
Fibrin Clot Properties in Atherosclerotic Vascular Disease: From Pathophysiology to Clinical Outcomes


Abstract
1. Introduction
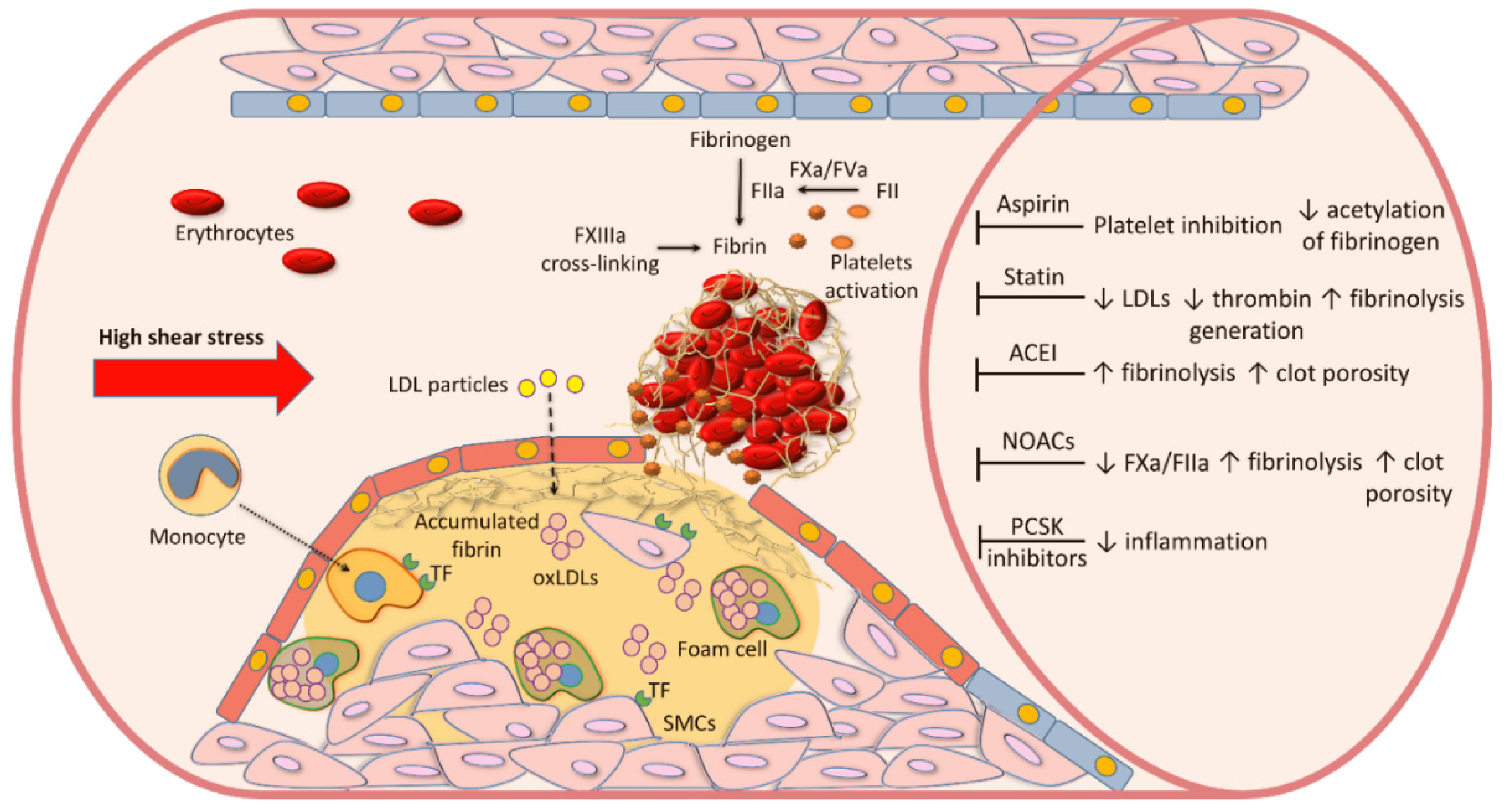
2. Atherosclerotic Plaque Formation
3. Blood Coagulation and Fibrin Formation in Atherosclerosis
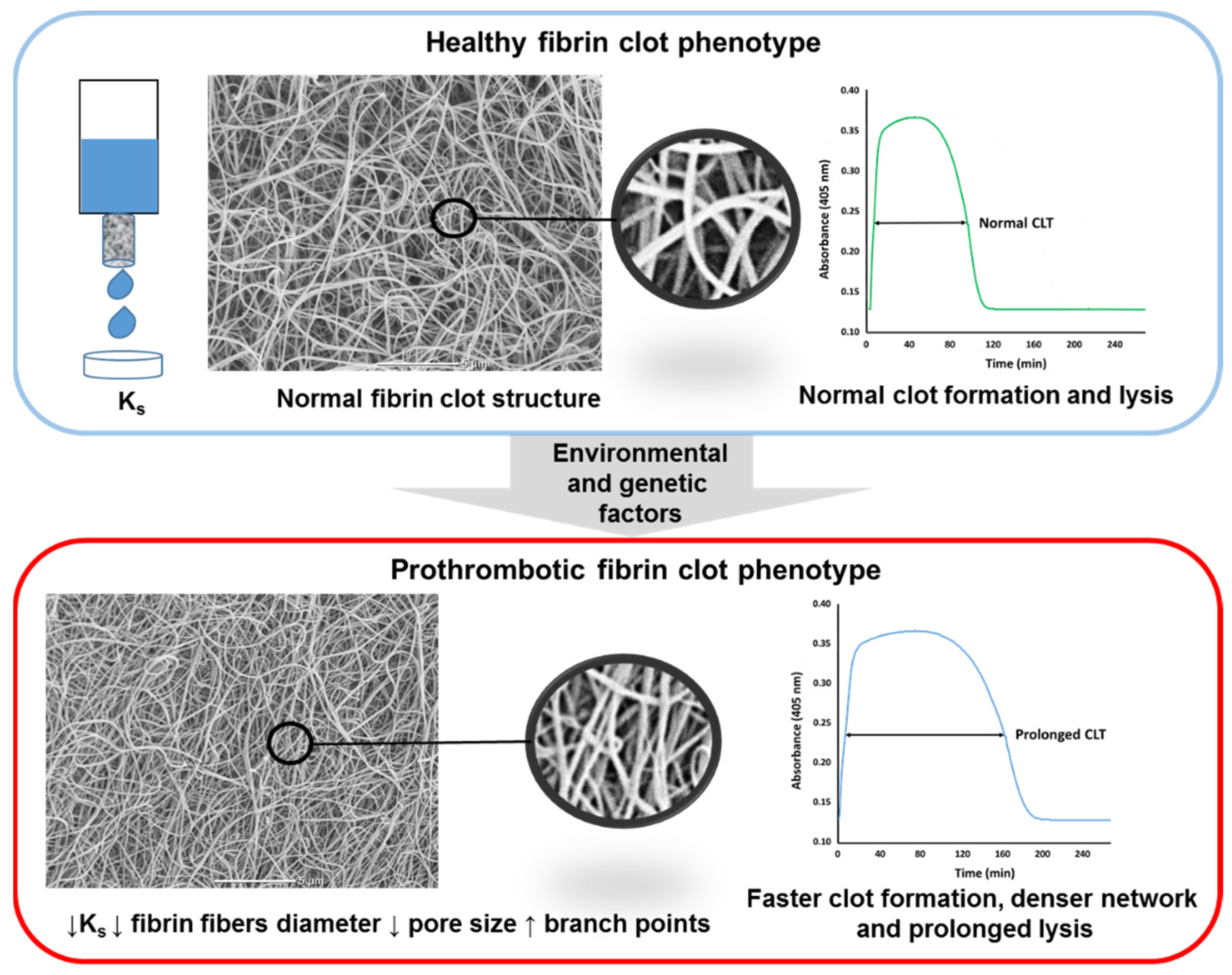
4. Measures of Fibrin Clot Properties
5. Cardiovascular Risk Factors
6. Coronary Artery Disease
6.1. Acute MI
6.2. Stable CAD
7. Peripheral Arterial Disease
8. Aortic Aneurysm
9. Pharmacological Treatment and Fibrin Clot Properties
9.1. Cholesterol-Lowering Agents
9.2. Aspirin
9.3. Angiotensin-Converting Enzyme Inhibitors (ACEI)
9.4. NOACs
10. Clinical Implications
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef]
- Schaftenaar, F.; Frodermann, V.; Kuiper, J.; Lutgens, E. Atherosclerosis: The interplay between lipids and immune cells. Curr. Opin. Lipidol. 2016, 27, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.J. Macrophages in atherosclerosis regression. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Jaipersad, A.S.; Lip, G.Y.; Silverman, S.; Shantsila, E. The role of monocytes in angiogenesis and atherosclerosis. J. Am. Coll. Cardiol. 2014, 63, 1–11. [Google Scholar] [CrossRef]
- Moreno, P.R.; Purushothaman, K.R.; Fuster, V.; Echeverri, D.; Truszczynska, H.; Sharma, S.K.; Badimon, J.J.; O’Connor, W.N. Plaque neovascularization is increased in ruptured atherosclerotic lesions of human aorta: Implications for plaque vulnerability. Circulation 2004, 110, 2032–2038. [Google Scholar] [CrossRef]
- Vergallo, R.; Porto, I.; D’Amario, D.; Annibali, G.; Galli, M.; Benenati, S.; Bendandi, F.; Migliaro, S.; Fracassi, F.; Aurigemma, C.; et al. Coronary atherosclerotic phenotype and plaque healing in patients with recurrent acute coronary syndromes compared with patients with long-term clinical stability: An in vivo optical coherence tomography study. JAMA Cardiol. 2019, 4, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Fracassi, F.; Crea, F.; Sugiyama, T.; Yamamoto, E.; Uemura, S.; Vergallo, R.; Porto, I.; Lee, H.; Fujimoto, J.; Fuster, V.; et al. Healed culprit plaques in patients with acute coronary syndromes. J. Am. Coll. Cardiol. 2019, 73, 2253–2263. [Google Scholar] [CrossRef]
- Mann, K.G.; Brummel, K.; Butenas, S. What is all that thrombin for? J. Thromb. Haemost. 2003, 1, 1504–1514. [Google Scholar] [CrossRef]
- Brown, R.A.; Shantsila, E.; Varma, C.; Lip, G.Y. Current understanding of atherogenesis. Am. J. Med. 2017, 130, 268–282. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.P.; Mackman, N. Tissue factor: An essential mediator of hemostasis and trigger of thrombosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 709–725. [Google Scholar] [CrossRef] [PubMed]
- Borissoff, J.I.; Spronk, H.M.; Heeneman, S.; ten Cate, H. Is thrombin a key player in the ‘coagulation-atherogenesis’ maze? Cardiovasc. Res. 2009, 82, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Borissoff, J.I.; Spronk, H.M.; ten Cate, H. The hemostatic system as a modulator of atherosclerosis. N. Engl. J. Med. 2011, 364, 1746–1760. [Google Scholar] [CrossRef] [PubMed]
- Finn, A.V.; Nakano, M.; Narula, J.; Kolodgie, F.D.; Virmani, R. Concept of vulnerable/unstable plaque. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1282–1292. [Google Scholar] [CrossRef]
- Borissoff, J.I.; Otten, J.J.T.; Heeneman, S.; Leenders, P.; van Oerle, R.; Soehnlein, O.; Loubele, S.T.B.G.; Hamulyák, K.; Hackeng, T.M.; Daemen, M.J.A.P.; et al. Genetic and pharmacological modifications of thrombin formation in apolipoprotein e-deficient mice determine atherosclerosis severity and atherothrombosis onset in a neutrophil-dependent manner. PLoS ONE. 2013, 8, e55784. [Google Scholar] [CrossRef]
- Borissoff, J.I.; Heeneman, S.; Kilinç, E.; Kassák, P.; Van Oerle, R.; Winckers, K.; Govers-Riemslag, J.W.; Hamulyák, K.; Hackeng, T.M.; Daemen, M.J.; et al. Early atherosclerosis exhibits an enhanced procoagulant state. Circulation 2010, 122, 821–830. [Google Scholar] [CrossRef]
