: A preliminary report. Eur. J. Vasc. Endovasc. Surg. 2008, 35, 111–118. [Google Scholar] [CrossRef]
- Choonhakarn, C.; Chaowattanapanit, S.; Julanon, N. Lipodermatosclerosis: A clinicopathologic correlation. Int. J. Dermatol. 2016, 55, 303–308. [Google Scholar] [CrossRef]
- Uhl, J.F.; Cornu-Thenard, A.; Satger, B.; Carpentier, P.H. Clinical analysis of the corona phlebectatica. J. Vasc. Surg. 2012, 55, 150–153. [Google Scholar] [CrossRef]
- Dean, S.M. Cutaneous Manifestations of Chronic Vascular Disease. Prog. Cardiovasc. Dis. 2018, 60, 567–579. [Google Scholar] [CrossRef]
- Senet, P.; Combemale, P.; Debure, C.; Baudot, N.; Machet, L.; Aout, M.; Vicaut, E.; Lok, C. Malignancy and chronic leg ulcers: The value of systematic wound biopsies: A prospective, multicenter, cross-sectional study. Arch. Dermatol. 2012, 148, 704–708. [Google Scholar] [CrossRef]
- Paul, J.C.; Pieper, B.; Templin, T.N. Itch: Association with chronic venous disease, pain, and quality of life. J. Wound Ostomy Cont. Nurs. 2011, 38, 46–54. [Google Scholar] [CrossRef]
- Jafferany, M.; Pastolero, P. Psychiatric and Psychological Impact of Chronic Skin Disease. Prim. Care Companion CNS Disord. 2018, 20, 17nr02247. [Google Scholar] [CrossRef]
- Sritharan, K.; Lane, T.R.A.; Davies, A.H. The burden of depression in patients with symptomatic varicose veins. Eur. J. Vasc. Endovasc. Surg. 2012, 43, 480–484. [Google Scholar] [CrossRef]
- Blättler, W.; Mendoza, E.; Zollmann, C.; Bendix, J.; Amsler, F. Homeostatic feelings—A novel explanation of vein symptoms derived from an experimental patient study. Vasa Eur. J. Vasc. Med. 2019, 48, 492–501. [Google Scholar] [CrossRef]
- Surmeli, M.; Ozdemir, O.C. Quality of Life in Venous Diseases of the Lower Limbs, Well-Being and Quality of Life—Medical Perspective; InTechOpen: Rijeka, Croatia, 2017. [Google Scholar] [CrossRef]
- Wu, Z.; Ma, Y. A narrative review of the quality of life scales specific for chronic venous diseases. Medicine 2021, 100, e25921. [Google Scholar] [CrossRef]
- Darvall, K.A.L.; Bate, G.R.; Adam, D.J.; Bradbury, A.W. Generic health-related quality of life is significantly worse in varicose vein patients with lower limb symptoms independent of CEAP clinical grade. Eur. J. Vasc. Endovasc. Surg. 2012, 44, 341–344. [Google Scholar] [CrossRef]
- Barstow, C.; Kassop, D. Cardiovascular Disease: Chronic Venous Insufficiency and Varicose Veins. FP Essent. 2019, 479, 16–20. [Google Scholar]
- Gloviczki, P.; Comerota, A.J.; Dalsing, M.C.; Eklof, B.G.; Gillespie, D.L.; Gloviczki, M.L.; Lohr, J.M.; McLafferty, R.B.; Meissner, M.H.; Murad, M.H.; et al. The care of patients with varicose veins and associated chronic venous diseases: Clinical practice guidelines of the Society for Vascular Surgery and the American Venous Forum. J. Vasc. Surg. 2011, 53, 2S–48S. [Google Scholar] [CrossRef]
- Eberhardt, R.T.; Raffetto, J.D. Chronic venous insufficiency. Circulation 2014, 130, 333–346. [Google Scholar] [CrossRef]
- Necas, M. Duplex ultrasound in the assessment of lower extremity venous insufficiency. Australas. J. Ultrasound Med. 2010, 13, 37–45. [Google Scholar] [CrossRef]
- Ruckley, C.V.; Evans, C.J.; Allan, P.L.; Lee, A.J.; Fowkes, F.G.R. Chronic venous insufficiency: Clinical and duplex correlations. The Edinburgh Vein Study of venous disorders in the general population. J. Vasc. Surg. 2002, 36, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Khilnani, N.M. Duplex ultrasound evaluation of patients with chronic venous disease of the lower extremities. Am. J. Roentgenol. 2014, 202, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Szary, C.; Wilczko, J.; Plucinska, D.; Pachuta, A.; Napierala, M.; Bodziony, A.; Zawadzki, M.; Grzela, T. The Number of Pregnancies and Deliveries and Their Association with Selected Morphological and Hemodynamic Parameters of the Pelvic and Abdominal Venous System. J. Clin. Med. 2021, 10, 736. [Google Scholar] [CrossRef]
- Dezotti, N.R.A.; Dalio, M.B.; Ribeiro, M.S.; Piccinato, C.E.; Joviliano, E.E. The clinical importance of air plethysmography in the assessment of chronic venous disease. J. Vasc. Bras. 2016, 15, 287–292. [Google Scholar] [CrossRef]
- Min, S.K.; Kim, S.Y.; Park, Y.J.; Lee, W.; Jung, I.M.; Lee, T.; Ha, J.; Kim, S.J. Role of three-dimensional computed tomography venography as a powerful navigator for varicose vein surgery. J. Vasc. Surg. 2010, 51, 893–899. [Google Scholar] [CrossRef]
- Lurie, F.; Passman, M.; Meisner, M.; Dalsing, M.; Masuda, E.; Welch, H.; Bush, R.L.; Blebea, J.; Carpentier, P.H.; De Maeseneer, M.; et al. The 2020 update of the CEAP classification system and reporting standards. J. Vasc. Surg. Venous Lymphat. Disord. 2020, 8, 342–352. [Google Scholar] [CrossRef]
- Meissner, M.H.; Gloviczki, P.; Bergan, J.; Kistner, R.L.; Morrison, N.; Pannier, F.; Pappas, P.J.; Rabe, E.; Raju, S.; Villavicencio, J.L. Primary chronic venous disorders. J. Vasc. Surg. 2007, 46. [Google Scholar] [CrossRef]
- Labropoulos, N.; Kang, S.S.; Mansour, M.A.; Giannoukas, A.D.; Buckman, J.; Baker, W.H. Primary superficial vein reflux with competent saphenous trunk. Eur. J. Vasc. Endovasc. Surg. 1999, 18, 201–206. [Google Scholar] [CrossRef]
- Labropoulos, N.; Tiongson, J.; Pryor, L.; Tassiopoulos, A.K.; Kang, S.S.; Mansour, M.A.; Baker, W.H. Nonsaphenous superficial vein reflux. J. Vasc. Surg. 2001, 34, 872–877. [Google Scholar] [CrossRef]
- Bergan, J.J.; Pascarella, L.; Schmid-Schönbein, G.W. Pathogenesis of primary chronic venous disease: Insights from animal models of venous hypertension. J. Vasc. Surg. 2008, 47, 183–192. [Google Scholar] [CrossRef]
- Fiebig, A.; Krusche, P.; Wolf, A.; Krawczak, M.; Timm, B.; Nikolaus, S.; Frings, N.; Schreiber, S. Heritability of chronic venous disease. Hum. Genet. 2010, 127, 669–674. [Google Scholar] [CrossRef]
- Jawien, A. The influence of environmental factors in chronic venous insufficiency. Angiology 2003, 54. [Google Scholar] [CrossRef]
- Saharay, M.; Shields, D.A.; Georgiannos, S.N.; Porter, J.B.; Scurr, J.H.; Coleridge Smith, P.D. Endothelial activation in patients with chronic venous disease. Eur. J. Vasc. Endovasc. Surg. 1998, 15, 342–349. [Google Scholar] [CrossRef]
- Bass, A. The effect of standing in the workplace and the development of chronic venous insufficiency. Harefuah 2007, 146, 675–676. [Google Scholar] [PubMed]
- Ropacka-Lesiak, M.; Kasperczak, J.; Breborowicz, G.H. Risk factors for the development of venous insufficiency of the lower limbs during pregnancy—Part 1. Ginekol. Pol. 2012, 83, 939–942. [Google Scholar] [PubMed]
- García-Honduvilla, N.; Asúnsolo, Á.; Ortega, M.A.; Sainz, F.; Leal, J.; Lopez-Hervas, P.; Pascual, G.; Buján, J. Increase and Redistribution of Sex Hormone Receptors in Premenopausal Women Are Associated with Varicose Vein Remodelling. Oxid. Med. Cell. Longev. 2018, 2018, 3974026. [Google Scholar] [CrossRef]
- Taylor, J.; Hicks, C.W.; Heller, J.A. The hemodynamic effects of pregnancy on the lower extremity venous system. J. Vasc. Surg. Venous Lymphat. Disord. 2018, 6, 246–255. [Google Scholar] [CrossRef]
- Matić, M.; Matić, A.; Gajinov, Z.; Golušin, Z.; Prćić, S.; Jeremić, B. Major risk factors for chronic venous disease development in women: Is childbirth among them? Women Health 2019, 59, 1118–1127. [Google Scholar] [CrossRef]
- Rodríguez-Nora, B.; Álvarez-Silvares, E. Actualización del tratamiento de la insuficiencia venosa en la gestación [An update on the treatment of venous insufficiency in pregnancy]. Semergen 2018, 44, 262–269. [Google Scholar] [CrossRef]
- Stone, J.; Hangge, P.; Albadawi, H.; Wallace, A.; Shamoun, F.; Knuttien, M.G.; Naidu, S.; Oklu, R. Deep vein thrombosis: Pathogenesis, diagnosis, and medical management. Cardiovasc. Diagn. Ther. 2017, 7, S276–S284. [Google Scholar] [CrossRef]
- Malkani, R.; Karia, R.; Thadani, S. A study of risk factors of chronic venous insufficiency and its association with features suggestive of preceding or present deep venous thrombosis. Indian J. Dermatol. 2019, 64, 366–371. [Google Scholar] [CrossRef]
- Waheed, S.M.; Kudaravalli, P.; Hotwagner, D.T. Deep Vein Thrombosis; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Meissner, M.H.; Eklof, B.; Smith, P.C.; Dalsing, M.C.; DePalma, R.G.; Gloviczki, P.; Moneta, G.; Neglén, P.; O’Donnell, T.; Partsch, H.; et al. Secondary chronic venous disorders. J. Vasc. Surg. 2007, 46. [Google Scholar] [CrossRef]
- Labropoulos, N.; Gasparis, A.P.; Pefanis, D.; Leon, L.R.; Tassiopoulos, A.K. Secondary chronic venous disease progresses faster than primary. J. Vasc. Surg. 2009, 49, 704–710. [Google Scholar] [CrossRef]
