
MDSC in Mice and Men: Mechanisms of Immunosuppression in Cancer

Abstract
:1. Introduction
2. Identification and Isolation
2.1. MDSCs in Mice
2.2. MDSCs in Humans
2.3. Common Challenges in Handling Murine and Human MDSCs
3. Immunosuppressive Mechanisms of MDSCs
3.1. Amino Acid Deprivation
3.2. Arginine Metabolism
3.3. Cysteine Metabolism
3.4. Tryptophan Metabolism
3.5. Generation of Oxidative Stress
3.6. MDSCs and the Accumulation of Regulatory T-Cells
3.7. Expression of Immune Checkpoint Receptors
4. Assessing Immunosuppression In Vitro and In Vivo
4.1. Immunosuppression Assays in Mice
4.2. Immunosuppression Assays in Humans
5. Roadmap to Future
5.1. How to Identify MDSCs?
5.2. MDSCs: State of Activation or Distinct Populations?
5.3. Relevance of MDSCs in the Clinic—Potential Therapeutic Targets?
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maman, S.; Witz, I.P. A History of Exploring Cancer in Context. Nat. Rev. Cancer 2018, 18, 359–376. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-Derived Suppressor Cells in the Era of Increasing Myeloid Cell Diversity. Nat. Rev. Immunol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Velders, M.P.; Sotomayor, E.M.; Kast, W.M. Mechanism of Immune Dysfunction in Cancer Mediated by Immature Gr-1 + Myeloid Cells. J. Immunol. 2001, 166, 5398–5406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almand, B.; Clark, J.I.; Nikitina, E.; van Beynen, J.; English, N.R.; Knight, S.C.; Carbone, D.P.; Gabrilovich, D.I. Increased Production of Immature Myeloid Cells in Cancer Patients: A Mechanism of Immunosuppression in Cancer. J. Immunol. 2001, 166, 678–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aarts, C.E.M.; Kuijpers, T.W. Neutrophils as Myeloid-Derived Suppressor Cells. Eur. J. Clin. Investig. 2018, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for Myeloid-Derived Suppressor Cell Nomenclature and Characterization Standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassetta, L.; Bruderek, K.; Skrzeczynska, J.; Osiecka, O.; Hu, X.; Rundgren, I.M.; Lin, A.; Tellez, T.G.; Taciak, B.; Gotic, M.; et al. Differential Expansion of Circulating Human MDSC Subsets in Patients with Cancer, Infection and Inflammation. J. Immunother. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.; Bruderek, K.; Kaspar, C.; Höing, B.; Kanaan, O.; Dominas, N.; Hussain, T.; Droege, F.; Eyth, C.; Hadaschik, B.; et al. Clinical Relevance and Suppressive Capacity of Human Myeloid-Derived Suppressor Cell Subsets. Clin. Cancer Res. 2018, 24, 4834–4844. [Google Scholar] [CrossRef] [Green Version]
- Sade-Feldman, M.; Kanterman, J.; Klieger, Y.; Ish-Shalom, E.; Olga, M.; Saragovi, A.; Shtainberg, H.; Lotem, M.; Baniyash, M. Clinical Significance of Circulating CD33+ CD11bHLA-DR Myeloid Cells in Patients with Stage IV Melanoma Treated with Ipilimumab. Clin. Cancer Res. 2016, 22, 5661–5672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.F.; Song, S.Y.; Wang, T.J.; Ji, W.J.; Li, S.W.; Liu, N.; Yan, C.X. Prognostic Role of Pretreatment Circulating MDSCs in Patients with Solid Malignancies: A Meta-Analysis of 40 Studies. Oncoimmunology 2018, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, M.K.; Zhu, L.; Harris-White, M.; Kar, U.; Huang, M.; Johnson, M.F.; Lee, J.M.; Elashoff, D.; Strieter, R.; Dubinett, S.; et al. Myeloid Suppressor Cell Depletion Augments Antitumor Activity in Lung Cancer. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Apodaca, M.C.; Wright, A.E.; Riggins, A.M.; Harris, W.P.; Yeung, R.S.; Yu, L.; Morishima, C. Characterization of a Whole Blood Assay for Quantifying Myeloid-Derived Suppressor Cells. J. Immunother. Cancer 2019, 7, 1–11. [Google Scholar] [CrossRef]
- Liu, C.; Yu, S.; Kappes, J.; Wang, J.; Grizzle, W.E.; Zinn, K.R.; Zhang, H.G. Expansion of Spleen Myeloid Suppressor Cells Represses NK Cell Cytotoxicity in Tumor-Bearing Host. Blood 2007, 109, 4336–4342. [Google Scholar] [CrossRef]
- Knaul, J.K.; Jörg, S.; Oberbeck-Mueller, D.; Heinemann, E.; Scheuermann, L.; Brinkmann, V.; Mollenkopf, H.J.; Yeremeev, V.; Kaufmann, S.H.E.; Dorhoi, A. Lung-Residing Myeloid-Derived Suppressors Display Dual Functionality in Murine Pulmonary Tuberculosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1053–1066. [Google Scholar] [CrossRef]
- Rieber, N.; Singh, A.; Öz, H.; Carevic, M.; Bouzani, M.; Amich, J.; Ost, M.; Ye, Z.; Ballbach, M.; Schäfer, I.; et al. Pathogenic Fungi Regulate Immunity by Inducing Neutrophilic Myeloid-Derived Suppressor Cells. Cell Host Microbe 2015, 17, 507–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Yu, Y.; Haarberg, K.; Fu, J.; Kaosaard, K.; Nagaraj, S.; Anasetti, C.; Gabrilovich, D.; Yu, X.Z. Dynamic Change and Impact of Myeloid-Derived Suppressor Cells in Allogeneic Bone Marrow Transplantation in Mice. Biol. Blood Marrow Transplant. 2013, 19, 692–702. [Google Scholar] [CrossRef] [Green Version]
- Cassetta, L.; Baekkevold, E.S.; Brandau, S.; Bujko, A.; Cassatella, M.A.; Dorhoi, A.; Krieg, C.; Lin, A.; Loré, K.; Marini, O.; et al. Deciphering Myeloid-Derived Suppressor Cells: Isolation and Markers in Humans, Mice and Non-Human Primates. Cancer Immunol. Immunother. 2019, 68, 687–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagiv, J.Y.; Michaeli, J.; Assi, S.; Mishalian, I.; Kisos, H.; Levy, L.; Damti, P.; Lumbroso, D.; Polyansky, L.; Sionov, R.V.; et al. Phenotypic Diversity and Plasticity in Circulating Neutrophil Subpopulations in Cancer. Cell Rep. 2015, 10, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-Expanded Myeloid-Derived Suppressor Cells Induce Anergy of NK Cells through Membrane-Bound TGF-Β1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Obermann, S.; Von Wasielewski, R.; Haile, L.; Manns, M.P.; Korangy, F.; Greten, T.F. Increase in Frequency of Myeloid-Derived Suppressor Cells in Mice with Spontaneous Pancreatic Carcinoma. Immunology 2009, 128, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Alicea-Torres, K.; Sanseviero, E.; Gui, J.; Chen, J.; Veglia, F.; Yu, Q.; Donthireddy, L.; Kossenkov, A.; Lin, C.; Fu, S.; et al. Immune Suppressive Activity of Myeloid-Derived Suppressor Cells in Cancer Requires Inactivation of the Type I Interferon Pathway. Nat. Commun. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, K.; Guilliams, M.; Van Den Bossche, J.; Van Den Bergh, R.; Gysemans, C.; Beschin, A.; De Baetselier, P.; Van Ginderachter, J.A. Identification of Discrete Tumor-Induced Myeloid-Derived Suppressor Cell Subpopulations with Distinct T Cell Suppressive Activity. Blood 2008, 111, 4233–4244. [Google Scholar] [CrossRef] [PubMed]