- Grover, S.P.; Mackman, N. Tissue factor in atherosclerosis and atherothrombosis. Atherosclerosis 2020, 307, 80–86. [Google Scholar] [CrossRef]
- Annex, B.H.; Denning, S.M.; Channon, K.M.; Sketch, M.H., Jr.; Stack, R.S.; Morrissey, J.H.; Peters, K.G. Differential expression of tissue factor protein in directional atherectomy specimens from patients with stable and unstable coronary syndromes. Circulation 1995, 91, 619–622. [Google Scholar] [CrossRef]
- Ragino, Y.I.; Striukova, E.V.; Murashov, I.S.; Polonskaya, Y.V.; Volkov, A.M.; Kurguzov, A.V.; Chernjavskii, A.M.; Kashtanova, E.V. Association of some hemostasis and endothelial dysfunction factors with probability of presence of vulnerable atherosclerotic plaques in patients with coronary atherosclerosis. BMC Res. Notes 2019, 12, 336. [Google Scholar] [CrossRef] [PubMed]
- Tavora, F.; Cresswell, N.; Li, L.; Ripple, M.; Burke, A. Immunolocalisation of fibrin in coronary atherosclerosis: Implications for necrotic core development. Pathology 2010, 42, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Dunn, E.J.; Ariëns, R.A.; de Lange, M.; Snieder, H.; Turney, J.H.; Spector, T.D.; Grant, P.J. Genetics of fibrin clot structure: A twin study. Blood 2004, 103, 1735–1740. [Google Scholar] [CrossRef]
- Gao, X.Y.; Zhou, B.Y.; Zhang, M.Z.; Zhao, X.; Qing, P.; Zhu, C.G.; Wu, N.Q.; Guo, Y.L.; Gao, Y.; Li, X.L.; et al. Association between fibrinogen level and the severity of coronary stenosis in 418 male patients with myocardial infarction younger than 35 years old. Oncotarget 2017, 8, 81361–81368. [Google Scholar] [CrossRef]
- Tatli, E.; Ozcelik, F.; Aktoz, M. Plasma fibrinogen level may predict critical coronary artery stenosis in young adults with myocardial infarction. Cardiol. J. 2009, 16, 317–320. [Google Scholar]
- Fibrinogen Studies Collaboration; Danesh, J.; Lewington, S.; Thompson, S.G.; Lowe, G.D.; Collins, R.; Kostis, J.B.; Wilson, A.C.; Folsom, A.R.; Wu, K.; et al. Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: An individual participant meta-analysis. JAMA 2005, 294, 1799–1809. [Google Scholar]
- Pieters, M.; Ferreira, M.; de Maat, M.P.M.; Ricci, C. Biomarker association with cardiovascular disease and mortality—The role of fibrinogen. A report from the NHANES study. Thromb. Res. 2021, 198, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Ward-Caviness, C.K.; de Vries, P.S.; Wiggins, K.L.; Huffman, J.E.; Yanek, L.R.; Bielak, L.F.; Giulianini, F.; Guo, X.; Kleber, M.E.; Kacprowski, T.; et al. Mendelian randomization evaluation of causal effects of fibrinogen on incident coronary heart disease. PLoS ONE 2019, 14, e0216222. [Google Scholar] [CrossRef]
- Smith, E.B.; Keen, G.A.; Grant, A.; Stirk, C. Fate of fibrinogen in human arterial intima. Arteriosclerosis 1990, 10, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Kleinegris, M.C.; ten Cate, H.; ten Cate-Hoek, A.J. D-dimer as a marker for cardiovascular and arterial thrombotic events in patients with peripheral arterial disease. A systematic review. Thromb. Haemost. 2013, 110, 233–243. [Google Scholar] [PubMed]
- Kohler, H.P.; Grant, P.J. Mechanisms of disease: Plasminogen-activator inhibitor type 1 and coronary artery disease. N. Engl. J. Med. 2000, 342, 1792–1801. [Google Scholar]
- Undas, A.; Casini, A. Congenital structural and functional fibrinogen disorders: A primer for internists. Pol. Arch. Intern. Med. 2019, 129, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Kryczka, K.E.; Płoski, R.; Księżycka, E.; Kruk, M.; Kostrzewa, G.; Kowalik, I.; Demkow, M.; Lubiszewska, B. The association between the insertion/deletion polymorphism of the angiotensin-converting enzyme gene and the plasma fibrinogen level in women and men with premature coronary artery atherosclerosis. Pol. Arch. Intern. Med. 2020, 130, 748–756. [Google Scholar] [CrossRef]
- Treliński, J.; Witkowski, M.; Chojnowski, K.; Neerman-Arbez, M.; Wypasek, E.; Undas, A. Fibrinogen Łódź: A new cause of dysfibrinogenemia associated with recurrent thromboembolic arterial events. Pol. Arch. Intern. Med. 2019, 129, 934–935. [Google Scholar] [CrossRef]