- Kahn, S.R.; M’Lan, C.E.; Lamping, D.L.; Kurz, X.; Bérard, A.; Abenhaim, L. The influence of venous thromboembolism on quality of life and severity of chronic venous disease. J. Thromb. Haemost. 2004, 2, 2146–2151. [Google Scholar] [CrossRef]
- Kahn, S.R. The post-thrombotic syndrome. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 413–418. [Google Scholar] [CrossRef]
- Huber, G.H.; Manna, B. Vascular Extremity Trauma; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Feliciano, D.V.; Moore, F.A.; Moore, E.E.; West, M.A.; Davis, J.W.; Cocanour, C.S.; Kozar, R.A.; McIntyre, R.C., Jr. Evaluation and management of peripheral vascular injury. Part 1. Western Trauma Association/critical decisions in trauma. J. Trauma 2011, 70, 1551–1556. [Google Scholar] [CrossRef]
- Meissner, M.H.; Wakefield, T.W.; Ascher, E.; Caprini, J.A.; Comerota, A.J.; Eklof, B.; Gillespie, D.L.; Greenfield, L.J.; He, A.R.; Henke, P.K.; et al. Acute venous disease: Venous thrombosis and venous trauma. J. Vasc. Surg. 2007, 46, 25S–53S. [Google Scholar] [CrossRef]
- Bermudez, K.M.; Knudson, M.M.; Nelken, N.A.; Shackleford, S.; Dean, C.L. Long-term results of lower-extremity venous injuries. Arch. Surg. 1997, 132, 963–968. [Google Scholar] [CrossRef]
- Bhatti, A.M.; Siddique, K.; Bashir, R.A.; Sajid, M.T.; Mustafa, Q.A.; Hussain, S.M.; Shukr, I.; Ahmed, M. Unusual causes of secondary varicose veins. J. Ayub Med. Coll. Abbottabad 2013, 25, 81–85. [Google Scholar]
- Ruskin, K.J. Deep vein thrombosis and venous thromboembolism in trauma. Curr. Opin. Anaesthesiol. 2018, 31, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Boisseau, M.-R. Chronic venous disease and the genetic influence—Servier—PhlebolymphologyServier—Phlebolymphology. Phlebolymphology 2013, 21, 100. [Google Scholar]
- Pistorius, M.A. Chronic venous insufficiency: The genetic influence. Angiology 2003, 54, S5–S12. [Google Scholar] [CrossRef] [PubMed]
- Vahidnezhad, H.; Youssefian, L.; Uitto, J. Klippel-Trenaunay syndrome belongs to the PIK3CA-related overgrowth spectrum (PROS). Exp. Dermatol. 2016, 25, 17–19. [Google Scholar] [CrossRef]
- Happle, R.; Aelvoet, G.E.; Jorens, P.G.; Roelen, L.M. Klippel-Trenauna syndrome: Is it a paradominant trait? Br. J. Dermatol. 1993, 128, 465. [Google Scholar] [CrossRef]
- Ceballos-Quintal, J.M.; Pinto-Escalante, D.; Castillo-Zapata, I. A new case of Klippel-Trenaunay-Weber (KTW) syndrome: Evidence of autosomal dominant inheritance. Am. J. Med. Genet. 1996, 63, 426–427. [Google Scholar] [CrossRef]
- Wang, Q.; Timur, A.A.; Szafranski, P.; Sadgephour, A.; Jurecic, V.; Cowell, J.; Baldini, A.; Driscoll, D.J. Identification and molecular characterization of de novo translocation t(8;14)(q22.3;q13) associated with a vascular and tissue overgrowth syndrome. Cytogenet. Cell Genet. 2002, 95, 183–188. [Google Scholar] [CrossRef]
- Whelan, A.J.; Watson, M.S.; Porter, F.D.; Steiner, R.D. Klippel-Trenaunay-Weber syndrome associated with a 5:11 balanced translocation. Am. J. Med. Genet. 1995, 59, 492–494. [Google Scholar] [CrossRef] [PubMed]
- John, P.R. Klippel-Trenaunay Syndrome. Tech. Vasc. Interv. Radiol. 2019, 22. [Google Scholar] [CrossRef] [PubMed]
- Asghar, F.; Aqeel, R.; Farooque, U.; Haq, A.; Taimur, M. Presentation and Management of Klippel-Trenaunay Syndrome: A Review of Available Data. Cureus 2020, 12, e8023. [Google Scholar] [CrossRef]
- Delis, K.T.; Gloviczki, P.; Wennberg, P.W.; Rooke, T.W.; Driscoll, D.J. Hemodynamic impairment, venous segmental disease, and clinical severity scoring in limbs with Klippel-Trenaunay syndrome. J. Vasc. Surg. 2007, 45, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Chagas, C.A.A.; Pires, L.A.S.; Babinski, M.A.; de Oliveira Leite, T.F. Klippel-Trenaunay and Parkes-Weber syndromes: Two case reports. J. Vasc. Bras. 2017, 16, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Bayrak-Toydemir, P.; Stevenson, D. Capillary Malformation-Arteriovenous Malformation Syndrome—GeneReviews®—NCBI Bookshelf; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Bojakowski, K.; Janusz, G.; Grabowska, I.; Zegrocka-Stendel, O.; Surowiecka-Pastewka, A.; Kowalewska, M.; Maciejko, D.; Koziak, K. Rat model of parkes weber syndrome. PLoS ONE 2015, 10, e0133752. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Banzic, I.; Brankovic, M.; Maksimović, Ž.; Davidović, L.; Marković, M.; Rančić, Z. Parkes Weber syndrome—Diagnostic and management paradigms: A systematic review. Phlebology 2017, 32, 371–383. [Google Scholar] [CrossRef]
- Anwar, M.A.; Georgiadis, K.A.; Shalhoub, J.; Lim, C.S.; Gohel, M.S.; Davies, A.H. A review of familial, genetic, and congenital aspects of primary varicose vein disease. Circ. Cardiovasc. Genet. 2012, 5, 460–466. [Google Scholar] [CrossRef]
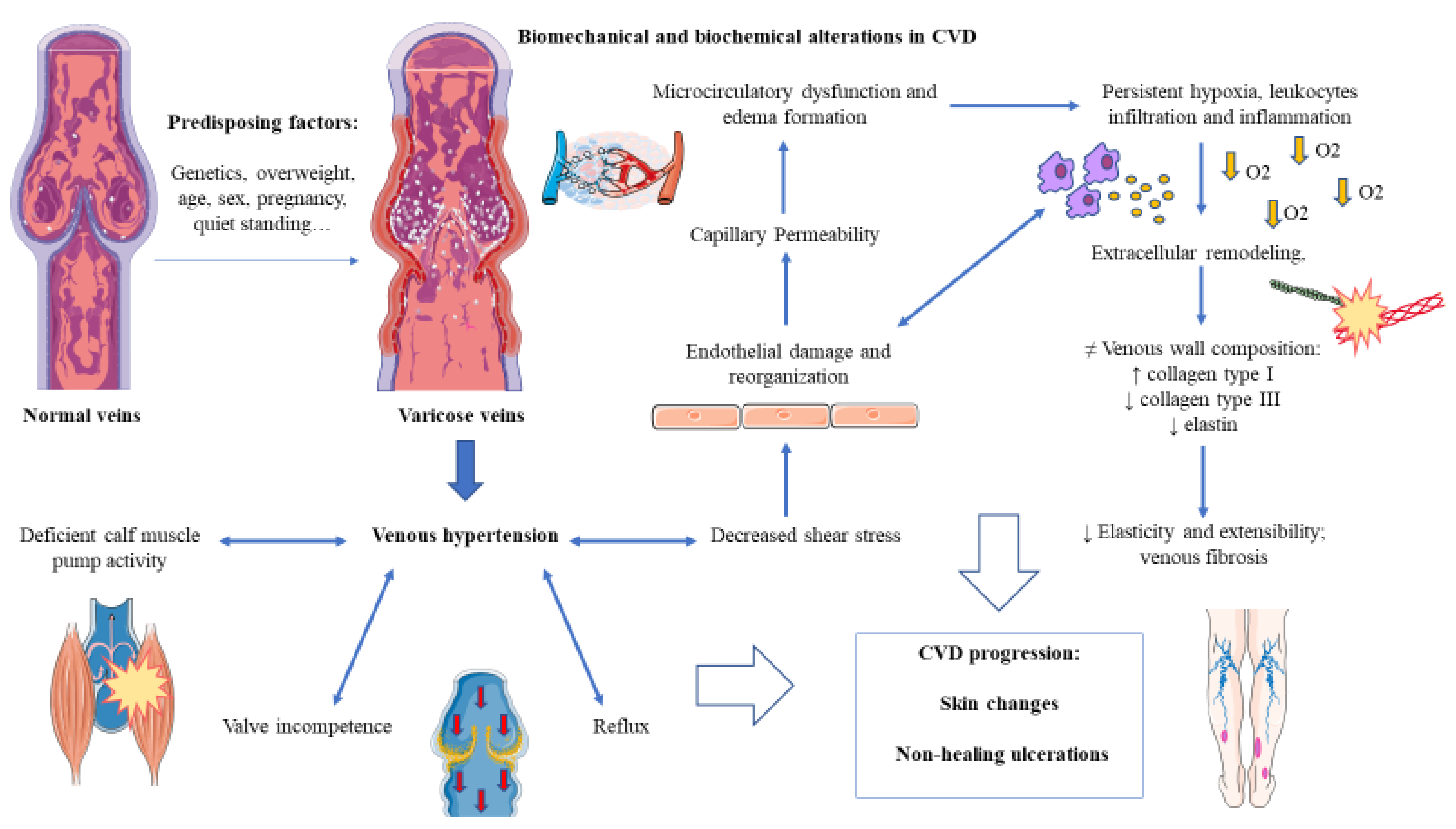
- Labropoulos, N. How Does Chronic Venous Disease Progress from the First Symptoms to the Advanced Stages? A Review. Adv. Ther. 2019, 36, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Raffetto, J.D. Pathophysiology of Chronic Venous Disease and Venous Ulcers. Surg. Clin. N. Am. 2018, 98, 337–347. [Google Scholar] [CrossRef]
- Raffetto, J.D.; Mannello, F. Pathophysiology of chronic venous disease. Int. Angiol. 2014, 33, 212–221. [Google Scholar]
- Ortega, M.A.; Romero, B.; Asúnsolo, Á.; Sola, M.; Álavrez-Rocha, M.J.; Sainz, F.; Álavrez-Mon, M.; Buján, J.; García-Honduvilla, N. Patients with incompetent valves in chronic venous insufficiency show increased systematic lipid peroxidation and cellular oxidative stress markers. Oxid. Med. Cell. Longev. 2019, 2019, 5164576. [Google Scholar] [CrossRef] [PubMed]
- Meissner, M.H.; Moneta, G.; Burnand, K.; Gloviczki, P.; Lohr, J.M.; Lurie, F.; Mattos, M.A.; McLafferty, R.B.; Mozes, G.; Rutherford, R.B.; et al. The hemodynamics and diagnosis of venous disease. J. Vasc. Surg. 2007, 46, S4–S24. [Google Scholar] [CrossRef]
- Raju, S.; Knepper, J.; May, C.; Knight, A.; Pace, N.; Jayaraj, A. Ambulatory venous pressure, air plethysmography, and the role of calf venous pump in chronic venous disease. J. Vasc. Surg. Venous Lymphat. Disord. 2019, 7, 428–440. [Google Scholar] [CrossRef]
- Baeyens, N.; Bandyopadhyay, C.; Coon, B.G.; Yun, S.; Schwartz, M.A. Endothelial fluid shear stress sensing in vascular health and disease. J. Clin. Investig. 2016, 126, 821–828. [Google Scholar] [CrossRef]
- Recek, C. Venous pressure gradients in the lower extremity and the hemodynamic consequences. Vasa J. Vasc. Dis. 2010, 39, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Raju, S.; Knight, A.; Lamanilao, L.; Pace, N.; Jones, T. Peripheral venous hypertension in chronic venous disease. J. Vasc. Surg. Venous Lymphat. Disord. 2019, 7, 706–714. [Google Scholar] [CrossRef]
- Recek, C.; Pojer, H. Ambulatory pressure gradient in the veins of the lower extremity. Vasa J. Vasc. Dis. 2000, 29, 187–190. [Google Scholar] [CrossRef]
- Senra Barros, B.; Kakkos, S.K.; De Maeseneer, M.; Nicolaides, A.N. Chronic venous disease: From symptoms to microcirculation. Int. Angiol. 2019, 38, 211–218. [Google Scholar] [CrossRef]
- Raffetto, J.D. Pathophysiology of wound healing and alterations in venous leg ulcers—Review. Phlebology 2016, 31, 56–62. [Google Scholar] [CrossRef]
- Sapelkin, S.V.; Timina, I.E.; Dudareva, A.S. Chronic venous diseases: Valvular function and leukocyte-endothelial interaction, possibilities of pharmacotherapy. Angiol. Sosud. Khir. 2017, 23, 89–96. [Google Scholar] [PubMed]
- Nicolaides, A.; Clark, H.; Labropoulos, N.; Geroulakos, G.; Lugli, M.; Maleti, O. Quantitation of reflux and outflow obstruction in patients with CVD and correlation with clinical severity. Int. Angiol. J. Int. Union Angiol. 2014, 33, 275–281. [Google Scholar]
- Simka, M. Calf muscle pump dysfunction in the patients with severe chronic venous insufficiency. Phlebolymphology 2004, 47, 298–303. [Google Scholar]
- Reček, Č. Conception of the venous hemodynamics in the lower extremity. Angiology 2006, 57, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Vekilov, D.P.; Grande-Allen, K.J. Mechanical Properties of Diseased Veins. Methodist Debakey Cardiovasc. J. 2018, 14, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Wali, M.A.; Eid, R.A. Changes of elastic and collagen fibers in varicose veins. Int. Angiol. J. Int. Union Angiol. 2002, 21, 337–343. [Google Scholar]
- Wali, M.A.; Dewan, M.; Eid, R.A. Histopathological changes in the wall of varicose veins. Int. Angiol. J. Int. Union Angiol. 2003, 22, 188–193. [Google Scholar]
- Coleridge Smith, P.D. Update on chronic-venous-insufficiency-induced inflammatory processes. Angiology 2001, 52, S35–S42. [Google Scholar] [CrossRef]