- Dross, S.E.; Munson, P.V.; Kim, S.E.; Bratt, D.L.; Tunggal, H.C.; Gervassi, A.L.; Fuller, D.H.; Horton, H. Kinetics of Myeloid-Derived Suppressor Cell Frequency and Function during Simian Immunodeficiency Virus Infection, Combination Antiretroviral Therapy, and Treatment Interruption. J. Immunol. 2017, 198, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.-I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.-I.; Collazo, M.; Shalova, I.N.; Biswas, S.K.; Gabrilovich, D.I. Characterization of the Nature of Granulocytic Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. J. Leukoc. Biol. 2012, 91, 167–181. [Google Scholar] [CrossRef] [Green Version]
- Alshetaiwi, H.; Pervolarakis, N.; McIntyre, L.L.; Ma, D.; Nguyen, Q.; Rath, J.A.; Nee, K.; Hernandez, G.; Evans, K.; Torosian, L.; et al. Defining the Emergence of Myeloid-Derived Suppressor Cells in Breast Cancer Using Single-Cell Transcriptomics. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty Acid Transport Protein 2 Reprograms Neutrophils in Cancer. Nature 2019, 569, 73–78. [Google Scholar] [CrossRef]
- Al-Khami, A.A.; Zheng, L.; Del Valle, L.; Hossain, F.; Wyczechowska, D.; Zabaleta, J.; Sanchez, M.D.; Dean, M.J.; Rodriguez, P.C.; Ochoa, A.C. Exogenous Lipid Uptake Induces Metabolic and Functional Reprogramming of Tumor-Associated Myeloid-Derived Suppressor Cells. Oncoimmunology 2017, 6. [Google Scholar] [CrossRef]
- Shaul, M.E.; Fridlender, Z.G. Tumour-Associated Neutrophils in Patients with Cancer. Nat. Rev. Clin. Oncol. 2019, 16, 601–620. [Google Scholar] [CrossRef]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Condamine, T.; Dominguez, G.A.; Youn, J.I.; Kossenkov, A.V.; Mony, S.; Alicea-Torres, K.; Tcyganov, E.; Hashimoto, A.; Nefedova, Y.; Lin, C.; et al. Lectin-Type Oxidized LDL Receptor-1 Distinguishes Population of Human Polymorphonuclear Myeloid-Derived Suppressor Cells in Cancer Patients. Sci. Immunol. 2016, 1, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Wu, D.; Ni, C.; Ye, J.; Chen, W.; Hu, G.; Wang, Z.; Wang, C.; Zhang, Z.; Xia, W.; et al. ΓδT17 Cells Promote the Accumulation and Expansion of Myeloid-Derived Suppressor Cells in Human Colorectal Cancer. Immunity 2014, 40, 785–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasnis, E.; Half, E.E. Intratumoral Myeloid-Derived Suppressor Cells Predict Response to Neoadjuvant Chemoradiotherapy in Locally Advanced Rectal Cancer. Front. Oncol. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Si, Y.; Merz, S.F.; Jansen, P.; Wang, B.; Bruderek, K.; Altenhoff, P.; Mattheis, S.; Lang, S.; Gunzer, M.; Klode, J.; et al. Multidimensional Imaging Provides Evidence for Down-Regulation of T Cell Effector Function by MDSC in Human Cancer Tissue. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]
- Jordan, K.R.; Kapoor, P.; Spongberg, E.; Tobin, R.P.; Gao, D.; Borges, V.F.; Mccarter, M.D. Immunosuppressive Myeloid-Derived Suppressor Cells Are Increased in Splenocytes from Cancer Patients. Cancer Immunol. Immunother. 2017, 66, 503–513. [Google Scholar] [CrossRef] [Green Version]
- Pinton, L.; Solito, S.; Damuzzo, V.; Francescato, S.; Pozzuoli, A.; Berizzi, A.; Mocellin, S.; Rossi, C.R.; Bronte, V.; Mandruzzato, S. Activated T Cells Sustain Myeloid-Derived Suppressor Cell-Mediated Immune Suppression. Oncotarget 2015, 7, 1168–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obermajer, N.; Muthuswamy, R.; Odunsi, K.; Edwards, R.P.; Kalinski, P. PGE 2-Induced CXCL 12 Production and CXCR4 Expression Controls the Accumulation of Human MDSCs in Ovarian Cancer Environment. Cancer Res. 2011, 71, 7463–7470. [Google Scholar] [CrossRef] [Green Version]
- Marigo, I.; Bosio, E.; Solito, S.; Mesa, C.; Fernandez, A.; Dolcetti, L.; Ugel, S.; Sonda, N.; Bicciato, S.; Falisi, E.; et al. Tumor-Induced Tolerance and Immune Suppression Depend on the C/EBPβ Transcription Factor. Immunity 2010, 32, 790–802. [Google Scholar] [CrossRef]
- Solito, S.; Falisi, E.; Diaz-Montero, C.M.; Doni, A.; Pinton, L.; Rosato, A.; Francescato, S.; Basso, G.; Zanovello, P.; Onicescu, G.; et al. A Human Promyelocytic-like Population Is Responsible for the Immune Suppression Mediated by Myeloid-Derived Suppressor Cells. Blood 2011, 118, 2254–2265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obermajer, N.; Kalinski, P. Generation of Myeloid-Derived Suppressor Cells Using Prostaglandin E2. Transplant. Res. 2012, 1, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Ubreva, J.; Català-Moll, F.; Obermajer, N.; Álvarez-Errico, D.; Ramirez, R.N.; Company, C.; Vento-Tormo, R.; Moreno-Bueno, G.; Edwards, R.P.; Mortazavi, A.; et al. Prostaglandin E2 Leads to the Acquisition of DNMT3A-Dependent Tolerogenic Functions in Human Myeloid-Derived Suppressor Cells. Cell Rep. 2017, 21, 154–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumitru, C.A.; Moses, K.; Trellakis, S.; Lang, S.; Brandau, S. Neutrophils and Granulocytic Myeloid-Derived Suppressor Cells: Immunophenotyping, Cell Biology and Clinical Relevance in Human Oncology. Cancer Immunol. Immunother. 2012, 61, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Nan, J.; Xing, Y.; Dong, H.; He, Y.; Cai, J.; Ma, X.; Chen, J.; Cai, X.; Lin, Z.; Wu, X. Endoplasmic Reticulum Stress Induced LOX-1 + CD15 + Polymorphonuclear Myeloid-Derived Suppressor Cells in Hepatocellular Carcinoma. Immunology 2017, 154, 144–155. [Google Scholar] [CrossRef] [Green Version]