- Byrnes, J.R.; Duval, C.; Wang, Y.; Hansen, C.E.; Ahn, B.; Mooberry, M.J.; Clark, M.A.; Johnsen, J.M.; Lord, S.T.; Lam, W.A.; et al. Factor XIIIa-dependent retention of red blood cells in clots is mediated by fibrin α-chain crosslinking. Blood 2015, 126, 1940–1948. [Google Scholar] [CrossRef]
- Cines, D.B.; Lebedeva, T.; Nagaswami, C.; Hayes, V.; Massefski, W.; Litvinov, R.I.; Rauova, L.; Lowery, T.J.; Weisel, J.W. Clot contraction: Compression of erythrocytes into tightly packed polyhedra and redistribution of platelets and fibrin. Blood 2014, 123, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, M.; Ząbczyk, M.; Undas, A. Coronary thrombus composition: Links with inflammation, platelet and endothelial markers. Atherosclerosis 2014, 237, 555–561. [Google Scholar] [CrossRef]
- Yunoki, K.; Naruko, T.; Sugioka, K.; Inaba, M.; Iwasa, Y.; Komatsu, R.; Itoh, A.; Haze, K.; Inoue, T.; Yoshiyama, M.; et al. Erythrocyte-rich thrombus aspirated from patients with ST-elevation myocardial infarction: Association with oxidative stress and its impact on myocardial reperfusion. Eur. Heart. J. 2012, 33, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Zalewski, J.; Bogaert, J.; Sadowski, M.; Woznicka, O.; Doulaptsis, K.; Ntoumpanaki, M.; Ząbczyk, M.; Nessler, J.; Undas, A. Plasma fibrin clot phenotype independently affects intracoronary thrombus ultrastructure in patients with acute myocardial infarction. Thromb. Haemost. 2015, 113, 1258–1269. [Google Scholar]
- Carr, M.E.; Shen, L.L.; Hermans, J. Mass-length ratio of fibrin fibers from gel permeation and light scattering. Biopolymers 1977, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Blombäck, B.; Okada, M. Fibrin gel structure and clotting time. Thromb. Res. 1982, 25, 51–70. [Google Scholar] [CrossRef]
- Mills, J.D.; Ariëns, R.A.; Mansfield, M.W.; Grant, P.J. Altered fibrin clot structure in the healthy relatives of patients with premature coronary artery disease. Circulation 2002, 106, 1938–1942. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.M.; Cymbalista, C.M.; Spector, T.D.; Grant, P.J.; EuroCLOT Investigators. Heritability of clot formation, morphology, and lysis: The EuroCLOT study. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2783–2789. [Google Scholar] [CrossRef]
- Collet, J.P.; Park, D.; Lesty, C.; Soria, J.; Soria, C.; Montalescot, G.; Weisel, J.W. Influence of fibrin network conformation and fibrin fiber diameter on fibrinolysis speed: Dynamic and structural approaches by confocal microscopy. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1354–1361. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.A.; Jacobson, L.J.; Miller, B.I.; Hathaway, W.E.; Manco-Johnson, M.J. A new euglobulin clot lysis assay for global fibrinolysis. Thromb. Res. 2003, 112, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Pieters, M.; Philippou, H.; Undas, A.; de Lange, Z.; Rijken, D.C.; Mutch, N.J.; Subcommittee on Factor XIII and Fibrinogen, and the Subcommittee on Fibrinolysis. An international study on the feasibility of a standardized combined plasma clot turbidity and lysis assay: Communication from the SSC of the ISTH. J. Thromb. Haemost. 2018, 16, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, M.E.; Doggen, C.J.; de Groot, P.G.; Rosendaal, F.R.; Lisman, T. Reduced plasma fibrinolytic capacity as a potential risk factor for a first myocardial infarction in young men. Br. J. Haematol. 2009, 145, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Siudut, J.; Iwaniec, T.; Plens, K.; Pieters, M.; Undas, A. Determinants of plasma fibrin clot lysis measured using three different assays in healthy subjects. Thromb. Res. 2021, 197, 1–7. [Google Scholar] [CrossRef]
- Swanepoel, A.C.; De Lange, Z.; Cockeran, M.; Pieters, M. Lifestyle influences changes in fibrin clot properties over a 10-year period on a population level. Thromb. Haemost. 2021. [Google Scholar] [CrossRef]
- de Lange, Z.; Pieters, M.; Jerling, J.C.; Kruger, A.; Rijken, D.C. Plasma clot lysis time and its association with cardiovascular risk factors in black Africans. PLoS ONE 2012, 7, e48881. [Google Scholar] [CrossRef] [PubMed]