- Danziger, N. Pathophysiology of pain in venous disease. J. Mal. Vasc. 2007, 32, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Zamboni, P.; Lanzara, S.; Mascoli, F.; Caggiati, A.; Liboni, A. Inflammation in venous disease. Int. Angiol. 2008, 27, 361–369. [Google Scholar]
- Davies, P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, 16–26. [Google Scholar] [CrossRef]
- Tisato, V.; Zauli, G.; Voltan, R.; Gianesini, S.; Iasio, M.G.; Volpi, I.; Fiorentini, G.; Zamboni, P.; Secchiero, P. Endothelial cells obtained from patients affected by chronic venous disease exhibit a pro-inflammatory phenotype. PLoS ONE 2012, 7, e39543. [Google Scholar] [CrossRef]
- Tarbell, J.M.; Cancel, L.M. The glycocalyx and its significance in human medicine. J. Intern. Med. 2016, 280, 97–113. [Google Scholar] [CrossRef]
- Komarów, W.; Hawro, P.; Lekston, A.; Urbanek, T.; Zagrodzki, P. Endothelial dysfunction in patients with chronic venous disease: An evaluation based on the flow-mediated dilatation test. Int. Angiol. 2015, 34, 36–42. [Google Scholar]
- Carrasco, O.F.; Ranero, A.; Hong, E.; Vidrio, H. Endothelial function impairment in chronic venous insufficiency: Effect of some cardiovascular protectant agents. Angiology 2009, 60, 763–771. [Google Scholar] [CrossRef]
- Mosmiller, L.T.; Steele, K.N.; Shrader, C.D.; Petrone, A.B. Evaluation of inflammatory cell biomarkers in chronic venous insufficiency. Phlebology 2017, 32, 634–640. [Google Scholar] [CrossRef]
- Ojdana, D.; Safiejko, K.; Lipska, A.; Sacha, P.; Wieczorek, P.; Radziwon, P.; Dadan, J.; Tryniszewska, E. The inflammatory reaction during chronic venous disease of lower limbs. Folia Histochem. Cytobiol. 2009, 47, 185–189. [Google Scholar] [CrossRef]
- Ono, T.; Bergan, J.J.; Schmid-Schonbein, G.W.; Takase, S. Monocyte infiltration into venous valves. J. Vasc. Surg. 1998, 27, 158–166. [Google Scholar] [CrossRef]
- Powell, C.C.; Rohrer, M.J.; Barnard, M.R.; Peyton, B.D.; Furman, M.I.; Michelson, A.D. Chronic venous insufficiency is associated with increased platelet and monocyte activation and aggregation. J. Vasc. Surg. 1999, 30, 844–853. [Google Scholar] [CrossRef][Green Version]
- Ferris, A.E.; Harding, K.G. An overview of the relationship between anaemia, iron, and venous leg ulcers. Int. Wound J. 2019, 16, 1323–1329. [Google Scholar] [CrossRef] [PubMed]
- Zamboni, P. The Big Idea: Iron-dependent inflammation in venous disease and proposed parallels in multiple sclerosis. J. R. Soc. Med. 2006, 99, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Ferris, A.E.; Harding, K.G. Does localized iron loss in venous disease lead to systemic iron deficiency? A descriptive pilot study. Wound Repair Regen. 2020, 28, 33–38. [Google Scholar] [CrossRef]
- Caggiati, A.; Rosi, C.; Casini, A.; Cirenza, M.; Petrozza, V.; Acconcia, M.C.; Zamboni, P. Skin iron deposition characterises lipodermatosclerosis and leg ulcer. Eur. J. Vasc. Endovasc. Surg. 2010, 40, 777–782. [Google Scholar] [CrossRef]
- Sindrilaru, A.; Peters, T.; Wieschalka, S.; Baican, C.; Baican, A.; Peter, H.; Hainzl, A.; Schatz, S.; Qi, Y.; Schlecht, A.; et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Investig. 2011, 121, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Nicolaides, A.N. Chronic venous disease and the leukocyte-endothelium interaction: From symptoms to ulceration. Proc. Angiol. 2005, 56, S11–S19. [Google Scholar] [CrossRef]
- Stvrtinova, V.; Jahnova, E.; Weissova, S.; Ferencik, H.M. Inflammatory mechanisms involving neutrophils in chronic venous insufficiency of lower limbs. Bratisl. Lek. Listy 2001, 102, 235–239. [Google Scholar]
- Bogachev, V.I.; Golovanova, O.V.; Sergeeva, N.A.; Kuznetsov, A.N. Participation of leucocytes in pathogenesis of primary forms of lower limb chronic venous disease. Angiol. Sosud. Khir. 2011, 17, 71–75. [Google Scholar] [PubMed]
- Whiston, R.J.; Hallett, M.B.; Davies, E.V.; Harding, K.G.; Lane, I.F. Inappropriate neutrophil activation in venous disease. Br. J. Surg. 1994, 81, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Takase, S.; Schmid-Schonbein, G.; Bergan, J.J. Leukocyte activation in patients with venous insufficiency. J. Vasc. Surg. 1999, 30, 148–156. [Google Scholar] [CrossRef]
- Grudzińska, E.; Czuba, Z.P. Immunological aspects of chronic venous disease pathogenesis. Cent. Eur. J. Immunol. 2014, 39, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Engin, M.; Goncu, M.T. The role of plateletcrit and neutrophil lymphocyte ratio in showing the clinical severity of the disease in patients with chronic venous insufficiency. Ann. Med. Res. 2020, 27, 1385–1390. [Google Scholar] [CrossRef]
- Sayer, G.L.; Smith, P.D.C. Immunocytochemical characterisation of the inflammatory cell infiltrate of varicose veins. Eur. J. Vasc. Endovasc. Surg. 2004, 28, 479–483. [Google Scholar] [CrossRef]
- Haviarová, Z.; Weismann, P.; Pavlíková, D.; Durdík, Š.; Kovác, P.; Štvrtinová, V.; Mráz, P. Mast cell infiltration in the wall of varicose veins. Acta Histochem. 2002, 104, 357–360. [Google Scholar] [CrossRef]
- Kakkos, S.; Zolota, V.; Peristeropoulou, P.; Apostolopoulou, A.; Geroukalos, G.; Tsolakis, I. Increased mast cell infiltration in familial varicose veins: Pathogenetic implications? Int. Angiol. 2003, 22, 43–49. [Google Scholar]
- Chu, H.B.; Yan, F.; Zhao, J.H.; Xu, Y.B.; Wang, T.; Guo, W.J. Assessment of the infiltration of inflammatory cells in the walls of thrombotic varicose veins. Angiology 2013, 64, 69–72. [Google Scholar] [CrossRef]
- Boisseau, M. Leukocyte involvement in the signs and symptoms of chronic venous disease. Perspectives for therapy. Clin. Hemorheol. Microcirc. 2007, 37, 277–290. [Google Scholar]
- Lintermans, L.L.; Stegeman, C.A.; Heeringa, P.; Abdulahad, W.H. T cells in vascular inflammatory diseases. Front. Immunol. 2014, 5, 504. [Google Scholar] [CrossRef] [PubMed]
- Gagliani, N.; Huber, S. Basic aspects of T helper cell differentiation. Methods Mol. Biol. 2017, 1514, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.V.; Connors, T.J.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, R.; Winkels, H.; Ley, K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 387–401. [Google Scholar] [CrossRef]
- Ojdana, D.; Safiejko, K.; Milewski, R.; Sacha, P.; Wieczorek, P.; Lipska, A.; Radziwon, P.; Dadan, J.; Tryniszewska, E. Evaluation of the memory CD4+ and CD8+ T cells homeostasis during chronic venous disease of lower limbs. Folia Histochem. Cytobiol. 2009, 47, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Buján, J.; Pascual, G.; Bellón, J. Interaction between ageing, inflammation process, and the occurence of varicose veins. Phlebolymphology 2008, 15, 123–130. [Google Scholar]
- Grudzinska, E.; Lekstan, A.; Szliszka, E.; Czuba, Z.P. Cytokines produced by lymphocytes in the incompetent great saphenous vein. Mediat. Inflamm. 2018, 2018, 7161346. [Google Scholar] [CrossRef]
- Lattimer, C.R.; Kalodiki, E.; Geroulakos, G.; Hoppensteadt, D.; Fareed, J. Are Inflammatory Biomarkers Increased in Varicose Vein Blood? Clin. Appl. Thromb. 2016, 22, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Guss, L.G.; Javvaji, S.; Case, J.; Barrick, B.; Schaefer, K.N. Differences in Inflammatory Cytokine Levels between Patients with Varying Severity of Chronic Venous Insufficiency. J. Vasc. Med. Surg. 2018, 6, 363. [Google Scholar] [CrossRef]
- Howlader, M.H.; Coleridge Smith, P.D. Symptoms of chronic venous disease and association with systemic inflammatory markers. J. Vasc. Surg. 2003, 38, 950–954. [Google Scholar] [CrossRef]
- Serralheiro, P.; Soares, A.; Costa Almeida, C.M.; Verde, I. TGF-β1 in Vascular Wall Pathology: Unraveling Chronic Venous Insufficiency Pathophysiology. Int. J. Mol. Sci. 2017, 18, 2534. [Google Scholar] [CrossRef]
- Serralheiro, P.; Novais, A.; Cairrão, E.; Maia, C.; Costa Almeida, C.M.; Verde, I. Variability of MMP/TIMP and TGF-β1 Receptors throughout the Clinical Progression of Chronic Venous Disease. Int. J. Mol. Sci. 2017, 19, 6. [Google Scholar] [CrossRef] [PubMed]
- Pastar, I.; Stojadinovic, O.; Krzyzanowska, A.; Barrientos, S.; Stuelten, C.; Zimmerman, K.; Blumenberg, M.; Brem, H.; Tomic-Canic, M. Attenuation of the transforming growth factor beta-signaling pathway in chronic venous ulcers. Mol. Med. 2010, 16, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Kowalewski, R.; Malkowski, A.; Sobolewski, K.; Gacko, M. Evaluation of transforming growth factor-beta signaling pathway in the wall of normal and varicose veins. Pathobiology 2010, 77, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.S.; Kiriakidis, S.; Sandison, A.; Paleolog, E.M.; Davies, A.H. Hypoxia-inducible factor pathway and diseases of the vascular wall. J. Vasc. Surg. 2013, 58, 219–230. [Google Scholar] [CrossRef]