- Tavukcuoglu, E. Tumor Immunology Human Splenic Polymorphonuclear Myeloid-Derived Suppressor Cells (PMN-MDSC) Are Strategically Located Immune Regulatory Cells in Cancer. Eur. J. Immunol. 2020, 1–8. [Google Scholar] [CrossRef]
- Bruger, A.M.; Vanhaver, C.; Bruderek, K.; Amodio, G.; Tavukçuoğlu, E.; Esendagli, G.; Gregori, S.; Brandau, S.; van der Bruggen, P. Protocol to Assess the Suppression of T-Cell Proliferation by Human MDSC. Methods Enzymol. 2019. [Google Scholar] [CrossRef]
- Quatromoni, J.G.; Singhal, S.; Bhojnagarwala, P.; Hancock, W.W.; Albelda, S.M.; Eruslanov, E. An Optimized Disaggregation Method for Human Lung Tumors That Preserves the Phenotype and Function of the Immune Cells. J. Leukoc. Biol. 2015, 97, 201–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef] [Green Version]
- Trellakis, S.; Bruderek, K.; Hütte, J.; Elian, M.; Hoffmann, T.K.; Lang, S.; Brandau, S. Granulocytic Myeloid-Derived Suppressor Cells Are Cryosensitive and Their Frequency Does Not Correlate with Serum Concentrations of Colony-Stimulating Factors in Head and Neck Cancer. Innate Immun. 2013, 19, 328–336. [Google Scholar] [CrossRef]
- Lahoz-Beneytez, J.; Elemans, M.; Zhang, Y.; Ahmed, R.; Salam, A.; Block, M.; Niederalt, C.; Asquith, B.; Macallan, D. Human Neutrophil Kinetics: Modeling of Stable Isotope Labeling Data Supports Short Blood Neutrophil Half-Lives. Blood 2016, 127, 3431–3438. [Google Scholar] [CrossRef] [Green Version]
- Bruger, A.M.; Dorhoi, A.; Esendagli, G.; Barczyk-Kahlert, K.; van der Bruggen, P.; Lipoldova, M.; Perecko, T.; Santibanez, J.; Saraiva, M.; van Ginderachter, J.A.; et al. How to Measure the Immunosuppressive Activity of MDSC: Assays, Problems and Potential Solutions. Cancer Immunol. Immunother. 2018, 68, 1–14. [Google Scholar] [CrossRef]
- Crook, K.R.; Jin, M.; Weeks, M.F.; Rampersad, R.R.; Baldi, R.M.; Glekas, A.S.; Shen, Y.; Esserman, D.A.; Little, P.; Schwartz, T.A.; et al. Myeloid-Derived Suppressor Cells Regulate T Cell and B Cell Responses during Autoimmune Disease. J. Leukoc. Biol. 2015, 97, 573–582. [Google Scholar] [CrossRef]
- Jaufmann, J.; Lelis, F.J.N.; Teschner, A.C.; Fromm, K.; Rieber, N.; Hartl, D.; Beer-Hammer, S. Human Monocytic Myeloid-Derived Suppressor Cells Impair B-Cell Phenotype and Function in Vitro. Eur. J. Immunol. 2020, 50, 33–47. [Google Scholar] [CrossRef] [Green Version]
- Goh, C.C.; Roggerson, K.M.; Lee, H.-C.; Golden-Mason, L.; Rosen, H.R.; Hahn, Y.S. Hepatitis C Virus–Induced Myeloid-Derived Suppressor Cells Suppress NK Cell IFN-γ Production by Altering Cellular Metabolism via Arginase-1. J. Immunol. 2016, 196, 2283–2292. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Sarhan, D.; Steven, A.; Seliger, B.; Kiessling, R.; Lundqvist, A. Inhibition of Tumor-Derived Prostaglandin-E2 Blocks the Induction of Myeloid-Derived Suppressor Cells and Recovers Natural Killer Cell Activity. Clin. Cancer Res. 2014, 20, 4096–4106. [Google Scholar] [CrossRef] [Green Version]
- Stiff, A.; Trikha, P.; Mundy-Bosse, B.; McMichael, E.; Mace, T.A.; Benner, B.; Kendra, K.; Campbell, A.; Gautam, S.; Abood, D.; et al. Nitric Oxide Production by Myeloid-Derived Suppressor Cells Plays a Role in Impairing Fc Receptor–Mediated Natural Killer Cell Function. Clin. Cancer Res. 2018, 24, 1891–1904. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I Production in the Tumor Microenvironment by Mature Myeloid Cells Inhibits T-Cell Receptor Expression and Antigen-Specific T-Cell Responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Hernandez, C.P.; Quiceno, D.; Dubinett, S.M.; Zabaleta, J.; Ochoa, J.B.; Gilbert, J.; Ochoa, A.C. Arginase I in Myeloid Suppressor Cells Is Induced by COX-2 in Lung Carcinoma. J. Exp. Med. 2005, 202, 931–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, P.C.; Ochoa, A.C. Arginine Regulation by Myeloid Derived Suppressor Cells and Tolerance in Cancer : Mechanisms and Therapeutic Perspectives. Immunol. Rev. 2008, 222, 180–191. [Google Scholar] [CrossRef]
- Miret, J.J.; Kirschmeier, P.; Koyama, S.; Zhu, M.; Li, Y.Y.; Naito, Y.; Wu, M.; Malladi, V.S.; Huang, W.; Walker, W.; et al. Suppression of Myeloid Cell Arginase Activity Leads to Therapeutic Response in a NSCLC Mouse Model by Activating Anti-Tumor Immunity. J. Immunother. Cancer 2019, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rotondo, R.; Bertolotto, M.; Barisione, G.; Astigiano, S.; Mandruzzato, S.; Ottonello, L.; Dallegri, F.; Bronte, V.; Ferrini, S.; Barbieri, O. Exocytosis of Azurophil and Arginase 1-Containing Granules by Activated Polymorphonuclear Neutrophils Is Required to Inhibit T Lymphocyte Proliferation. J. Leukoc. Biol. 2011, 89, 721–727. [Google Scholar] [CrossRef]
- Zea, A.H.; Rodriguez, P.C.; Atkins, M.B.; Hernandez, C.; Signoretti, S.; Zabaleta, J.; McDermott, D.; Quiceno, D.; Youmans, A.; O’Neill, A.; et al. Arginase-Producing Myeloid Suppressor Cells in Renal Cell Carcinoma Patients: A Mechanism of Tumor Evasion. Cancer Res. 2005, 65, 3044–3048. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Ernstoff, M.S.; Hernandez, C.; Atkins, M.; Zabaleta, J.; Sierra, R.; Ochoa, A.C. Arginase I-Producing Myeloid-Derived Suppressor Cells in Renal Cell Carcinoma Are a Subpopulation of Activated Granulocytes. Cancer Res. 2009, 69, 1553–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czystowska-Kuzmicz, M.; Sosnowska, A.; Nowis, D.; Ramji, K.; Szajnik, M.; Chlebowska-Tuz, J.; Wolinska, E.; Gaj, P.; Grazul, M.; Pilch, Z.; et al. Small Extracellular Vesicles Containing Arginase-1 Suppress T-Cell Responses and Promote Tumor Growth in Ovarian Carcinoma. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-Arginine Availability Regulates T-Lymphocyte Cell-Cycle Progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.Y.; Wang, Y.M.; Wang, C.L.; Feng, P.H.; Ko, H.W.; Liu, Y.H.; Wu, Y.C.; Chu, Y.; Chung, F.T.; Kuo, C.H.; et al. Population Alterations of L-Arginase- and Inducible Nitric Oxide Synthase-Expressed CD11b+/CD14-/CD15+/CD33+ Myeloid-Derived Suppressor Cells and CD8+ T Lymphocytes in Patients with Advanced-Stage Non-Small Cell Lung Cancer. J. Cancer Res. Clin. Oncol. 2010, 136, 35–45. [Google Scholar] [CrossRef]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cells Inhibit T-Cell Activation by Depleting Cystine and Cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-Derived Suppressor Cells Suppress Antitumor Immune Responses through IDO Expression and Correlate with Lymph Node Metastasis in Patients with Breast Cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raber, P.L.; Thevenot, P.; Sierra, R.; Wyczechowska, D.; Halle, D.; Ramirez, M.E.; Ochoa, A.C.; Fletcher, M.; Velasco, C.; Wilk, A.; et al. Subpopulations of Myeloid-Derived Suppressor Cells Impair T Cell Responses through Independent Nitric Oxide-Related Pathways. Int. J. Cancer 2014, 134, 2853–2864. [Google Scholar] [CrossRef] [PubMed]
- Corzo, C.A.; Cotter, M.J.; Cheng, P.; Cheng, F.; Kusmartsev, S.; Sotomayor, E.; Padhya, T.; McCaffrey, T.V.; McCaffrey, J.C.; Gabrilovich, D.I. Mechanism Regulating Reactive Oxygen Species in Tumor-Induced Myeloid-Derived Suppressor Cells. J. Immunol. 2009, 182, 5693–5701. [Google Scholar] [CrossRef]
- Lu, T.; Ramakrishnan, R.; Altiok, S.; Youn, J.-I.; Cheng, P.; Celis, E.; Pisarev, V.; Sherman, S.; Sporn, M.B.; Gabrilovich, D. Tumor-Infiltrating Myeloid Cells Induce Tumor Cell Resistance to Cytotoxic T Cells in Mice. J. Clin. Investig. 2011, 121, 4015–4029. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered Recognition of Antigen Is a Mechanism of CD8+ T Cell Tolerance in Cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortin, C.; Huang, X.; Yang, Y. NK Cell Response to Vaccinia Virus Is Regulated by Myeloid-Derived Suppressor Cells. J. Immunol. 2012, 189, 1843–1849. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+CD115+ Immature Myeloid Suppressor Cells Mediate the Development of Tumor-Induced T Regulatory Cells and T-Cell Anergy in Tumor-Bearing Host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Serafini, P.; Mgebroff, S.; Noonan, K.; Borrello, I. Myeloid-Derived Suppressor Cells Promote Cross-Tolerance in B-Cell Lymphoma by Expanding Regulatory T Cells. Cancer Res. 2008, 68, 5439–5449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, P.Y.; Ma, G.; Weber, K.J.; Ozao-Choy, J.; Wang, G.; Yin, B.; Divino, C.M.; Chen, S.H. Immune Stimulatory Receptor CD40 Is Required for T-Cell Suppression and T Regulatory Cell Activation Mediated by Myeloid-Derived Suppressor Cells in Cancer. Cancer Res. 2010, 70, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Hoechst, B.; Ormandy, L.A.; Ballmaier, M.; Lehner, F.; Krüger, C.; Manns, M.P.; Greten, T.F.; Korangy, F. A New Population of Myeloid-Derived Suppressor Cells in Hepatocellular Carcinoma Patients Induces CD4+CD25+Foxp3+ T Cells. Gastroenterology 2008, 135, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Schlecker, E.; Stojanovic, A.; Eisen, C.; Quack, C.; Falk, C.S.; Umansky, V.; Cerwenka, A. Tumor-Infiltrating Monocytic Myeloid-Derived Suppressor Cells Mediate CCR5-Dependent Recruitment of Regulatory T Cells Favoring Tumor Growth. J. Immunol. 2012, 189, 5602–5611. [Google Scholar] [CrossRef] [Green Version]
- Luan, Y.; Mosheir, E.; Menon, M.C.; Wilson, D.; Woytovich, C.; Ochando, J.; Murphy, B. Monocytic Myeloid-Derived Suppressor Cells Accumulate in Renal Transplant Patients and Mediate CD4(+) Foxp3(+) Treg Expansion. Am. J. Transplant 2013, 13, 3123–3131. [Google Scholar] [CrossRef]
- Lu, C.; Redd, P.S.; Lee, J.R.; Savage, N.; Liu, K. The Expression Profiles and Regulation of PD-L1 in Tumor-Induced Myeloid-Derived Suppressor Cells. Oncoimmunology 2016, 5, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 Is a Novel Direct Target of HIF-1α, and Its Blockade under Hypoxia Enhanced MDSC-Mediated T Cell Activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Yamauchi, Y.; Safi, S.; Blattner, C.; Rathinasamy, A.; Umansky, L.; Juenger, S.; Warth, A.; Eichhorn, M.; Muley, T.; Herth, F.J.F.; et al. Circulating and Tumor Myeloid-Derived Suppressor Cells in Resectable Non-Small Cell Lung Cancer. Am. J. Respir. Crit. Care Med. 2018, 198, 777–787. [Google Scholar] [CrossRef]
- Strauss, L.; Mahmoud, M.A.A.; Weaver, J.D.; Tijaro-Ovalle, N.M.; Christofides, A.; Wang, Q.; Pal, R.; Yuan, M.; Asara, J.; Patsoukis, N.; et al. Targeted Deletion of PD-1 in Myeloid Cells Induces Antitumor Immunity. Sci. Immunol. 2020, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, C.; Sevko, A.; Jiang, H.; Lichtenberger, R.; Reith, M.; Tarnanidis, K.; Holland-Letz, T.; Umansky, L.; Beckhove, P.; Sucker, A.; et al. Myeloid Cells and Related Chronic Inflammatory Factors as Novel Predictive Markers in Melanoma Treatment with Ipilimumab. Clin. Cancer Res. 2015, 21, 5453–5459. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Powis De Tenbossche, C.G.; Cané, S.; Colau, D.; Van Baren, N.; Lurquin, C.; Schmitt-Verhulst, A.M.; Liljeström, P.; Uyttenhove, C.; Van Den Eynde, B.J. Resistance to Cancer Immunotherapy Mediated by Apoptosis of Tumor-Infiltrating Lymphocytes. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinclair, L.V.; Rolf, J.; Emslie, E.; Shi, Y.B.; Taylor, P.M.; Cantrell, D.A. Control of Amino-Acid Transport by Antigen Receptors Coordinates the Metabolic Reprogramming Essential for T Cell Differentiation. Nat. Immunol. 2013, 14, 500–508. [Google Scholar] [CrossRef] [Green Version]