- Eksteen, P.; Pieters, M.; de Lange, Z.; Kruger, H.S. The association of clot lysis time with total obesity is partly independent from the association of PAI-1 with central obesity in African adults. Thromb. Res. 2015, 136, 415–421. [Google Scholar] [CrossRef]
- Barua, R.S.; Sy, F.; Srikanth, S.; Huang, G.; Javed, U.; Buhari, C.; Margosan, D.; Ambrose, J.A. Effects of cigarette smoke exposure on clot dynamics and fibrin structure: An ex vivo investigation. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Undas, A.; Topór-Madry, R.; Tracz, W.; Pasowicz, M. Effect of cigarette smoking on plasma fibrin clot permeability and susceptibility to lysis. Thromb. Haemost. 2009, 102, 1289–1291. [Google Scholar]
- Pieters, M.; Guthold, M.; Nunes, C.M.; de Lange, Z. Interpretation and validation of maximum absorbance data obtained from turbidimetry analysis of plasma clots. Thromb. Haemost. 2020, 120, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Dunn, E.J.; Ariëns, R.A.; Grant, P.J. The influence of type 2 diabetes on fibrin structure and function. Diabetologia 2005, 48, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Pieters, M.; Covic, N.; van der Westhuizen, F.H.; Nagaswami, C.; Baras, Y.; Toit Loots, D.; Jerling, J.C.; Elgar, D.; Edmondson, K.S.; van Zyl, D.G.; et al. Glycaemic control improves fibrin network characteristics in type 2 diabetes—A purified fibrinogen model. Thromb. Haemost. 2008, 99, 691–700. [Google Scholar]
- Rajzer, M.; Wojciechowska, W.; Kawecka-Jaszcz, K.; Undas, A. Plasma fibrin clot properties in arterial hypertension and their modification by antihypertensive medication. Thromb. Res. 2012, 130, 99–103. [Google Scholar] [CrossRef]
- Ząbczyk, M.; Hońdo, Ł.; Krzek, M.; Undas, A. High-density cholesterol and apolipoprotein AI as modifiers of plasma fibrin clot properties in apparently healthy individuals. Blood Coagul. Fibrinolysis 2013, 24, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef]
- Undas, A.; Szułdrzynski, K.; Stepien, E.; Zalewski, J.; Godlewski, J.; Tracz, W.; Pasowicz, M.; Zmudka, K. Reduced clot permeability and susceptibility to lysis in patients with acute coronary syndrome: Effects of inflammation and oxidative stress. Atherosclerosis 2008, 196, 551–557. [Google Scholar] [CrossRef]
- Leander, K.; Blombäck, M.; Wallén, H.; He, S. Impaired fibrinolytic capacity and increased fibrin formation associate with myocardial infarction. Thromb. Haemost. 2012, 107, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Gajos, G.; Siniarski, A.; Natorska, J.; Ząbczyk, M.; Siudut, J.; Malinowski, K.P.; Gołębiowska-Wiatrak, R.; Rostoff, P.; Undas, A. Polyhedrocytes in blood clots of type 2 diabetic patients with high cardiovascular risk: Association with glycemia, oxidative stress and platelet activation. Cardiovasc. Diabetol. 2018, 17, 146. [Google Scholar] [CrossRef]
- Ząbczyk, M.; Natorska, J.; Zalewski, J.; Undas, A. Fibrin biofilm can be detected on intracoronary thrombi aspirated from patients with acute myocardial infarction. Cardiovasc. Res. 2019, 115, 1026–1028. [Google Scholar] [CrossRef]
- Macrae, F.L.; Duval, C.; Papareddy, P.; Baker, S.R.; Yuldasheva, N.; Kearney, K.J.; McPherson, H.R.; Asquith, N.; Konings, J.; Casini, A.; et al. A fibrin biofilm covers blood clots and protects from microbial invasion. J. Clin. Investig. 2018, 128, 3356–3368. [Google Scholar] [CrossRef]
- Ząbczyk, M.; Stachowicz, A.; Natorska, J.; Olszanecki, R.; Wiśniewski, J.R.; Undas, A. Plasma fibrin clot proteomics in healthy subjects: Relation to clot permeability and lysis time. J. Proteom. 2019, 208, 103487. [Google Scholar] [CrossRef]
- Suski, M.; Siudut, J.; Ząbczyk, M.; Korbut, R.; Olszanecki, R.; Undas, A. Shotgun analysis of plasma fibrin clot-bound proteins in patients with acute myocardial infarction. Thromb. Res. 2015, 135, 754–759. [Google Scholar] [CrossRef]