- Kachlík, D.; Lametschwandtner, A.; Rejmontová, J.; Stingl, J.; Vaněk, I. Vasa vasorum of the human great saphenous vein. Surg. Radiol. Anat. 2002, 24, 377–381. [Google Scholar] [CrossRef]
- Lim, C.S.; Gohel, M.S.; Shepherd, A.C.; Paleolog, E.; Davies, A.H. Venous hypoxia: A poorly studied etiological factor of varicose veins. J. Vasc. Res. 2011, 48, 185–194. [Google Scholar] [CrossRef]
- Lim, C.S.; Kiriakidis, S.; Paleolog, E.M.; Davies, A.H. Increased activation of the hypoxia-inducible factor pathway in varicose veins. J. Vasc. Surg. 2012, 55, 1427–1439. [Google Scholar] [CrossRef]
- Michiels, C.; Arnould, T.; Remacle, J. Endothelial cell responses to hypoxia: Initiation of a cascade of cellular interactions. Biochim. Biophys. Acta Mol. Cell Res. 2000, 1497, 1–10. [Google Scholar] [CrossRef]
- Kachlík, D.; Stingl, J.; Sosna, B.; Straka, Z.; Lametschwandtner, A.; Minnich, B.; Fára, P. Morphological features of vasa vasorum in pathologically changed human great saphenous vein and its tributaries. Vasa J. Vasc. Dis. 2008, 37, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Bei, Y.; Li, Y.; Chu, H. Phenotypic and functional transformation in smooth muscle cells derived from varicose veins. J. Vasc. Surg. Venous Lymphat. Disord. 2017, 5, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Wali, M.A.; Eid, R.A. Smooth muscle changes in varicose veins: An ultrastructural study. J. Smooth Muscle Res. 2001, 37, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Somers, P.; Knaapen, M. The histopathology of varicose vein disease. Angiology 2006, 57, 546–555. [Google Scholar] [CrossRef]
- Xiao, Y.; Huang, Z.; Yin, H.; Lin, Y.; Wang, S. In vitro differences between smooth muscle cells derived from varicose veins and normal veins. J. Vasc. Surg. 2009, 50, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.A.; Romero, B.; Asúnsolo, Á.; Sainz, F.; Martinez-Vivero, C.; Álvarez-Mon, M.; Buján, J.; Garc-a-Honduvilla, N. Behavior of smooth muscle cells under hypoxic conditions: Possible implications on the varicose vein endothelium. BioMed Res. Int. 2018, 2018, 7156150. [Google Scholar] [CrossRef] [PubMed]
- Bujan, J.; Jimenez-Cossio, J.A.; Jurado, F.; Gimeno, M.J.; Pascual, G.; Garcia-Honduvilla, N.; Dominguez, B.; Bellon, J.M. Evaluation of the smooth muscle cell component and apoptosis in the varicose vein wall. Histol. Histopathol. 2000, 15, 745–752. [Google Scholar] [CrossRef]
- Lim, C.S.; Davies, A.H. Pathogenesis of primary varicose veins. Br. J. Surg. 2009, 96, 1231–1242. [Google Scholar] [CrossRef]
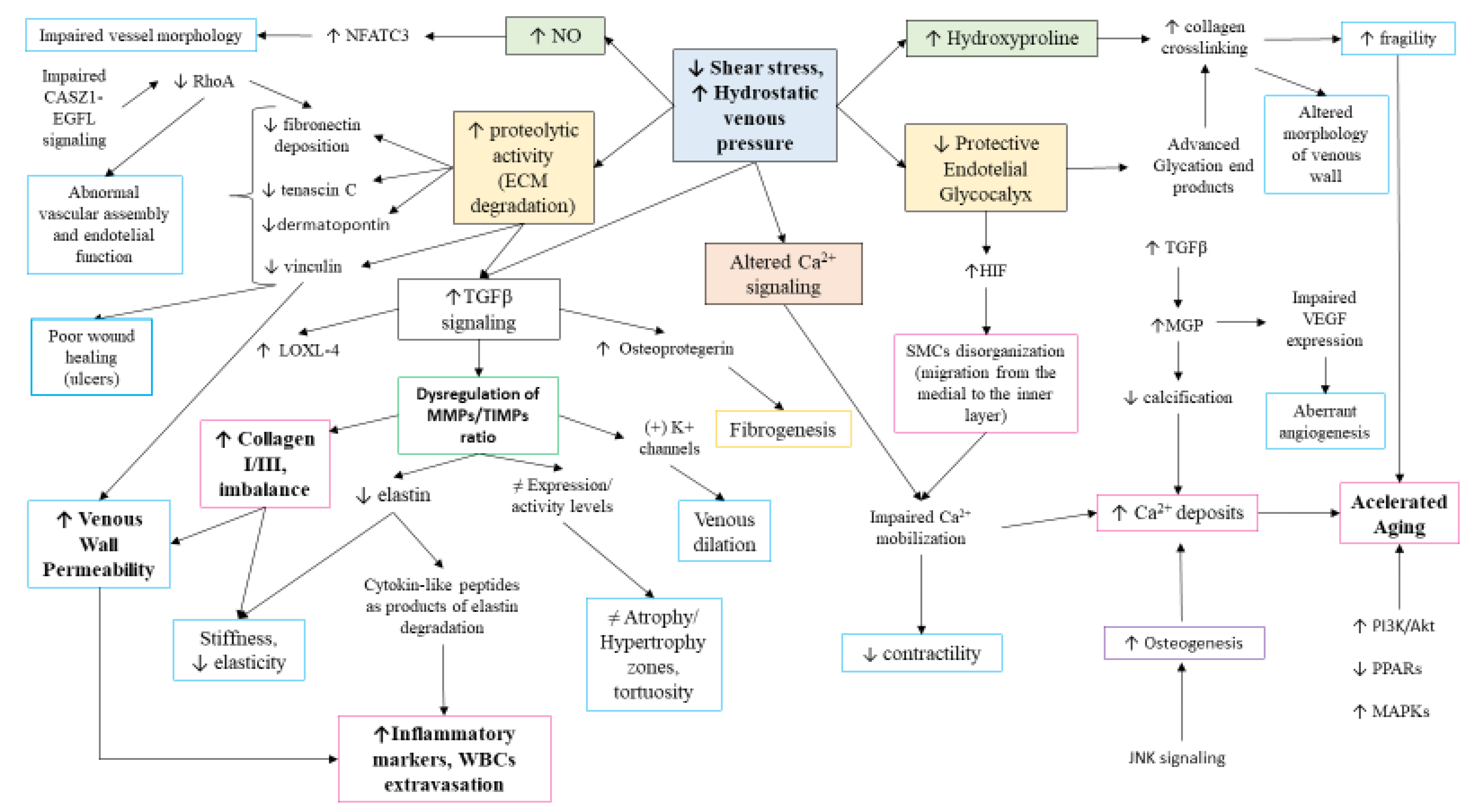
- Atta, H.M. Varicose veins: Role of mechanotransduction of venous hypertension. Int. J. Vasc. Med. 2012, 2012, 538627. [Google Scholar] [CrossRef]
- Bergan, J. Molecular Mechanisms in Chronic Venous Insufficiency. Ann. Vasc. Surg. 2007, 21, 260–266. [Google Scholar] [CrossRef]
- Saberianpour, S.; modaghegh, M.H.S.; Rahimi, H.; Kamyar, M.M. Role of mechanosignaling on pathology of varicose vein. Biophys. Rev. 2021, 13, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Bruczko-Goralewska, M.; Romanowicz, L.; Baczyk, J.; Wolańska, M.; Sobolewski, K.; Kowalewski, R. Peptide growth factors and their receptors in the vein wall. J. Investig. Med. 2019, 67, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.A.; Fraile-Martínez, O.; Asúnsolo, Á.; Martínez-Vivero, C.; Pekarek, L.; Coca, S.; Guijarro, L.G.; Álvarez-Mon, M.; Buján, J.; García-Honduvilla, N.; et al. Chronic Venous Disease Patients Showed Altered Expression of IGF-1/PAPP-A/STC-2 Axis in the Vein Wall. BioMed Res. Int. 2020, 2020, 6782659. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Mendieta, C.; García-Honduvilla, N.; Corrales, C.; Bellón, J.M.; Buján, J. TGF-β1 upregulation in the aging varicose vein. J. Vasc. Res. 2007, 44, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.C.; Kim, H.T.; Park, S.H.; Cha, J.S.; Yufit, T.; Kim, S.J.; Falanga, V. Fibroblasts from chronic wounds show altered TGF-beta-signaling and decreased TGF-beta Type II receptor expression. J. Cell Physiol. 2003, 195, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.A.; Fraile-Martínez, O.; Asúnsolo, Á.; Buján, J.; García-Honduvilla, N.; Coca, S. Signal Transduction Pathways in Breast Cancer: The Important Role of PI3K/Akt/mTOR. J. Oncol. 2020, 2020, 9258396. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.A.; Asúnsolo, Á.; Leal, J.; Romero, B.; Alvarez-Rocha, M.J.; Sainz, F.; Álvarez-Mon, M.; Buján, J.; García-Honduvilla, N. Implication of the PI3K/Akt/mTOR pathway in the process of incompetent valves in patients with chronic venous insufficiency and the relationship with aging. Oxid. Med. Cell. Longev. 2018, 2018, 1495170. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.A.; Asúnsolo, Á.; Romero, B.; Álvarez-Rocha, M.J.; Sainz, F.; Leal, J.; Álvarez-Mon, M.; Buján, J.; García-Honduvilla, N. Unravelling the role of mapks (erk1/2) in venous reflux in patients with chronic venous disorder. Cells Tissues Organs 2019, 206, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.A.; Fraile-Martínez, O.; Pekarek, L.; Alvarez-Mon, M.A.; Asúnsolo, Á.; Sanchez-Trujillo, L.; Coca, S.; Buján, J.; Álvarez-Mon, M.; García-Honduvilla, N.; et al. Defective expression of the peroxisome regulators PPARα receptors and lysogenesis with increased cellular senescence in the venous wall of chronic venous disorder. Histol. Histopathol. 2021, 18322. [Google Scholar] [CrossRef]
- Ortega, M.A.; Asúnsolo, Á.; Pekarek, L.; Alvarez-Mon, M.A.; Delforge, A.; Sáez, M.A.; Coca, S.; Sainz, F.; Álvarez-Mon, M.; Buján, J.; et al. Histopathological study of JNK in venous wall of patients with chronic venous insufficiency related to osteogenesis process. Int. J. Med. Sci. 2021, 18, 1921–1934. [Google Scholar] [CrossRef]
- Schuller-Petrovic, S.; Stessel, H.; Brunner, F. Ca2+ mobilization in saphenous vein smooth muscle cells derived from patients with primary varicosity. Eur. J. Clin. Investig. 2002, 32, 649–656. [Google Scholar] [CrossRef]
- Cario-Toumaniantz, C.; Evellin, S.; Maury, S.; Baron, O.; Pacaud, P.; Loirand, G. Role of Rho kinase signalling in healthy and varicose human saphenous veins. Br. J. Pharmacol. 2002, 137, 205–212. [Google Scholar] [CrossRef]
- Charpentier, M.S.; Christine, K.S.; Amin, N.M.; Dorr, K.M.; Kushner, E.J.; Bautch, V.L.; Taylor, J.M.; Conlon, F.L. CASZ1 Promotes Vascular Assembly and Morphogenesis through the Direct Regulation of an EGFL7/RhoA-Mediated Pathway. Dev. Cell 2013, 25, 132–143. [Google Scholar] [CrossRef]
- Oh-hora, M.; Rao, A. The calcium/NFAT pathway: Role in development and function of regulatory T cells. Microbes Infect. 2009, 11, 612–619. [Google Scholar] [CrossRef]
- Graef, I.A.; Chen, F.; Chen, L.; Kuo, A.; Crabtree, G.R. Signals transduced by Ca2+/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell 2001, 105, 863–875. [Google Scholar] [CrossRef]
- Gonzalez Bosc, L.V.; Wilkerson, M.K.; Bradley, K.N.; Eckman, D.M.; Hill-Eubanks, D.C.; Nelson, M.T. Intraluminal pressure is a stimulus for NFATc3 nuclear accumulation: Role of calcium, endothelium-derived nitric oxide, and cGMP-dependent protein kinase. J. Biol. Chem. 2004, 279, 10702–10709. [Google Scholar] [CrossRef] [PubMed]
- Feldo, M.; Woźniak, M.; Wójciak-Kosior, M.; Sowa, I.; Kot-Waśik, A.; Aszyk, J.; Bogucki, J.; Zubilewicz, T.; Bogucka-Kocka, A. Influence of Diosmin Treatment on the Level of Oxidative Stress Markers in Patients with Chronic Venous Insufficiency. Oxid. Med. Cell. Longev. 2018, 2018, 2561705. [Google Scholar] [CrossRef] [PubMed]
- Cario-Toumaniantz, C.; Boularan, C.; Schurgers, L.J.; Heymann, M.F.; Le Cunff, M.; Léger, J.; Loirand, G.; Pacaud, P. Identification of differentially expressed genes in human varicose veins: Involvement of matrix Gla protein in extracellular matrix remodeling. J. Vasc. Res. 2007, 44, 444–459. [Google Scholar] [CrossRef] [PubMed]
- Jaminon, A.M.G.; Dai, L.; Qureshi, A.R.; Evenepoel, P.; Ripsweden, J.; Söderberg, M.; Witasp, A.; Olauson, H.; Schurgers, L.J.; Stenvinkel, P. Matrix Gla protein is an independent predictor of both intimal and medial vascular calcification in chronic kidney disease. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Bjørklund, G.; Svanberg, E.; Dadar, M.; Card, D.J.; Chirumbolo, S.; Harrington, D.J.; Aaseth, J. The Role of Matrix Gla Protein (MGP) in Vascular Calcification. Curr. Med. Chem. 2019, 27, 1647–1660. [Google Scholar] [CrossRef] [PubMed]
- Boström, K.; Zebbondj, A.F.; Yao, Y.; Lin, T.S.; Torres, A. Matrix GLA protein stimulates VEGF expression through increased transforming growth factor-β1 activity in endothelial cells. J. Biol. Chem. 2004, 279, 52904–52913. [Google Scholar] [CrossRef]