- Siska, P.J.; Rathmell, J.C. T Cell Metabolic Fitness in Antitumor Immunity. Trends Immunol. 2015, 36, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Carr, E.L.; Kelman, A.; Wu, G.S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A.M.; Frauwirth, K.A. Glutamine Uptake and Metabolism Are Coordinately Regulated by ERK/MAPK during T Lymphocyte Activation. J. Immunol. 2010, 185, 1037–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfson, R.L.; Sabatini, D.M. The Dawn of the Age of Amino Acid Sensors for the MTORC1 Pathway. Cell Metab. 2017, 26, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Battu, S.; Minhas, G.; Mishra, A.; Khan, N. Amino Acid Sensing via General Control Nonderepressible-2 Kinase and Immunological Programming. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-Tumor Activity. Cell 2016, 167, 829–842. [Google Scholar] [CrossRef] [Green Version]
- Werner, A.; Amann, E.; Schnitzius, V.; Habermeier, A.; Luckner-Minden, C.; Leuchtner, N.; Rupp, J.; Closs, E.I.; Munder, M. Induced Arginine Transport via Cationic Amino Acid Transporter-1 Is Necessary for Human T-Cell Proliferation. Eur. J. Immunol. 2016, 46, 92–103. [Google Scholar] [CrossRef] [Green Version]
- Vonwirth, V.; Bülbül, Y.; Werner, A.; Echchannaoui, H.; Windschmitt, J.; Habermeier, A.; Ioannidis, S.; Shin, N.; Conradi, R.; Bros, M.; et al. Inhibition of Arginase 1 Liberates Potent T Cell Immunostimulatory Activity of Human Neutrophil Granulocytes. Front. Immunol. 2021, 11, 1–16. [Google Scholar] [CrossRef]
- Carriche, G.M.; Almeida, L.; Stüve, P.; Velasquez, L.; Dhillon-LaBrooy, A.; Roy, U.; Lindenberg, M.; Strowig, T.; Plaza-Sirvent, C.; Schmitz, I.; et al. Regulating T-Cell Differentiation through the Polyamine Spermidine. J. Allergy Clin. Immunol. 2021, 147, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, P.C.; Ochoa, A.C. T Cell Dysfunction in Cancer: Role of Myeloid Cells and Tumor Cells Regulating Amino Acid Availability and Oxidative Stress. Semin. Cancer Biol. 2006, 16, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Dolcetti, L.; Peranzoni, E.; Ugel, S.; Marigo, I.; Gomez, A.F.; Mesa, C.; Geilich, M.; Winkels, G.; Traggiai, E.; Casati, A.; et al. Hierarchy of Immunosuppressive Strength among Myeloid-Derived Suppressor Cell Subsets Is Determined by GM-CSF. Eur. J. Immunol. 2010, 40, 22–35. [Google Scholar] [CrossRef]
- Perrotta, C.; Cervia, D.; Di Renzo, I.; Moscheni, C.; Bassi, M.T.; Campana, L.; Martelli, C.; Catalani, E.; Giovarelli, M.; Zecchini, S.; et al. Nitric Oxide Generated by Tumor-Associated Macrophages Is Responsible for Cancer Resistance to Cisplatin and Correlated with Syntaxin 4 and Acid Sphingomyelinase Inhibition. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Bian, Z.; Abdelaal, A.M.; Shi, L.; Liang, H.; Xiong, L.; Kidder, K.; Venkataramani, M.; Culpepper, C.; Zen, K.; Liu, Y. Arginase-1 Is Neither Constitutively Expressed in nor Required for Myeloid-Derived Suppressor Cell-Mediated Inhibition of T-Cell Proliferation. Eur. J. Immunol. 2018, 48, 1046–1058. [Google Scholar] [CrossRef] [Green Version]
- Munder, M.; Mollinedo, F.; Calafat, J.; Canchado, J.; Gil-Lamaignere, C.; Fuentes, J.M.; Luckner, C.; Doschko, G.; Soler, G.; Eichmann, K.; et al. Arginase I Is Constitutively Expressed in Human Granulocytes and Participates in Fungicidal Activity. Blood 2005, 105, 2549–2556. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The Cystine/Glutamate Antiporter System Xc- in Health and Disease: From Molecular Mechanisms to Novel Therapeutic Opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Zou, W. Amino Acids and Their Transporters in T Cell Immunity and Cancer Therapy. Mol. Cell 2020, 80, 384–395. [Google Scholar] [CrossRef]
- Levring, T.B.; Hansen, A.K.; Nielsen, B.L.; Kongsbak, M.; Von Essen, M.R.; Woetmann, A.; Ødum, N.; Bonefeld, C.M.; Geisler, C. Activated Human CD4 + T Cells Express Transporters for Both Cysteine and Cystine. Sci. Rep. 2012, 2, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Platten, M.; Wick, W.; Van Den Eynde, B.J. Tryptophan Catabolism in Cancer: Beyond IDO and Tryptophan Depletion. Cancer Res. 2012, 72, 5435–5440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An Interaction between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Frumento, G.; Rotondo, R.; Tonetti, M.; Damonte, G.; Benatti, U.; Ferrara, G.B. Tryptophan-Derived Catabolites Are Responsible for Inhibition of T and Natural Killer Cell Proliferation Induced by Indoleamine 2,3-Dioxygenase. J. Exp. Med. 2002, 196, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Della Chiesa, M.; Carlomagno, S.; Frumento, G.; Balsamo, M.; Cantoni, C.; Conte, R.; Moretta, L.; Moretta, A.; Vitale, M. The Tryptophan Catabolite L-Kynurenine Inhibits the Surface Expression of NKp46- and NKG2D-Activating Receptors and Regulates NK-Cell Function. Blood 2006, 108, 4118–4125. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.M.; Nam, G.; Lee, Y.K.; Jung, M.; Chang, H.; Kim, W.; Shon, W.J.; Lim, J.Y.; Kim, J.Y.; Chang, J.; et al. IDO1 Scavenges Reactive Oxygen Species in Myeloid-Derived Suppressor Cells to Prevent Graft-versus-Host Disease. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Belikov, A.V.; Schraven, B.; Simeoni, L. T Cells and Reactive Oxygen Species. J. Biomed. Sci. 2015, 22, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Cheng, X.; Zhang, L.; Lu, X.; Chaudhary, S.; Teng, R.; Frederickson, C.; Champion, M.M.; Zhao, R.; Cheng, L.; et al. Myeloid-Derived Suppressor Cells Inhibit T Cell Activation through Nitrating LCK in Mouse Cancers. Proc. Natl. Acad. Sci. USA 2018, 115, 10094–10099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cemerski, S.; Cantagrel, A.; Van Meerwijk, J.P.M.; Romagnoli, P. Reactive Oxygen Species Differentially Affect T Cell Receptor-Signaling Pathways. J. Biol. Chem. 2002, 277, 19585–19593. [Google Scholar] [CrossRef] [Green Version]