- Sumaya, W.; Wallentin, L.; James, S.K.; Siegbahn, A.; Gabrysch, K.; Bertilsson, M.; Himmelmann, A.; Ajjan, R.A.; Storey, R.F. Fibrin clot properties independently predict adverse clinical outcome following acute coronary syndrome: A PLATO substudy. Eur. Heart J. 2018, 39, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Undas, A. Fibrin clot properties and their modulation in thrombotic disorders. Thromb. Haemost. 2014, 112, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.; Gram, J.B.; Sidelmann, J.J.; Dey, D.; Kusk, M.W.; Nørgaard, B.L.; Sand, N.P.R. Sex difference in fibrin clot lysability: Association with coronary plaque composition. Thromb. Res. 2019, 174, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Greilich, P.E.; Carr, M.E.; Zekert, S.L.; Dent, R.M. Quantitative assessment of platelet function and clot structure in patients with severe coronary artery disease. Am. J. Med. Sci. 1994, 307, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.P.; Allali, Y.; Lesty, C.; Tanguy, M.L.; Silvain, J.; Ankri, A.; Blanchet, B.; Dumaine, R.; Gianetti, J.; Payot, L.; et al. Altered fibrin architecture is associated with hypofibrinolysis and premature coronary atherothrombosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2567–2573. [Google Scholar] [CrossRef]
- Undas, A.; Stepien, E.; Tracz, W.; Szczeklik, A. Lipoprotein(a) as a modifier of fibrin clot permeability and susceptibility to lysis. J. Thromb. Haemost. 2006, 4, 973–975. [Google Scholar] [CrossRef]
- Undas, A.; Plicner, D.; Stepień, E.; Drwiła, R.; Sadowski, J. Altered fibrin clot structure in patients with advanced coronary artery disease: A role of C-reactive protein, lipoprotein(a) and homocysteine. J. Thromb. Haemost. 2007, 5, 1988–1990. [Google Scholar] [CrossRef]
- Neergaard-Petersen, S.; Hvas, A.M.; Kristensen, S.D.; Grove, E.L.; Larsen, S.B.; Phoenix, F.; Kurdee, Z.; Grant, P.J.; Ajjan, R.A. The influence of type 2 diabetes on fibrin clot properties in patients with coronary artery disease. Thromb. Haemost. 2014, 112, 1142–1150. [Google Scholar] [CrossRef]
- Winther-Larsen, A.; Christiansen, M.K.; Larsen, S.B.; Nyegaard, M.; Neergaard-Petersen, S.; Ajjan, R.A.; Würtz, M.; Grove, E.L.; Jensen, H.K.; Kristensen, S.D.; et al. The ABO locus is associated with increased fibrin network formation in patients with stable coronary artery disease. Thromb. Haemost. 2020, 120, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Neergaard-Petersen, S.; Larsen, S.B.; Grove, E.L.; Kristensen, S.D.; Ajjan, R.A.; Hvas, A.M. Imbalance between fibrin clot formation and fibrinolysis predicts cardiovascular events in patients with stable coronary artery disease. Thromb. Haemost. 2020, 120, 75–82. [Google Scholar] [CrossRef]
- Bhasin, N.; Parry, D.J.; Scott, D.J.; Ariëns, R.A.; Grant, P.J.; West, R.M. Regarding “Altered fibrin clot structure and function in individuals with intermittent claudication”. J. Vasc. Surg. 2009, 49, 1088–1089. [Google Scholar] [CrossRef] [PubMed]
- Bhasin, N.; Ariëns, R.A.; West, R.M.; Parry, D.J.; Grant, P.J.; Scott, D.J. Altered fibrin clot structure and function in the healthy first-degree relatives of subjects with intermittent claudication. J. Vasc. Surg. 2008, 48, 1497–1503. [Google Scholar] [CrossRef][Green Version]
- Olinic, D.M.; Stanek, A.; Tătaru, D.A.; Homorodean, C.; Olinic, M. Acute limb ischemia: An update on diagnosis and management. J. Clin. Med. 2019, 8, 1215. [Google Scholar] [CrossRef]
- Karpińska, I.A.; Nowakowski, T.; Wypasek, E.; Plens, K.; Undas, A. A prothrombotic state and denser clot formation in patients following acute limb ischemia of unknown cause. Thromb. Res. 2020, 187, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Okraska-Bylica, A.; Wilkosz, T.; Słowik, L.; Bazanek, M.; Konieczyńska, M.; Undas, A. Altered fibrin clot properties in patients with premature peripheral artery disease. Pol. Arch. Med. Wewn. 2012, 122, 608–615. [Google Scholar] [CrossRef]
- Nowakowski, T.; Malinowski, K.P.; Niżankowski, R.; Iwaniec, T.; Undas, A. Restenosis is associated with prothrombotic plasma fibrin clot characteristics in endovascularly treated patients with critical limb ischemia. J. Thromb. Thrombolysis 2019, 47, 540–549. [Google Scholar] [CrossRef]