- Harraz, O.F.; Jensen, L.J. Aging, calcium channel signaling and vascular tone. Mech. Ageing Dev. 2020, 191, 111336. [Google Scholar] [CrossRef]
- Horecka, A.; Hordyjewska, A.; Biernacka, J.; Dąbrowski, W.; Zubilewicz, T.; Malec, A.; Musik, I.; Kurzepa, J. Intense remodeling of extracellular matrix within the varicose vein: The role of gelatinases and vascular endothelial growth factor. Ir. J. Med. Sci. 2021, 190, 255–259. [Google Scholar] [CrossRef]
- Chen, Y.; Peng, W.; Raffetto, J.D.; Khalil, R.A. Matrix Metalloproteinases in Remodeling of Lower Extremity Veins and Chronic Venous Disease. Prog. Mol. Biol. Transl. Sci. 2017, 147, 267–299. [Google Scholar] [CrossRef] [PubMed]
- MacColl, E.; Khalil, R.A. Matrix metalloproteinases as regulators of vein structure and function: Implications in chronic venous disease. J. Pharmacol. Exp. Ther. 2015, 355, 410–428. [Google Scholar] [CrossRef] [PubMed]
- Raffetto, J.D.; Ross, R.L.; Khalil, R.A. Matrix metalloproteinase 2-induced venous dilation via hyperpolarization and activation of K+ channels: Relevance to varicose vein formation. J. Vasc. Surg. 2007, 45, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Dorland, Y.L.; Huveneers, S. Cell–cell junctional mechanotransduction in endothelial remodeling. Cell. Mol. Life Sci. 2016, 74, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Faringthon, R.; Sosa, V. Chronic venous insufficiency and structural changes in the walls of the veins. Rev. Méd. Sinerg. 2019, 4, 3–20. [Google Scholar]
- Boisseau, M.R. Recent findings in the pathogenesis of venous wall degradation. Phlebolymphology 2007, 14, 59–73. [Google Scholar]
- Sansilvestri-Morel, P.; Rupin, A.; Badier-Commander, C.; Fabiani, J.N.; Verbeuren, T.J. Chronic venous insufficiency: Dysregulation of collagen synthesis. Angiology 2003, 54, S13–S18. [Google Scholar] [CrossRef]
- Antonicelli, F.; Bellon, G.; Debelle, L.; Hornebeck, W. Elastin-Elastases and Inflamm-Aging. Curr. Top. Dev. Biol. 2007, 79, 99–155. [Google Scholar] [CrossRef] [PubMed]
- Kanta, J.; Zavadakova, A. Role of fibronectin in chronic venous diseases: A review. Vasc. Med. 2020, 25, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Barallobre-Barreiro, J.; Oklu, R.; Lynch, M.; Fava, M.; Baig, F.; Yin, X.; Barwari, T.; Potier, D.N.; Albadawi, H.; Jahangiri, M.; et al. Extracellular matrix remodelling in response to venous hypertension: Proteomics of human varicose veins. Cardiovasc. Res. 2016, 110, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, O.; Fujiwara, S. Dermatopontin, a novel player in the biology of the extracellular matrix. Connect. Tissue Res. 2006, 47, 177–189. [Google Scholar] [CrossRef]
- Shibuya, H.; Okamoto, O.; Fujiwara, S. The bioactivity of transforming growth factor-β1 can be regulated via binding to dermal collagens in mink lung epithelial cells. J. Dermatol. Sci. 2006, 41, 187–195. [Google Scholar] [CrossRef]
- Imanaka-Yoshida, K.; Yoshida, T.; Miyagawa-Tomita, S. Tenascin-C in development and disease of blood vessels. Anat. Rec. 2014, 297, 1747–1757. [Google Scholar] [CrossRef]
- Huveneers, S.; Oldenburg, J.; Spanjaard, E.; van der Krogt, G.; Grigoriev, I.; Akhmanova, A.; Rehmann, H.; de Rooij, J. Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J. Cell Biol. 2012, 196, 641–652. [Google Scholar] [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef]
- Barallobre-Barreiro, J.; Baig, F.; Fava, M.; Yin, X.; Mayr, M. Glycoproteomics of the extracellular matrix: A method for intact glycopeptide analysis using mass spectrometry. J. Vis. Exp. 2017, 2017, 55674. [Google Scholar] [CrossRef]
- Ellinghaus, E.; Ellinghaus, D.; Krusche, P.; Greiner, A.; Schreiber, C.; Nikolaus, S.; Gieger, C.; Strauch, K.; Lieb, W.; Rosenstiel, P.; et al. Genome-wide association analysis for chronic venous disease identifies EFEMP1 and KCNH8 as susceptibility loci. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Serra, R.; Ssempijja, L.; Provenzano, M.; Andreucci, M. Genetic biomarkers in chronic venous disease. Biomark. Med. 2020, 14, 75–80. [Google Scholar] [CrossRef]
- Xu, H.M.; Zhao, Y.; Zhang, X.M.; Zhu, T.; Fu, W.G. Polymorphisms in MMP-9 and TIMP-2 in Chinese patients with varicose veins. J. Surg. Res. 2011, 168, e143–e148. [Google Scholar] [CrossRef]
- Shadrina, A.S.; Sharapov, S.Z.; Shashkova, T.I.; Tsepilov, Y.A. Varicose veins of lower extremities: Insights from the first large-scale genetic study. PLoS Genet. 2019, 15, e1008110. [Google Scholar] [CrossRef]
- Jones, G.T.; Marsman, J.; Pardo, L.M.; Nijsten, T.; De Maeseneer, M.; Phillips, V.; Lynch-Sutherland, C.; Horsfield, J.; Krysa, J.; van Rij, A.M. A variant of the castor zinc finger 1 (CASZ1) gene is differentially associated with the clinical classification of chronic venous disease. Sci. Rep. 2019, 9, 1–7. [Google Scholar] [CrossRef]
- Douguet, D.; Patel, A.; Xu, A.; Vanhoutte, P.M.; Honoré, E. Piezo Ion Channels in Cardiovascular Mechanobiology. Trends Pharmacol. Sci. 2019, 40, 956–970. [Google Scholar] [CrossRef]
- Nonomura, K.; Lukacs, V.; Sweet, D.T.; Goddard, L.M.; Kanie, A.; Whitwam, T.; Ranade, S.S.; Fujimori, T.; Kahn, M.L.; Patapoutian, A. Mechanically activated ion channel PIEZO1 is required for lymphatic valve formation. Proc. Natl. Acad. Sci. USA 2018, 115, 12817–12822. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hou, B.; Tumova, S.; Muraki, K.; Bruns, A.; Ludlow, M.J.; Sedo, A.; Hyman, A.J.; McKeown, L.; Young, R.S.; et al. Piezo1 integration of vascular architecture with physiological force. Nature 2014, 515, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Alper, S.L. Genetic Diseases of PIEZO1 and PIEZO2 Dysfunction. Curr. Top. Membr. 2017, 79, 97–134. [Google Scholar] [CrossRef] [PubMed]
- Vilagos, B.; Hoffmann, M.; Souabni, A.; Sun, Q.; Werner, B.; Medvedovic, J.; Bilic, I.; Minnich, M.; Axelsson, E.; Jaritz, M.; et al. Essential role of EBF1 in the generation and function of distinct mature B cell types. J. Exp. Med. 2012, 209, 775–792. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.-U.-R.; Wiberg MRCS, A.; Ng, M.; Wang, W.; Auton, A. Genome-wide association analysis and replication in 810,625 individuals identifies novel therapeutic targets for varicose veins. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kazenwadel, J.; Betterman, K.L.; Chong, C.E.; Stokes, P.H.; Lee, Y.K.; Secker, G.A.; Agalarov, Y.; Demir, C.S.; Lawrence, D.M.; Sutton, D.L.; et al. GATA2 is required for lymphatic vessel valve development and maintenance. J. Clin. Investig. 2015, 125, 2879–2994. [Google Scholar] [CrossRef]
- Smetanina, M.A.; Shevela, A.I.; Gavrilov, K.A.; Filipenko, M.L. The genetic constituent of varicose vein pathogenesis as a key for future treatment option development. Vessel Plus 2021, 5. [Google Scholar] [CrossRef]
- Lyons, O.; Saha, P.; Seet, C.; Kuchta, A.; Arnold, A.; Grover, S.; Rashbrook, V.; Sabine, A.; Vizcay-Barrena, G.; Patel, A.; et al. Human venous valve disease caused by mutations in FOXC2 and GJC2. J. Exp. Med. 2017, 214, 2437–2452. [Google Scholar] [CrossRef]
- Sabine, A.; Bovay, E.; Demir, C.S.; Kimura, W.; Jaquet, M.; Agalarov, Y.; Zangger, N.; Scallan, J.P.; Graber, W.; Gulpinar, E.; et al. FOXC2 and fluid shear stress stabilize postnatal lymphatic vasculature. J. Clin. Investig. 2015, 125, 3861–3877. [Google Scholar] [CrossRef]
- Berna-Erro, A.; Jardin, I.; Salido, G.M.; Rosado, J.A. Role of STIM2 in cell function and physiopathology. J. Physiol. 2017, 595, 3111–3128. [Google Scholar] [CrossRef] [PubMed]
- Barton, J.C.; Edwards, C.Q.; Acton, R.T. HFE gene: Structure, function, mutations, and associated iron abnormalities. Gene 2015, 574, 179–192. [Google Scholar] [CrossRef]
- Gaenzer, H.; Marschang, P.; Sturm, W.; Neumayr, G.; Vogel, W.; Patsch, J.; Weiss, G. Association between increased iron stores and impaired endothelial function in patients with hereditary hemochromatosis. J. Am. Coll. Cardiol. 2002, 40, 2189–2194. [Google Scholar] [CrossRef]
- Zamboni, P.; Izzo, M.; Tognazzo, S.; Carandina, S.; De Palma, M.; Catozzi, L.; Caggiati, A.; Scapoli, G.; Gemmati, D. The overlapping of local iron overload and HFE mutation in venous leg ulcer pathogenesis. Free Radic. Biol. Med. 2006, 40, 1869–1873. [Google Scholar] [CrossRef] [PubMed]
- Augstein, A.; Mierke, J.; Poitz, D.M.; Strasser, R.H. Sox9 is increased in arterial plaque and stenosis, associated with synthetic phenotype of vascular smooth muscle cells and causes alterations in extracellular matrix and calcification. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2526–2537. [Google Scholar] [CrossRef] [PubMed]