- Cemerski, S.; van Meerwijk, J.P.M.; Romagnoli, P. Oxidative-Stress-Induced T Lymphocyte Hyporesponsiveness Is Caused by Structural Modification Rather than Proteasomal Degradation of Crucial TCR Signaling Molecules. Eur. J. Immunol. 2003, 33, 2178–2185. [Google Scholar] [CrossRef] [PubMed]
- Mougiakakos, D.; Johansson, C.C.; Kiessling, R. Naturally Occurring Regulatory T Cells Show Reduced Sensitivity toward Oxidative Stress-Induced Cell Death. Blood 2009, 113, 3542–3545. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Hanson, M.G.V.; Norell, H.R.; Havelka, A.M.; Kono, K.; Malmberg, K.-J.; Kiessling, R.V.R. Preferential Cell Death of CD8+ Effector Memory (CCR7− CD45RA−) T Cells by Hydrogen Peroxide-Induced Oxidative Stress. J. Immunol. 2005, 174, 6080–6087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Huang, X.; Yang, Y. Myeloid-Derived Suppressor Cells Regulate Natural Killer Cell Response to Adenovirus-Mediated Gene Transfer. J. Virol. 2012, 86, 13689–13696. [Google Scholar] [CrossRef] [Green Version]
- Molon, B.; Ugel, S.; Del Pozzo, F.; Soldani, C.; Zilio, S.; Avella, D.; De Palma, A.; Mauri, P.; Monegal, A.; Rescigno, M.; et al. Chemokine Nitration Prevents Intratumoral Infiltration of Antigen-Specific T Cells. J. Exp. Med. 2011, 208, 1949–1962. [Google Scholar] [CrossRef]
- Aarts, C.E.M.; Hiemstra, I.H.; Béguin, E.P.; Hoogendijk, A.J.; Bouchmal, S.; van Houdt, M.; Tool, A.T.J.; Mul, E.; Jansen, M.H.; Janssen, H.; et al. Activated Neutrophils Exert Myeloid-Derived Suppressor Cell Activity Damaging T Cells beyond Repair. Blood Adv. 2019, 3, 3562–3574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, M.; Fanelli, G.; Albany, C.J.; Giganti, G.; Lombardi, G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular Mechanisms Oftreg-Mediatedt Cell Suppression. Front. Immunol. 2012, 3, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Wang, J.; Ioan-facsinay, A.; Voort, E.I.H.; Van Der Huizinga, T.W.J.; Toes, R.E.M. Transient Expression of FOXP3 in Human Activated Nonregulatory CD4+ T Cells. Eur. J. Immunol. 2007, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Pillai, V.; Ortega, S.B.; Wang, C.K.; Karandikar, N.J. Transient Regulatory T-Cells: A State Attained by All Activated Human T-Cells. Clin. Immunol. 2007, 123, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Polansky, J.K.; Kretschmer, K.; Freyer, J.; Floess, S.; Garbe, A.; Baron, U.; Olek, S.; Hamann, A.; von Boehmer, H.; Huehn, J. DNA Methylation Controls Foxp3 Gene Expression. Eur. J. Immunol. 2008, 38, 1654–1663. [Google Scholar] [CrossRef]
- Li, J.; Wang, L.; Chen, X.; Li, L.; Li, Y.; Ping, Y.; Huang, L.; Yue, D.; Zhang, Z.; Wang, F.; et al. CD39/CD73 Upregulation on Myeloid-Derived Suppressor Cells via TGF-β-MTOR-HIF-1 Signaling in Patients with Non-Small Cell Lung Cancer. Oncoimmunology 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, L.; Li, J.; Fan, Z.; Yang, L.; Zhang, Z.; Zhang, C.; Yue, D.; Qin, G.; Zhang, T.; et al. Metformin-Induced Reduction of CD39 and CD73 Blocks Myeloid-Derived Suppressor Cell Activity in Patients with Ovarian Cancer. Cancer Res. 2018, 78, 1779–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Haskó, G. CD39 and CD73 in Immunity and Inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juneja, V.R.; McGuire, K.A.; Manguso, R.T.; LaFleur, M.W.; Collins, N.; Nicholas Haining, W.; Freeman, G.J.; Sharpe, A.H. PD-L1 on Tumor Cells Is Sufficient for Immune Evasion in Immunogenic Tumors and Inhibits CD8 T Cell Cytotoxicity. J. Exp. Med. 2017, 214, 895–904. [Google Scholar] [CrossRef]
- Johnson, D.B.; Peng, C.; Sosman, J.A. Nivolumab in Melanoma: Latest Evidence and Clinical Potential. Ther. Adv. Med. Oncol. 2015, 7, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.J.; Moore, E.C.; Clavijo, P.E.; Friedman, J.; Cash, H.; Chen, Z.; Silvin, C.; Van Waes, C.; Allen, C. Anti-PD-L1 Efficacy Can Be Enhanced by Inhibition of Myeloid-Derived Suppressor Cells with a Selective Inhibitor of PI3Kd/G. Cancer Res. 2017, 77, 2607–2619. [Google Scholar] [CrossRef] [Green Version]
- Brudecki, L.; Ferguson, D.A.; McCall, C.E.; Gazzar, M.E. Myeloid-Derived Suppressor Cells Evolve during Sepsis and Can Enhance or Attenuate the Systemic Inflammatory Response. Infect. Immun. 2012, 80, 2026–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, M.; Zimara, N.; Wege, A.K.; Ritter, U. Myeloid-Derived Suppressor Cell Functionality and Interaction with Leishmania Major Parasites Differ in C57BL/6 and BALB/c Mice. Eur. J. Immunol. 2014, 44, 3295–3306. [Google Scholar] [CrossRef] [PubMed]
- Su, N.; Yue, Y.; Xiong, S. Monocytic Myeloid-Derived Suppressor Cells from Females, but Not Males, Alleviate CVB3-Induced Myocarditis by Increasing Regulatory and CD4+ IL-10+ T Cells. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Carretero-Iglesia, L.; Bouchet-Delbos, L.; Louvet, C.; Drujont, L.; Segovia, M.; Merieau, E.; Chiffoleau, E.; Josien, R.; Hill, M.; Cuturi, M.C.; et al. Comparative Study of the Immunoregulatory Capacity of in Vitro Generated Tolerogenic Dendritic Cells, Suppressor Macrophages, and Myeloid-Derived Suppressor Cells. Transplantation 2016, 100, 2079–2089. [Google Scholar] [CrossRef]
- Sierra, R.A.; Thevenot, P.; Raber, P.L.; Cui, Y.; Parsons, C.; Ochoa, A.C.; Trillo-Tinoco, J.; Del Valle, L.; Rodriguez, P.C. Rescue of Notch-1 Signaling in Antigen-Specific CD8+ T Cells Overcomes Tumor-Induced T-Cell Suppression and Enhances Immunotherapy in Cancer. Cancer Immunol. Res. 2014, 2, 800–811. [Google Scholar] [CrossRef] [Green Version]