- Scott, D.J.; Prasad, P.; Philippou, H.; Rashid, S.T.; Sohrabi, S.; Whalley, D.; Kordowicz, A.; Tang, Q.; West, R.M.; Johnson, A.; et al. Clot architecture is altered in abdominal aortic aneurysms and correlates with aneurysm size. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 3004–3010. [Google Scholar] [CrossRef] [PubMed]
- Sundermann, A.C.; Saum, K.; Conrad, K.A.; Russell, H.M.; Edwards, T.L.; Mani, K.; Björck, M.; Wanhainen, A.; Owens, A.P. Prognostic value of D-dimer and markers of coagulation for stratification of abdominal aortic aneurysm growth. Blood Adv. 2018, 2, 3088–3096. [Google Scholar] [CrossRef] [PubMed]
- Undas, A.; Brummel-Ziedins, K.E.; Mann, K.G. Anticoagulant effects of statins and their clinical implications. Thromb. Haemost. 2014, 111, 392–400. [Google Scholar]
- Undas, A.; Celinska-Löwenhoff, M.; Löwenhoff, T.; Szczeklik, A. Statins, fenofibrate, and quinapril increase clot permeability and enhance fibrinolysis in patients with coronary artery disease. J. Thromb. Haemost. 2006, 4, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Undas, A.; Topór-Madry, R.; Tracz, W. Simvastatin increases clot permeability and susceptibility to lysis in patients with LDL cholesterol below 3.4 mmol/l. Pol. Arch. Med. Wewn. 2009, 119, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.A.; Mellis, S.; Yancopoulos, G.D.; Stahl, N.; Logan, D.; Smith, W.B.; Lisbon, E.; Gutierrez, M.; Webb, C.; Wu, R.; et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N. Engl. J. Med. 2012, 366, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Hovingh, G.K.; Kastelein, J.J.; van Deventer, S.J.; Round, P.; Ford, J.; Saleheen, D.; Rader, D.J.; Brewer, H.B.; Barter, P.J. Cholesterol ester transfer protein inhibition by TA-8995 in patients with mild dyslipidaemia (TULIP): A randomised, double-blind, placebo-controlled phase 2 trial. Lancet 2015, 386, 452–460. [Google Scholar] [CrossRef]
- Tsimikas, S.; Viney, N.J.; Hughes, S.G.; Singleton, W.; Graham, M.J.; Baker, B.F.; Burkey, J.L.; Yang, Q.; Marcovina, S.M.; Geary, R.S.; et al. Antisense therapy targeting apolipoprotein(a): A randomised, double-blind, placebo-controlled phase 1 study. Lancet 2015, 386, 1472–1483. [Google Scholar] [CrossRef]
- Viney, N.J.; van Capelleveen, J.C.; Geary, R.S.; Xia, S.; Tami, J.A.; Yu, R.Z.; Marcovina, S.M.; Hughes, S.G.; Graham, M.J.; Crooke, R.M.; et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): Two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016, 388, 2239–2253. [Google Scholar] [CrossRef]
- Williams, S.; Fatah, K.; Ivert, T.; Blombäck, M. The effect of acetyl salicylic acid on fibrin gel lysis by tissue plasminogen activator. Blood Coagul. Fibrinolysis 1995, 6, 718–725. [Google Scholar] [CrossRef]
- Ajjan, R.A.; Standeven, K.F.; Khanbhai, M.; Phoenix, F.; Gersh, K.C.; Weisel, J.W.; Kearney, M.T.; Ariëns, R.A.; Grant, P.J. Effects of aspirin on clot structure and fibrinolysis using a novel in vitro cellular system. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 712–717. [Google Scholar] [CrossRef]
- Svensson, J.; Bergman, A.C.; Adamson, U.; Blombäck, M.; Wallén, H.; Jörneskog, G. Acetylation and glycation of fibrinogen in vitro occur at specific lysine residues in a concentration dependent manner: A mass spectrometric and isotope labeling study. Biochem. Biophys. Res. Commun. 2012, 421, 335–342. [Google Scholar] [CrossRef]
- Williams, S.; Fatah, K.; Hjemdahl, P.; Blombäck, M. Better increase in fibrin gel porosity by low dose than intermediate dose acetylsalicylic acid. Eur. Heart. J. 1998, 19, 1666–1672. [Google Scholar] [CrossRef]
- He, S.; Bark, N.; Wang, H.; Svensson, J.; Blombäck, M. Effects of acetylsalicylic acid on increase of fibrin network porosity and the consequent upregulation of fibrinolysis. J. Cardiovasc. Pharmacol. 2009, 53, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Fatah, K.; Beving, H.; Albåge, A.; Ivert, T.; Blombäck, M. Acetylsalicylic acid may protect the patient by increasing fibrin gel porosity. Is withdrawing of treatment harmful to the patient? Eur. Heart J. 1996, 17, 1362–1366. [Google Scholar] [CrossRef]