- Hanley, K.P.; Oakley, F.; Sugden, S.; Wilson, D.I.; Mann, D.A.; Hanley, N.A. Ectopic SOX9 mediates extracellular matrix deposition characteristic of organ fibrosis. J. Biol. Chem. 2008, 283, 14063–14071. [Google Scholar] [CrossRef]
- Jin, Y.; Xu, G.; Huang, J.; Zhou, D.; Huang, X.; Shen, L. Analysis of the association between an insertion/deletion polymorphism within the 3′ untranslated region of COL1A2 and chronic venous insufficiency. Ann. Vasc. Surg. 2013, 27, 959–963. [Google Scholar] [CrossRef]
- Zöller, B.; Ji, J.; Sundquist, J.; Sundquist, K. Venous thromboembolism and varicose veins share familial susceptibility: A nationwide family study in Sweden. J. Am. Heart Assoc. 2014, 3, e000850. [Google Scholar] [CrossRef] [PubMed]
- Poredos, P.; Spirkoska, A.; Rucigaj, T.; Fareed, J.; Jezovnik, M.K. Do blood constituents in varicose veins differ from the systemic blood constituents? Eur. J. Vasc. Endovasc. Surg. 2015, 50, 250–256. [Google Scholar] [CrossRef]
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef]
- Moosavi, A.; Ardekani, A.M. Role of epigenetics in biology and human diseases. Iran. Biomed. J. 2016, 20, 246–258. [Google Scholar] [CrossRef]
- Al Aboud, N.M.; Jialal, I. Genetics, Epigenetic Mechanism; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Alegría-Torres, J.A.; Baccarelli, A.; Bollati, V. Epigenetics and lifestyle. Epigenomics 2011, 3, 267–277. [Google Scholar] [CrossRef]
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef] [PubMed]
- Sahar, S.; Sassone-Corsi, P. The epigenetic language of circadian clocks. Handb. Exp. Pharmacol. 2013, 217, 29–44. [Google Scholar] [CrossRef]
- Ordovás, J.M.; Smith, C.E. Epigenetics and cardiovascular disease. Nat. Rev. Cardiol. 2010, 7, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Ku, K.H.; Subramaniam, N.; Marsden, P.A. Epigenetic determinants of flow-mediated vascular endothelial gene expression. Hypertension 2019, 74, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.; Simmons, R.; Thabet, S.; Jo, H. The role of epigenetics in the endothelial cell shear stress response and atherosclerosis. Int. J. Biochem. Cell Biol. 2015, 67, 167–176. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Sethi, G. Role of epigenetics in in flammation-associated diseases. Subcell. Biochem. 2013, 61, 627–657. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Shi, X.; Darabi, R.; Li, Y. Hypoxia in Cell Reprogramming and the Epigenetic Regulations. Front. Cell Dev. Biol. 2021, 9, 609984. [Google Scholar] [CrossRef]
- Edwards, J.R.; Yarychkivska, O.; Boulard, M.; Bestor, T.H. DNA methylation and DNA methyltransferases. Epigenet. Chromatin 2017, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Sallustio, F.; Gesualdo, L.; Gallone, A. New findings showing how DNA methylation influences diseases. World J. Biol. Chem. 2019, 10, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.J.; McKay, G.J.; Maxwell, A.P.; McKnight, A.J. DNA hypermethylation and DNA hypomethylation is present at different loci in chronic kidney disease. Epigenetics 2013, 9, 366–376. [Google Scholar] [CrossRef]
- Derecka, M.; Herman, J.S.; Cauchy, P.; Ramamoorthy, S.; Lupar, E.; Grün, D.; Grosschedl, R. EBF1-deficient bone marrow stroma elicits persistent changes in HSC potential. Nat. Immunol. 2020, 21, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Wilmanns, C.; Cooper, A.; Wockner, L.; Katsandris, S.; Glaser, N.; Meyer, A.; Bartsch, O.; Binder, H.; Walter, P.K.; Zechner, U. Morphology and Progression in Primary Varicose Vein Disorder Due to 677C>T and 1298A>C Variants of MTHFR. EBioMedicine 2015, 2, 158–164. [Google Scholar] [CrossRef]
- Xu, J.; Li, K.; Zhou, W. Relationship between genetic polymorphism of MTHFR C677T and lower extremities deep venous thrombosis. Hematology 2019, 24, 108–111. [Google Scholar] [CrossRef]
- Smetanina, M.A.; Kel, A.E.; Sevost’ianova, K.S.; Maiborodin, I.V.; Shevela, A.I.; Zolotukhin, I.A.; Stegmaier, P.; Filipenko, M.L. DNA methylation and gene expression profiling reveal MFAP5 as a regulatory driver of extracellular matrix remodeling in varicose vein disease. Epigenomics 2018, 10, 1103–1119. [Google Scholar] [CrossRef]
- Jiang, H.; Lun, Y.; Wu, X.; Xia, Q.; Zhang, X.; Xin, S.; Zhang, J. Association between the hypomethylation of osteopontin and integrin β3 promoters and vascular smooth muscle cell phenotype switching in great saphenous varicose veins. Int. J. Mol. Sci. 2014, 15, 18747–18761. [Google Scholar] [CrossRef]
- Beermann, J.; Piccoli, M.T.; Viereck, J.; Thum, T. Non-coding rnas in development and disease: Background, mechanisms, and therapeutic approaches. Physiol. Rev. 2016, 96, 1297–1325. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Cui, C.; Liu, G.; Huang, Y.; Lu, X.; Lu, M.; Huang, X.; Li, W. MicroRNA Profiling in GSV Tissues of Patients with CVI MicroRNA Profiling in Great Saphenous Vein Tissues of Patients with Chronic Venous Insufficiency. Tohoku J. Exp. Med. 2012, 228, 341–350. [Google Scholar] [CrossRef]
- Barwari, T.; Joshi, A.; Mayr, M. MicroRNAs in Cardiovascular Disease. J. Am. Coll. Cardiol. 2016, 68, 2577–2584. [Google Scholar] [CrossRef] [PubMed]
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. miRNAs as Biomarkers in Disease: Latest Findings Regarding Their Role in Diagnosis and Prognosis. Cells 2020, 9, 276. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.A.; Adesina-Georgiadis, K.N.; Spagou, K.; Vorkas, P.A.; Li, J.V.; Shalhoub, J.; Holmes, E.; Davies, A.H. A comprehensive characterisation of the metabolic profile of varicose veins; implications in elaborating plausible cellular pathways for disease pathogenesis. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Zalewski, D.P.; Ruszel, K.P.; Stępniewski, A.; Gałkowski, D.; Bogucki, J.; Komsta, Ł.; Kołodziej, P.; Chmiel, P.; Zubilewicz, T.; Feldo, M.; et al. Dysregulations of MicroRNA and Gene Expression in Chronic Venous Disease. J. Clin. Med. 2020, 9, 1251. [Google Scholar] [CrossRef]
- Schmitz, S.U.; Grote, P.; Herrmann, B.G. Mechanisms of long noncoding RNA function in development and disease. Cell. Mol. Life Sci. 2016, 73, 2491–2509. [Google Scholar] [CrossRef]
- Li, X.; Jiang, X.Y.; Ge, J.; Wang, J.; Chen, G.J.; Xu, L.; Xie, D.Y.; Yuan, T.Y.; Zhang, D.S.; Zhang, H.; et al. Aberrantly expressed lncRNAs in primary varicose great saphenous veins. PLoS ONE 2014, 9, e86156. [Google Scholar] [CrossRef]
- Biranvand, A.S.; Khosravi, M.; Esfandiari, G.; Poursaleh, A.; Hosseini-Fard, S.R.; Amirfarhangi, A.; Najafi, M. Associations between miR-661, miR-1202, lncRNA-HOTAIR, lncRNA-GAS5 and MMP9 in differentiated M2-macrophages of patients with varicose veins. Int. Angiol. 2018, 37, 451–456. [Google Scholar] [CrossRef]
- Smith, R.K.; Golledge, J. A systematic review of circulating markers in primary chronic venous insufficiency. Phlebology 2014, 29, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Gude, N.M.; Roberts, C.T.; Kalionis, B.; King, R.G. Growth and function of the normal human placenta. Thromb. Res. 2004, 114, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Latendresse, G.; Founds, S. The Fascinating and Complex Role of the Placenta in Pregnancy and Fetal Well-being. J. Midwifery Women’s Health 2015, 60, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Fowden, A.L.; Thornburg, K.L. Placental origins of chronic disease. Physiol. Rev. 2016, 96, 1509–1565. [Google Scholar] [CrossRef] [PubMed]
- García-Honduvilla, N.; Ortega, M.A.; Asúnsolo, Á.; Álvarez-Rocha, M.J.; Romero, B.; De León-Luis, J.; Álvarez-Mon, M.; Buján, J. Placentas from women with pregnancy-associated venous insufficiency show villi damage with evidence of hypoxic cellular stress. Hum. Pathol. 2018, 77, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.A.; Saez, M.Á.; Asúnsolo, Á.; Romero, B.; Bravo, C.; Coca, S.; Sainz, F.; Álvarez-Mon, M.; Buján, J.; García-Honduvilla, N. Upregulation of VEGF and PEDF in Placentas of Women with Lower Extremity Venous Insufficiency during Pregnancy and Its Implication in Villous Calcification. BioMed Res. Int. 2019, 2019, 5320902. [Google Scholar] [CrossRef]
- Ortega, M.A.; Saez, M.A.; Sainz, F.; Fraile-Martínez, O.; García-Gallego, S.; Pekarek, L.; Bravo, C.; Coca, S.; Álvarez-Mon, M.; Buján, J.; et al. Lipidomic profiling of chorionic villi in the placentas of women with chronic venous disease. Int. J. Med. Sci. 2020, 17, 2790–2798. [Google Scholar] [CrossRef]
- Ortega, M.A.; Saez, M.A.; Fraile-Martínez, O.; Asúnsolo, Á.; Pekarek, L.; Bravo, C.; Coca, S.; Sainz, F.; Álvarez-Mon, M.; Buján, J.; et al. Increased angiogenesis and lymphangiogenesis in the placental villi of women with chronic venous disease during pregnancy. Int. J. Mol. Sci. 2020, 21, 2487. [Google Scholar] [CrossRef]