- Clavijo, P.E.; Moore, E.C.; Chen, J.; Davis, R.J.; Friedman, J.; Kim, Y.; Van Waes, C.; Chen, Z.; Allen, C.T. Resistance to CTLA-4 Checkpoint Inhibition Reversed through Selective Elimination of Granulocytic Myeloid Cells. Oncotarget 2017, 8, 55804–55820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moses, K.; Klein, J.C.; Männ, L.; Klingberg, A.; Gunzer, M.; Brandau, S. Survival of Residual Neutrophils and Accelerated Myelopoiesis Limit the Efficacy of Antibody-Mediated Depletion of Ly-6G + Cells in Tumor-Bearing Mice. J. Leukoc. Biol. 2016, 99, 811–823. [Google Scholar] [CrossRef] [Green Version]
- Toor, S.M.; Syed Khaja, A.S.; El Salhat, H.; Faour, I.; Kanbar, J.; Quadri, A.A.; Albashir, M.; Elkord, E. Myeloid Cells in Circulation and Tumor Microenvironment of Breast Cancer Patients. Cancer Immunol. Immunother. 2017, 66, 753–764. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.Y.; Chung, J.S.; Teshima, T.; Feigenbaum, L.; Cruz, P.D.; Jacobe, H.T.; Chong, B.F.; Ariizumi, K. Myeloid-Derived Suppressor Cells in Psoriasis Are an Expanded Population Exhibiting Diverse T-Cell–Suppressor Mechanisms. J. Investig. Dermatol. 2016, 136, 1801–1810. [Google Scholar] [CrossRef] [Green Version]
- Wesolowski, R.; Markowitz, J.; Carson, W.E. Myeloid Derived Suppressor Cells—A New Therapeutic Target in the Treatment of Cancer. J. Immunother. Cancer 2013, 1, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized Mouse Models of Clinical Disease. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 187–215. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, B.E.; Brown, M.E.; Duffin, B.M.; Maufort, J.P.; Vereide, D.T.; Slukvin, I.I.; Thomson, J.A. Nonirradiated NOD,B6.SCID Il2rγ-/- KitW41/W41 (NBSGW) Mice Support Multilineage Engraftment of Human Hematopoietic Cells. Stem Cell Rep. 2015, 4, 171–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, T.X.; Kryczek, I.; Zhao, L.; Zhao, E.; Kuick, R.; Roh, M.H.; Vatan, L.; Szeliga, W.; Mao, Y.; Thomas, D.G.; et al. Myeloid-Derived Suppressor Cells Enhance Stemness of Cancer Cells by Inducing MicroRNA101 and Suppressing the Corepressor CTBP2. Immunity 2013, 39, 611–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.; Hoechst, B.; Duffy, A.; Gamrekelashvili, J.; Fioravanti, S.; Manns, M.P.; Greten, T.F.; Korangy, F. S100A9 a New Marker for Monocytic Human Myeloid-Derived Suppressor Cells. Immunology 2012, 136, 176–183. [Google Scholar] [CrossRef]
- Ortiz, M.L.; Lu, L.; Ramachandran, I.; Gabrilovich, D.I. Myeloid-Derived Suppressor Cells in the Development of Lung Cancer. Cancer Immunol. Res. 2014, 2, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Pillay, J.; Tak, T.; Kamp, V.M.; Koenderman, L. Immune Suppression by Neutrophils and Granulocytic Myeloid-Derived Suppressor Cells: Similarities and Differences. Cell. Mol. Life Sci. 2013, 70, 3813–3827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandau, S.; Moses, K.; Lang, S. The Kinship of Neutrophils and Granulocytic Myeloid-Derived Suppressor Cells in Cancer: Cousins, Siblings or Twins? Semin. Cancer Biol. 2013, 23, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Bronte, V.; Chen, S.H.; Colombo, M.P.; Ochoa, A.; Ostrand-Rosenberg, S.; Schreiber, H. The Terminology Issue for Myeloid-Derived Suppressor Cells. Cancer Res. 2007, 67, 425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, A.; Regev, A.; Yosef, N. Revealing the Vectors of Cellular Identity with Single-Cell Genomics. Nat. Biotechnol. 2016, 34, 1145–1160. [Google Scholar] [CrossRef] [Green Version]
- Villani, A.C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-Cell RNA-Seq Reveals New Types of Human Blood Dendritic Cells, Monocytes, and Progenitors. Science 2017, 356. [Google Scholar] [CrossRef] [Green Version]
- See, P.; Dutertre, C.A.; Chen, J.; Günther, P.; McGovern, N.; Irac, S.E.; Gunawan, M.; Beyer, M.; Händler, K.; Duan, K.; et al. Mapping the Human DC Lineage through the Integration of High-Dimensional Techniques. Science 2017, 356. [Google Scholar] [CrossRef] [Green Version]
- Mercer, T.R.; Clark, M.B.; Crawford, J.; Brunck, M.E.; Gerhardt, D.J.; Taft, R.J.; Nielsen, L.K.; Dinger, M.E.; Mattick, J.S. Targeted Sequencing for Gene Discovery and Quantification Using RNA CaptureSeq. Nat. Protoc. 2014, 9, 989–1009. [Google Scholar] [CrossRef] [PubMed]
- Curion, F.; Handel, A.E.; Attar, M.; Gallone, G.; Bowden, R.; Cader, M.Z.; Clark, M.B. Targeted RNA Sequencing Enhances Gene Expression Profiling of Ultra-Low Input Samples. RNA Biol. 2020, 17, 1741–1753. [Google Scholar] [CrossRef] [PubMed]
- Rongvaux, A.; Willinger, T.; Martinek, J.; Strowig, T.; Gearty, S.V.; Teichmann, L.L.; Saito, Y.; Marches, F.; Halene, S.; Palucka, A.K.; et al. Development and Function of Human Innate Immune Cells in a Humanized Mouse Model. Nat. Biotechnol. 2014, 32, 364–372. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α Regulates Function and Differentiation of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.; Borin, T.F.; Ara, R.; Piranlioglu, R.; Achyut, B.R.; Korkaya, H.; Liu, Y.; Arbab, A.S. Critical Immunosuppressive Effect of MDSC-Derived Exosomes in the Tumor Microenvironment. Oncol. Rep. 2021, 45, 1171–1181. [Google Scholar] [CrossRef]
- Fawkner-Corbett, D.; Antanaviciute, A.; Parikh, K.; Jagielowicz, M.; Gerós, A.S.; Gupta, T.; Ashley, N.; Khamis, D.; Fowler, D.; Morrissey, E.; et al. Spatiotemporal Analysis of Human Intestinal Development at Single-Cell Resolution. Cell 2021, 184, 810–826. [Google Scholar] [CrossRef]
- Zhu, C.; Preissl, S.; Ren, B. Single-Cell Multimodal Omics: The Power of Many. Nat. Methods 2020, 17, 11–14. [Google Scholar] [CrossRef]
- Yuki, K.; Cheng, N.; Nakano, M.; Kuo, C.J. Organoid Models of Tumor Immunology. Trends Immunol. 2020, 41, 652–664. [Google Scholar] [CrossRef]