- Scott, E.M.; Ariëns, R.A.S.; Grant, P.J. Genetic and environmental determinants of fibrin structure and function. Relevance to clinical disease. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1558–1566. [Google Scholar] [CrossRef]
- Franchini, M.; Mannucci, P.M. Direct oral anticoagulants and venous thromboembolism. Eur. Respir. Rev. 2016, 25, 295–302. [Google Scholar] [CrossRef]
- Gauer, J.S.; Riva, N.; Page, E.M.; Philippou, H.; Makris, M.; Gatt, A.; Ariëns, R.A.S. Effect of anticoagulants on fibrin clot structure: A comparison between vitamin K antagonists and factor Xa inhibitors. Res. Pract. Thromb. Haemost. 2020, 4, 1269–1281. [Google Scholar] [CrossRef] [PubMed]
- Varin, R.; Mirshahi, S.; Mirshahi, P.; Klein, C.; Jamshedov, J.; Chidiac, J.; Perzborn, E.; Mirshahi, M.; Soria, C.; Soria, J. Whole blood clots are more resistant to lysis than plasma clots—Greater efficacy of rivaroxaban. Thromb. Res. 2013, 131, e100–e109. [Google Scholar] [CrossRef] [PubMed]
- Janion-Sadowska, A.; Natorska, J.; Siudut, J.; Ząbczyk, M.; Stanisz, A.; Undas, A. Plasma fibrin clot properties in the G20210A prothrombin mutation carriers following venous thromboembolism: The effect of rivaroxaban. Thromb. Haemost. 2017, 117, 1739–1749. [Google Scholar] [CrossRef]
- Connolly, S.J.; Eikelboom, J.W.; Bosch, J.; Dagenais, G.; Dyal, L.; Lanas, F.; Metsarinne, K.; O’Donnell, M.; Dans, A.L.; Ha, J.W.; et al. Rivaroxaban with or without aspirin in patients with stable coronary artery disease: An international, randomised, double-blind, placebo-controlled trial. Lancet 2018, 391, 205–218. [Google Scholar] [CrossRef]
- Desperak, P.; Hudzik, B.; Gąsior, M. Assessment of patients with coronary artery disease who may benefit from the use of rivaroxaban in the real world: Implementation of the COMPASS trial criteria in the TERCET registry population. Pol. Arch. Intern. Med. 2019, 129, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Posthuma, J.J.; Posma, J.J.N.; van Oerle, R.; Leenders, P.; van Gorp, R.H.; Jaminon, A.M.G.; Mackman, N.; Heitmeier, S.; Schurgers, L.J.; Ten Cate, H.; et al. Targeting Coagulation factor Xa promotes regression of advanced atherosclerosis in apolipoprotein-E deficient mice. Sci. Rep. 2019, 9, 3909. [Google Scholar] [CrossRef] [PubMed]
- Pieters, M.; Undas, A.; Marchi, R.; De Maat, M.P.; Weisel, J.; Ariëns, R.A.; Factor XIII and Fibrinogen Subcommittee of the Scientific and Standardisation Committee of the International Society for Thrombosis and Haemostasis. An international study on the standardization of fibrin clot permeability measurement: Methodological considerations and implications for healthy control values. J. Thromb. Haemost. 2012, 10, 2179–2181. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cardiovascular Risk Factor | Study Design | No. of Subjects | Measure | Reference |
|---|---|---|---|---|
| Age | Case-control | 642 controls (and 421 MI patients) | No clear effect on CLT in controls | [44] |
| Cohort study | 80 healthy controls | ↑ CLT and Lys50 with increasing age | [45] | |
| Cohort study | 2010 healthy controls | ↑ clot turbidity and CLT with increasing age | [46] | |
| Cross-sectional study | 2000 healthy controls | No clear effect on CLT | [47] | |
| Body-mass index (BMI) | Cohort study | 1288 healthy subjects | BMI positively associated with CLT in men and women | [48] |
| Family history of coronary artery disease | Case-control | 100 healthy male relatives of patients with premature coronary artery disease and 100 healthy controls | ↓ Ks and ↑ clot turbidity in relatives of patients | [39] |
| Current smoking | Case-control | 642 controls (and 421 MI patients) | No clear effect on CLT | [44] |
| Cross-sectional study | 2000 healthy controls | No clear effect on CLT | [47] | |
| Case-control | 34 healthy male smokers and 34 nonsmokers | ↑ clot strength, ↑ clot turbidity, ↓ fibrin fiber diameter in smokers | [49] | |