- Ortega, M.A.; Asúnsolo, Á.; Álvarez-Rocha, M.J.; Romero, B.; De León-Luis, J.; Álvarez-Mon, M.; Buján, J.; García-Honduvilla, N. Remodelling of collagen fibres in the placentas of women with venous insufficiency during pregnancy. Histol. Histopathol. 2018, 33, 567–576. [Google Scholar] [CrossRef]
- Ortega, M.A.; Romero, B.; Asúnsolo, Á.; Martínez-Vivero, C.; Sainz, F.; Bravo, C.; De León-Luis, J.; Álvarez-Mon, M.; Buján, J.; García-Honduvilla, N. Pregnancy-associated venous insufficiency course with placental and systemic oxidative stress. J. Cell. Mol. Med. 2020, 24, 4157–4170. [Google Scholar] [CrossRef]
- Jena, M.K.; Sharma, N.R.; Petitt, M.; Maulik, D.; Nayak, N.R. Pathogenesis of preeclampsia and therapeutic approaches targeting the placenta. Biomolecules 2020, 10, 953. [Google Scholar] [CrossRef]
- Urbanek, T.; Juśko, M.; Kuczmik, W.B. Compression therapy for leg oedema in patients with heart failure. ESC Heart Fail. 2020, 7, 2012–2020. [Google Scholar] [CrossRef]
- Pappas, P.J.; Lakhanpal, S.; Nguyen, K.Q.; Vanjara, R. The Center for Vein Restoration Study on presenting symptoms, treatment modalities, and outcomes in Medicare-eligible patients with chronic venous disorders. J. Vasc. Surg. Venous Lymphat. Disord. 2018, 6, 13–24. [Google Scholar] [CrossRef]
- Dissemond, J.; Storck, M.; Kröger, K.; Stücker, M. Indications and contraindications for modern compression therapy. Wien. Med. Wochenschr. 2018, 168, 228–235. [Google Scholar] [CrossRef]
- Shingler, S.; Robertson, L.; Boghossian, S.; Stewart, M. Compression stockings for the initial treatment of varicose veins in patients without venous ulceration. Cochrane Database Syst. Rev. 2013, 2013. [Google Scholar] [CrossRef]
- Konschake, W.; Riebe, H.; Pediaditi, P.; Haase, H.; Jünger, M.; Lutze, S. Compression in the treatment of chronic venous insufficiency: Efficacy depending on the length of the stocking. Clin. Hemorheol. Microcirc. 2016, 64, 425–434. [Google Scholar] [CrossRef]
- Omeara, S.; Martyn-St James, M. Foam dressings for venous leg ulcers. Cochrane Database Syst. Rev. 2013, 2013. [Google Scholar] [CrossRef]
- O’Hare, J.L.; Stephens, J.; Parkin, D.; Earnshaw, J.J. Randomized clinical trial of different bandage regimens after foam sclerotherapy for varicose veins. Br. J. Surg. 2010, 97, 650–656. [Google Scholar] [CrossRef]
- Huang, T.W.; Chen, S.L.; Bai, C.H.; Wu, C.H.; Tam, K.W. The optimal duration of compression therapy following varicose vein surgery: A meta-analysis of randomized controlled trials. Eur. J. Vasc. Endovasc. Surg. 2013, 45, 397–402. [Google Scholar] [CrossRef]
- Rabe, E.; Partsch, H.; Morrison, N.; Meissner, M.H.; Mosti, G.; Lattimer, C.R.; Carpentier, P.H.; Gaillard, S.; Jünger, M.; Urbanek, T.; et al. Risks and contraindications of medical compression treatment—A critical reappraisal. An international consensus statement. Phlebology 2020, 35, 447–460. [Google Scholar] [CrossRef]
- Gohel, M.; Davies, A. Pharmacological Agents in the Treatment of Venous Disease: An Update of the Available Evidence. Curr. Vasc. Pharmacol. 2009, 7, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Zapata, M.J.; Vernooij, R.W.; Uriona Tuma, S.M.; Stein, A.T.; Moreno, R.M.; Vargas, E.; Capellà, D.; Bonfill Cosp, X. Phlebotonics for venous insufficiency. Cochrane Database Syst. Rev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Andreozzi, G.M. Sulodexide in the treatment of chronic venous disease. Am. J. Cardiovasc. Drugs 2012, 12, 73–81. [Google Scholar] [CrossRef]
- Bush, R.; Comerota, A.; Meissner, M.; Raffetto, J.D.; Hahn, S.R.; Freeman, K. Recommendations for the medical management of chronic venous disease: The role of Micronized Purified Flavanoid Fraction (MPFF): Recommendations from the Working Group in Chronic Venous Disease (CVD) 2016. Phlebology 2017, 32, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Mansilha, A.; Sousa, J. Pathophysiological mechanisms of chronic venous disease and implications for venoactive drug therapy. Int. J. Mol. Sci. 2018, 19, 1669. [Google Scholar] [CrossRef]
- Perrin, M.; Ramelet, A.A. Pharmacological treatment of primary chronic venous disease: Rationale, results and unanswered questions. Eur. J. Vasc. Endovasc. Surg. 2011, 41, 117–125. [Google Scholar] [CrossRef]
- Wittens, C.; Davies, A.H.; Bækgaard, N.; Broholm, R.; Cavezzi, A.; Chastanet, S.; De Wolf, M.; Eggen, C.; Giannoukas, A.; Gohel, M.; et al. Editor’s choice—Management of chronic venous disease: Clinical practice guidelines of the European Society for Vascular Surgery (ESVS). Eur. J. Vasc. Endovasc. Surg. 2015, 49, 678–737. [Google Scholar] [CrossRef]
- Coleridge-Smith, P.; Lok, C.; Ramelet, A.A. Venous leg ulcer: A meta-analysis of adjunctive therapy with micronized purified flavonoid fraction. Eur. J. Vasc. Endovasc. Surg. 2005, 30, 198–208. [Google Scholar] [CrossRef]
- De-Abreu, G.C.G.; De Camargo Júnior, O.; De-Abreu, M.F.M.; De-Aquino, J.L.B. Ultrasound-guided foam sclerotherapy for severe chronic venous insufficiency. Rev. Col. Bras. Cir. 2017, 44, 511–520. [Google Scholar] [CrossRef]
- Rasmussen, L.H.; Lawaetz, M.; Bjoern, L.; Vennits, B.; Blemings, A.; Eklof, B. Randomized clinical trial comparing endovenous laser ablation, radiofrequency ablation, foam sclerotherapy and surgical stripping for great saphenous varicose veins. Br. J. Surg. 2011, 98, 1079–1087. [Google Scholar] [CrossRef]
- Rigby, K.A.; Palfreyman, S.S.; Beverley, C.; Michaels, J.A. Surgery versus sclerotherapy for the treatment of varicose veins. Cochrane Database Syst. Rev. 2004. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.R.; Slim, F.J.A.; Emerson, L.G.; Davies, C.; Bulbulia, R.A.; Whyman, M.R.; Poskitt, K.R. Effect of foam sclerotherapy on healing and long-term recurrence in chronic venous leg ulcers. Phlebology 2013, 28, 140–146. [Google Scholar] [CrossRef]
- Lin, F.; Zhang, S.; Sun, Y.; Ren, S.; Liu, P. The management of varicose veins. Int. Surg. 2015, 100, 185–189. [Google Scholar] [CrossRef]
- Gujja, K.; Wiley, J.; Krishnan, P. Chronic venous insufficiency. Interv. Cardiol. Clin. 2014, 3, 593–605. [Google Scholar] [CrossRef]
- Brar, R.; Nordon, I.M.; Hinchliffe, R.J.; Loftus, I.M.; Thompson, M.M. Surgical management of varicose veins: Meta-analysis. Vascular 2010, 18, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Puggioni, A.; Kalra, M.; Gloviczki, P. Superficial vein surgery and SEPS for chronic venous insufficiency. Semin. Vasc. Surg. 2005, 18, 41–48. [Google Scholar] [CrossRef]
- Van de Bos, R.R.; de Maeseneer, M.M. Endovenous thermal ablation for varicose veins: Strengths and weaknesses. Phlebolymphology 2012, 19, 163–169. [Google Scholar]
- Attaran, R. Latest Innovations in the Treatment of Venous Disease. J. Clin. Med. 2018, 7, 77. [Google Scholar] [CrossRef]
- Balint, R.; Farics, A.; Parti, K.; Vizsy, L.; Batorfi, J.; Menyhei, G.; Balint, I.B. Which endovenous ablation method does offer a better long-term technical success in the treatment of the incompetent great saphenous vein? Review. Vascular 2016, 24, 649–657. [Google Scholar] [CrossRef]
- Van den Bos, R.; Arends, L.; Kockaert, M.; Neumann, M.; Nijsten, T. Endovenous therapies of lower extremity varicosities: A meta-analysis. J. Vasc. Surg. 2009, 49, 230–239. [Google Scholar] [CrossRef]
- Lane, R.J.; Cuzzilla, M.L.; Coroneos, J.C. The treatment of varicose veins with external stenting to the saphenofemoral junction. Vasc. Endovasc. Surg. 2002, 36, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Meghdadi, A.; Jones, S.A.; Patel, V.A.; Lewis, A.L.; Millar, T.M.; Carugo, D. Foam-in-vein: A review of rheological properties and characterization methods for optimization of sclerosing foams. J. Biomed. Mater. Res. Part B Appl. Biomater. 2021, 109, 69–91. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Colino, A.; Jockenhoevel, S. Advances in Engineering Venous Valves: The Pursuit of a Definite Solution for Chronic Venous Disease. Tissue Eng. Part B Rev. 2020. [Google Scholar] [CrossRef]
- Brandt, A.H. Evaluation of new ultrasound techniques for clinical imaging in selected liver and vascular applications. Dan. Med. J. 2018, 65, B5455. [Google Scholar]
- García-Montero, C.; Fraile-Martínez, O.; Gómez-Lahoz, A.M.; Pekarek, L.; Castellanos, A.J.; Noguerales-Fraguas, F.; Coca, S.; Guijarro, L.G.; García-Honduvilla, N.; Asúnsolo, A.; et al. Nutritional components in western diet versus mediterranean diet at the gut microbiota-immune system interplay implications for health and disease. Nutrients 2021, 13, 699. [Google Scholar] [CrossRef]