- Chen, J.; Sun, H.W.; Yang, Y.Y.; Chen, H.T.; Yu, X.J.; Wu, W.C.; Xu, Y.T.; Jin, L.L.; Wu, X.J.; Xu, J.; et al. Reprogramming Immunosuppressive Myeloid Cells by Activated T Cells Promotes the Response to Anti-PD-1 Therapy in Colorectal Cancer. Signal Transduct. Target. Ther. 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.F.; Schoonderwoerd, M.; Knopf, P.; Camps, M.G.; Hawinkels, L.J.; Kneilling, M.; van Hall, T.; Ossendorp, F. Tumor-Draining Lymph Nodes Are Pivotal in PD-1/PD-L1 Checkpoint Therapy. JCI Insight 2018, 3, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Shim, S.; Belanger, M.C.; Harris, A.R.; Munson, J.M.; Pompano, R.R. Two-Way Communication between Ex Vivo Tissues on a Microfluidic Chip: Application to Tumor-Lymph Node Interaction. Lab Chip 2019, 19, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente, A.; Zilio, S.; Caroli, J.; van Simaeys, D.; Mazza, E.M.C.; Ince, T.A.; Bronte, V.; Bicciato, S.; Weed, D.T.; Serafini, P. Aptamers against Mouse and Human Tumor-Infiltrating Myeloid Cells as Reagents for Targeted Chemotherapy. Sci. Transl. Med. 2020, 12, 1–13. [Google Scholar] [CrossRef]
- Pinton, L.; Magri, S.; Masetto, E.; Vettore, M.; Schibuola, I.; Ingangi, V.; Marigo, I.; Matha, K.; Benoit, J.P.; Della Puppa, A.; et al. Targeting of Immunosuppressive Myeloid Cells from Glioblastoma Patients by Modulation of Size and Surface Charge of Lipid Nanocapsules. J. Nanobiotechnol. 2020, 18, 1–12. [Google Scholar] [CrossRef]
- Sasso, M.S.; Lollo, G.; Pitorre, M.; Solito, S.; Pinton, L.; Valpione, S.; Bastiat, G.; Mandruzzato, S.; Bronte, V.; Marigo, I.; et al. Low Dose Gemcitabine-Loaded Lipid Nanocapsules Target Monocytic Myeloid-Derived Suppressor Cells and Potentiate Cancer Immunotherapy. Biomaterials 2016, 96, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Zilio, S.; Vella, J.L.; De la Fuente, A.C.; Daftarian, P.M.; Weed, D.T.; Kaifer, A.; Marigo, I.; Leone, K.; Bronte, V.; Serafini, P. 4PD Functionalized Dendrimers: A Flexible Tool for In Vivo Gene Silencing of Tumor-Educated Myeloid Cells. J. Immunol. 2017, 198, 4166–4177. [Google Scholar] [CrossRef]
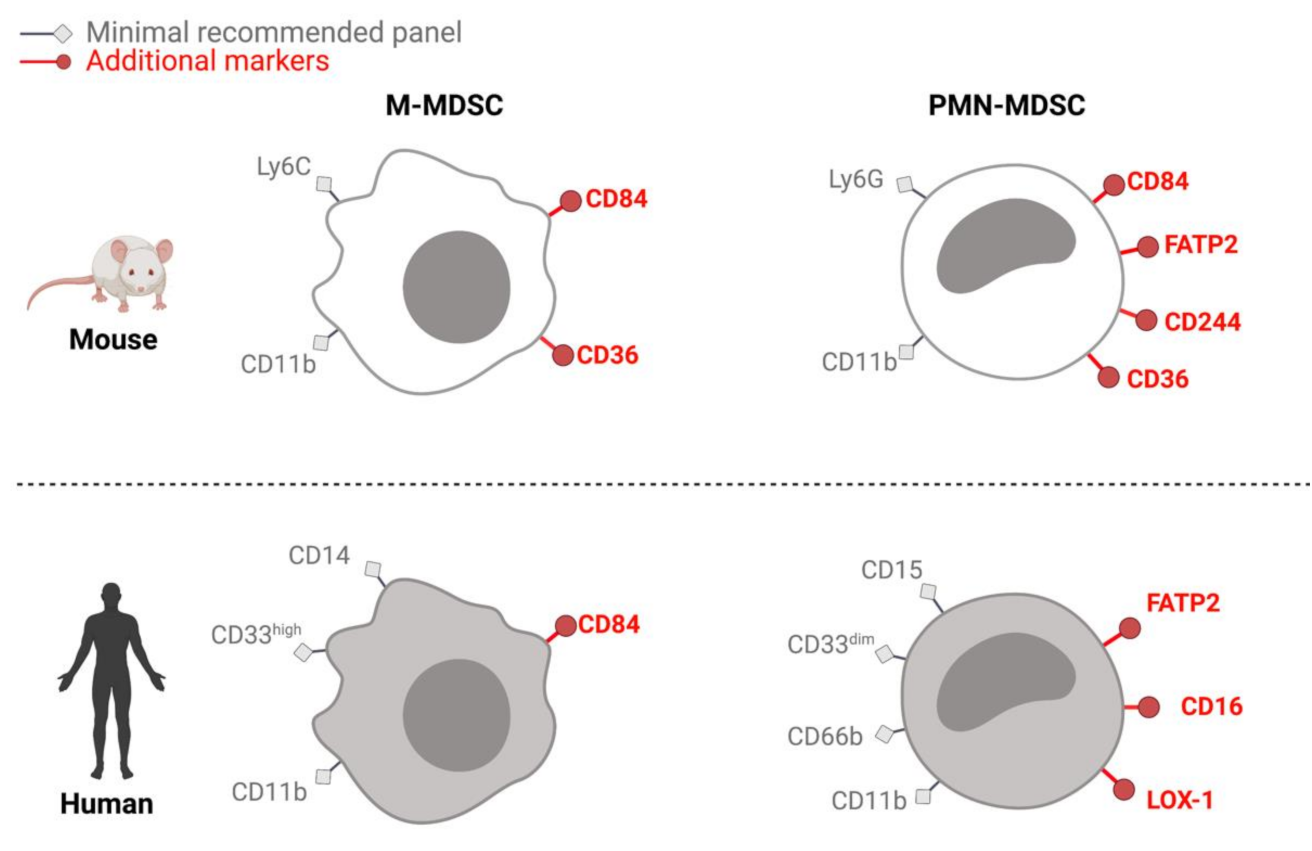

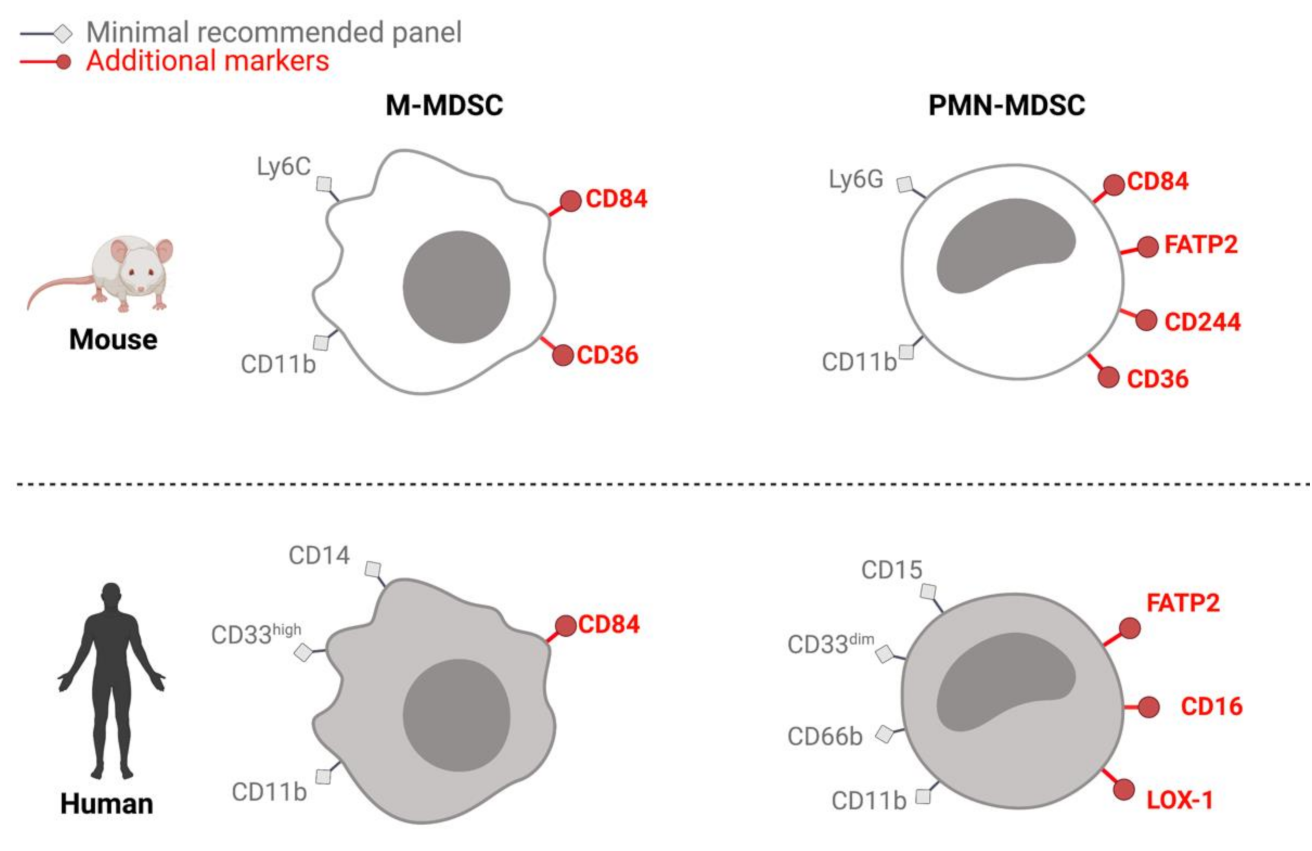{kind=link}
| Mechanisms of Suppression | Mouse | Ref. | Human | Ref. | |
|---|---|---|---|---|---|
| Amino acid depletion | L-Arginine | PMN-MDSCs express arginase-1 a. Through arginase-1, PMN-MDSCs inhibit proliferation, cytokine secretion and decrease CD3ζ expression in T-cells during in vitro co-culture. | [56,57,58,59] | PMN-MDSCs and activated PMN express arginase-1. T-cells activated in the presence of arginase-1 or L-arginine-depleted medium arrest the cell-cycle. | [60,61,62,63,64] |
| Tumors regress in vivo after inhibition of arginase-1. | [61] | The presence of arginase-1-expressing PMN-MDSCs in the blood of cancer patients is correlated to PBMCs losing CD3ζ expression. | [58,59,62,65] | ||
| L-Cysteine | M- and PMN-MDSCs sequester cysteine. Cysteine concentrations are lower in the serum of tumor-bearing mice. | [66] | |||
| L-Tryptophan | Bone marrow (in vitro)-derived M-MDSCs express IDO. Inhibiting IDO restores T-cell proliferation. | [36] | E-MDSCs isolated from blood and metastasis of cancer patients express IDO. IDO expression is correlated with Foxp3+ T-cell expansion. | [67] | |
| Oxidative stress | PMN-MDSCs produce ROS (peroxynitrite) by NOX2. M-MDSCs produce NO by iNOS. Inhibiting ROS or ROS-producing enzyme blocks suppressive capacities of M- and PMN-MDSCs. | [68,69,70,71] | PMN-MDSCs isolated from cancer patients produce ROS and increased levels of nitrotyrosine are present in CD8+ T-cell. | [66,68] | |
| M- and PMN-MDSCs mediate nitration of the TCR in vivo and in vitro, which disrupts pMHC binding to CD8+ T-cells. | [71] | T-cells co-cultured with iNOS-expressing PMN-MDSCs isolated from cancer patients express diminished levels of CD3ζ. | [65] | ||
| M- and PMN-MDSCs produce ROS, which inhibits proliferation, cytokine secretion, ADCC and granzyme-B production by NK cells. | [72] | M- and PMN-MDSCs from cancer patients inhibit cytokine secretion and ADCC of NK cell through NO production by iNOS. | [55] | ||
| Treg | M-MDSCs produce IL-10 and TGF-β. Foxp3+ Treg expand in vivo and in vitro in IL-10-, TGF-β-, arginase- and CD40L-dependant manners. | [73,74,75] | M-MDSCs induce Treg in an IL-10-dependant manner. | [76] | |
| M-MDSCs produce CCR5-ligand in vivo and in vitro, which recruits of Treg. | [77] | M- and PMN-MDSCs that express CD39 and CD73 generate adenosine and inhibit T-cell and NK cell proliferation and cytokine secretion. | [75,76] | ||
| Accumulation of M-MDSCs correlate with increased frequency of circulating Foxp3+ T-cells in renal transplant patients. | [78] | ||||
| Direct MDSC/ T-cell interaction | Immune checkpoint blockade | PD-L1+ M- and PMN-MDSCs present at higher frequency in tumor microenvironment compared to peripheral blood. | [79,80] | PD-L1+ M- and PMN-MDSCs accumulate in blood of cancer patients. PD-L1+ PMN-MDSCs accumulate in TME. | [81] |
| Myeloid-specific ablation of PD-1 decreases tumor growth. | [82] | PMN-MDSCs from non-responding cancer patients (ipilimumab) express more PD-L1+. | [83] | ||
| Cell death ligand | FasL-expressing PMN-MDSCs induce CD8+ T-cell apoptosis. | [84] | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanhaver, C.; van der Bruggen, P.; Bruger, A.M. MDSC in Mice and Men: Mechanisms of Immunosuppression in Cancer. J. Clin. Med. 2021, 10, 2872. https://doi.org/10.3390/jcm10132872
Vanhaver C, van der Bruggen P, Bruger AM. MDSC in Mice and Men: Mechanisms of Immunosuppression in Cancer. Journal of Clinical Medicine. 2021; 10(13):2872. https://doi.org/10.3390/jcm10132872
Chicago/Turabian StyleVanhaver, Christophe, Pierre van der Bruggen, and Annika M. Bruger. 2021. "MDSC in Mice and Men: Mechanisms of Immunosuppression in Cancer" Journal of Clinical Medicine 10, no. 13: 2872. https://doi.org/10.3390/jcm10132872
APA StyleVanhaver, C., van der Bruggen, P., & Bruger, A. M. (2021). MDSC in Mice and Men: Mechanisms of Immunosuppression in Cancer. Journal of Clinical Medicine, 10(13), 2872. https://doi.org/10.3390/jcm10132872