| Case-control | 44 male cigarette smokers and 44 nonsmokers | ↓ Ks and ↑ clot lysis time | [50] | |
| Cohort study | 30 healthy subjects | No clear effect on CLT | [51] | |
| Lipid profile | Cohort study | 30 healthy subjects | Low-density lipoprotein cholesterol level positively associated with CLT | [51] |
| Diabetes | Case-control | 642 controls (and 421 MI patients) | No clear effect on CLT | [44] |
| Case-control | 150 patients with type 2 diabetes and 50 controls | ↓ Ks and ↑ clot turbidity associated with glycated hemoglobin levels | [52] | |
| Interventional study | 20 type 2 diabetes subjects | ↑ Ks after achievement of glycemic control; Ks associated with glycated hemoglobin levels | [53] | |
| Arterial hypertension | Cohort study | 61 patients with essential arterial hypertension | ↑ Ks, ↓ clot lysis time, ↓ clot resistance to lysis at 6 months of antihypertensive treatment | [54] |
| Author (Year of Publication) | Study Type | Sample Size, Condition | Main Findings |
|---|---|---|---|
| Sadowski et al. (2014) [34] | Cohort study | 40 acute MI patients | Plasma levels of platelet activation markers correlated with thrombus fibrin content |
| Zalewski et al. (2015) [36] | Cohort study | 80 acute MI patients | Low Ks was independently associated with high fibrin content within the intracoronary thrombi |
| Sumaya W et al. (2018) [64] | Cohort study | 4354 acute coronary syndrome patients | Prolonged lysis time was associated with cardiovascular death/MI |
| Ramanathan R et al. (2018) [66] | Cross-sectional study | 138 individuals without known cardiovascular disease | Women with coronary plaques had reduced fibrin clot lysability compared to women or men without coronary plaques |
| Neergaard-Petersen S et al. (2014) [71] | Cohort study | 581 CAD patients, including 148 subjects with type 2 diabetes | Type 2 diabetes in CAD patients was associated with prothrombotic fibrin clot compared to non-diabetic CAD patients |
| Winther-Larsen A et al. (2020) [72] | Cohort study | 773 patients with stable CAD | The ABO risk allele (rs495828) was associated with a more compact fibrin network in stable CAD patients |
| Neergaard-Petersen S et al. (2020) [73] | Cohort study | 786 patients with stable CAD | Increased area under the curve of clot formation and lysis predicted cardiovascular events |
| Karpińska I et al. (2020) [77] | Case-control study | 43 patients with a history of acute limb ischemia, 43 patients with cryptogenic stroke, 43 controls | Increased clot density and hypofibrinolysis characterized patients with acute limb ischemia compared to controls |
| Nowakowski et al. (2019) [79] | Case-control study | 85 patients with critical limb ischemia and restenosis and 47 PAD patients | Restenosis compared to PAD was associated with reduced Ks and prolonged CLT |
| Scott DJ et al. (2011) [80] | Case-control study | 42 patients with large AAA, 40 patients with small AAA, and 49 controls | Patients with AAA compared to controls formed denser plasma clots, which were more resistant to lysis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ząbczyk, M.; Natorska, J.; Undas, A. Fibrin Clot Properties in Atherosclerotic Vascular Disease: From Pathophysiology to Clinical Outcomes. J. Clin. Med. 2021, 10, 2999. https://doi.org/10.3390/jcm10132999
Ząbczyk M, Natorska J, Undas A. Fibrin Clot Properties in Atherosclerotic Vascular Disease: From Pathophysiology to Clinical Outcomes. Journal of Clinical Medicine. 2021; 10(13):2999. https://doi.org/10.3390/jcm10132999
Chicago/Turabian StyleZąbczyk, Michał, Joanna Natorska, and Anetta Undas. 2021. "Fibrin Clot Properties in Atherosclerotic Vascular Disease: From Pathophysiology to Clinical Outcomes" Journal of Clinical Medicine 10, no. 13: 2999. https://doi.org/10.3390/jcm10132999
APA StyleZąbczyk, M., Natorska, J., & Undas, A. (2021). Fibrin Clot Properties in Atherosclerotic Vascular Disease: From Pathophysiology to Clinical Outcomes. Journal of Clinical Medicine, 10(13), 2999. https://doi.org/10.3390/jcm10132999