- McDaniel, J.C.; Kemmner, K.G.; Rusnak, S. Nutritional profile of older adults with chronic venous leg ulcers: A pilot study. Geriatr. Nurs. 2015, 36, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Volpe, E.F.T.; Resqueti, V.R.; Da Silva, A.A.M.; Gualdi, L.P.; Fregonezi, G.A.F. Supervised exercise protocol for lower limbs in subjects with chronic venous disease: An evaluator-blinded, randomized clinical trial. Trials 2020, 21, 414. [Google Scholar] [CrossRef]
- Orr, L.; Klement, K.A.; McCrossin, L.; Drombolis, D.O.; Houghton, P.E.; Spaulding, S.; Burke, S. A systematic review and meta-Analysis of exercise intervention for the treatment of calf muscle pump impairment in individuals with chronic venous insufficiency. Ostomy Wound Manag. 2017, 63, 30–43. [Google Scholar] [CrossRef]
- Padberg, F.T.; Johnston, M.V.; Sisto, S.A.; Burnand, K.G.; Wakefield, T.W.; Perkowski, P. Structured exercise improves calf muscle pump function in chronic venous insufficiency: A randomized trial. J. Vasc. Surg. 2004, 39, 79–87. [Google Scholar] [CrossRef]
- Mutlak, O.; Slam, M.A.; Dfield, N.S. The influence of exercise on ulcer healing in patients with chronic venous insufficiency. Proc. Inter. Angiol. 2018, 37, 160–167. [Google Scholar] [CrossRef]
- Brown, G.C. Living too long. EMBO Rep. 2015, 16, 137–141. [Google Scholar] [CrossRef]
- Bozkurt, A.K.; Balkanay, O.O. Approach to venous diseases in the elderly. Turk Kardiyol. Dern. Ars. 2017, 45, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.M. Improving nutrition to support healthy ageing: What are the opportunities for intervention? Proc. Nutr. Soc. 2018, 77, 257–264. [Google Scholar] [CrossRef]
- Shlisky, J.; Bloom, D.E.; Beaudreault, A.R.; Tucker, K.L.; Keller, H.H.; Freund-Levi, Y.; Fielding, R.A.; Cheng, F.W.; Jensen, G.L.; Wu, D.; et al. Nutritional considerations for healthy aging and reduction in age-related chronic disease. Adv. Nutr. 2017, 8, 17–26. [Google Scholar] [CrossRef]
- Guillermo, J.; Avila, O.; Aguilar De Plata, A.C. Impact of oxidative stress during pregnancy on fetal epigenetic patterns and early origin of vascular diseases. Nutr. Rev. 2015, 73, 12–21. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical (C) Classification | Etiologic (E) Classification | Anatomic (A) Classification | Pathophysiologic (P) Classification |
|---|---|---|---|
|
|
An No venous anatomic location identified |
|
| Gene | Whole Name | Chromosome and Location | Variant(s) (rs ID) | Risk Allele | Mutation | Highlighted Associated Function | (Possible) Implication in CVD Etiology/Pathogenesis | References |
|---|---|---|---|---|---|---|---|---|
| KCNH8 | Potassium voltage-gated channel subfamily H member 8 | chr3:19293203 (3p24.3) | rs727139 | A > G | - | Smooth muscle contraction | Venous dilation and VV formation | [237] |
| EFEMP1 | EGF containing fibulin extracellular matrix protein 1 | chr2:55868859 | rs17278665 | C > G | - | Cell adhesion and migration | Remodeling of ECM components and changes in vessel elasticity by altering the expression of MMPs and TIMPs | [238] |
| MMP-9 | Matrix Metallopeptidase 9 | chr20q11.2-q13.1 | - | - | ✓ | ECM degradation | Collagen type I degradation entailing stiffness of vein | [239] |
| TIMP-2 | Tissue inhibitor of metalloproteinases 2 | chr17q25.3. | - | - | ✓ | Inhibition of MMPs | Lower expression implies higher collagen degradation by MMPs | [239] |
| CASZ1 | Castor zinc finger 1 | chr1:10765520 Intron 1 | rs11121615 | C > T | - | Transcription factor for EGFLD7 | Angiogenesis stimulation and aberrant vascular assembly | [241] |
| PIEZO1 | Piezo type mechanosensitive ion channel component 1 | chr16:88769137 (16q24.3) | rs2911463 | G > A/G > C/G > T | ✓ | Shear stress sensing by Ca2+ | Impaired function implies aberrant vascular structure, ECs reorganization and edema | [242,243,244,245] |
| PPP3R1 | Protein phosphatase 3 regulatory subunit B, alpha | chr 2:68262089 (2p14) Intergenic | rs2861819 | G > A/G > C | - | Ca2+ sensitivity | Abnormal vascular integrity | [43] |
| EBF1 | Early B Cell Factor Transcription Factor 1 | chr5:158803005 | rs11135046 | G > A/G > T | - | Adhesion and migration in early B lymphopoiesis | Possible epigenetic reprogramming and B cells activation | [247,248] |
| GATA2 | GATA Binding Protein 2 | chr3:128578726 | rs9880192 | G > A/G > C | - | Lymphatic vessel valve development | Impaired function implies lymphedema | [240,249] |
| NFATC2 | Nuclear Factor of Activated T cells 2 | chr20:51541298 chr20:51538108 | rs3787184 and rs12625547 | A > G T > G | - | Induces immune response or inflammation in vascular remodeling | Not well understood; Acting in consonance with FOXC2 and GJC2. | [248,250] |
| FOXC2 | Fork-head box protein C2 | 16q24 | - | - | ✓ | Critical product in developmental processes | Inactivation implies abnormal shear stress sensing and valve incompetence | [251,252] |
| GJC2 | Gap junction gamma-2 | 1q41-q42 | - | - | ✓ | Implicated in the gap junctions between cells | Cell-cell junction defects and valve incompetence | [251] |
| STIM2 | Stromal interaction molecule 2 | 4:26857601-27025381 (4p15.2) Intergenic variant Mapped gene(s): STIM2, TBC1D19 | rs28558138 | G > C | - | Controls Ca2+ concentration in cytosol | Higher Ca2+ deposition | [250,253] |
| HFE | Homeostatic iron regulator | chr6:26090951 chr6:26267527 | rs1799945 and rs7773004 | C > G C > T A > C/A > G/A > T | ✓ | Regulates hepcidin expression, involved in iron storage | Iron overload implies endothelial dysfunction. Moreover, activation of MMPs and inhibition of TIMPs, deposits of iron, RBCs extravasation | [254,255,256] |
| SOX9 | SRY-Box Transcription Factor 9 | 17:72032304 17q24.3 | rs2241173 | A > C A > G | - | ECM remodeling | Influenced by TGF-β1may involve higher Ca2+ deposition | [24,257,258] |
| COL2A1 | Collagen type II alpha 1 chain | 12:47793818 (12q13.11) | rs73107980 | C > G C > T | - | Coding collagen type II alpha 1 | Abnormal modelling of ECM | [239] |
| COL1A2 | Collagen type I alpha 2 chain | chr7:94431047-94431048 | rs3917 | (indels) | - | Coding collagen type I alpha 2 | Collagen dysregulation, higher susceptibility to CVI | [259] |
| THBD | Thrombomodulin | 20p11.21 | - | - | ✓ | Related with thromboembolic diseases | Prothrombotic markers | [43] |
| MTHFR | Methylenetetrahydrofolate reductase | 1p36.22 | - | - | ✓ | Related with thromboembolic diseases | Prothrombotic markers | [43] |
| Treatment | Uses | Level of Evidence and Recommendation |
|---|---|---|
| Compression therapy (stockings, bandages, adjustable compression wraps) | Initial therapeutical method of CVD A powerful tool in CVI ulceration Increase the efficacy after interventional procedures | IB [303] IB [305] IA [307,308] |
| Pharmacological therapies | Venotonics to ameliorate early symptoms (Pain, edema) Flavonoids and derivates as complement therapy with compression stockings in venous ulcers | IIaA [310] IIaA [311,316] |
| Sclerotherapy | Second-line treatment for patients who are not candidates for endovenous ablation or surgery First-line therapy in patients with recurrent CVD and fragile patients with venous ulceration | IA [319] IIaB [320] |
| Endovenous thermal ablation | First line therapy for patients with CVD and great saphenous vein reflux | IA [318,328] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortega, M.A.; Fraile-Martínez, O.; García-Montero, C.; Álvarez-Mon, M.A.; Chaowen, C.; Ruiz-Grande, F.; Pekarek, L.; Monserrat, J.; Asúnsolo, A.; García-Honduvilla, N.; et al. Understanding Chronic Venous Disease: A Critical Overview of Its Pathophysiology and Medical Management. J. Clin. Med. 2021, 10, 3239. https://doi.org/10.3390/jcm10153239
Ortega MA, Fraile-Martínez O, García-Montero C, Álvarez-Mon MA, Chaowen C, Ruiz-Grande F, Pekarek L, Monserrat J, Asúnsolo A, García-Honduvilla N, et al. Understanding Chronic Venous Disease: A Critical Overview of Its Pathophysiology and Medical Management. Journal of Clinical Medicine. 2021; 10(15):3239. https://doi.org/10.3390/jcm10153239
Chicago/Turabian StyleOrtega, Miguel A., Oscar Fraile-Martínez, Cielo García-Montero, Miguel A. Álvarez-Mon, Chen Chaowen, Fernando Ruiz-Grande, Leonel Pekarek, Jorge Monserrat, Angel Asúnsolo, Natalio García-Honduvilla, and et al. 2021. "Understanding Chronic Venous Disease: A Critical Overview of Its Pathophysiology and Medical Management" Journal of Clinical Medicine 10, no. 15: 3239. https://doi.org/10.3390/jcm10153239
APA StyleOrtega, M. A., Fraile-Martínez, O., García-Montero, C., Álvarez-Mon, M. A., Chaowen, C., Ruiz-Grande, F., Pekarek, L., Monserrat, J., Asúnsolo, A., García-Honduvilla, N., Álvarez-Mon, M., & Bujan, J. (2021). Understanding Chronic Venous Disease: A Critical Overview of Its Pathophysiology and Medical Management. Journal of Clinical Medicine, 10(15), 3239. https://doi.org/10.3390/jcm10153239