
Whole-Exome Sequencing to Identify Potential Genetic Risk in Substance Use Disorders: A Pilot Feasibility Study

 ,
,  and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Eligibility Criteria
2.3. Data Collection
2.4. Substance Use Data Collection
2.4.1. Alcohol Use Disorder and Opioid Use Disorder
2.4.2. Nicotine Dependence: Fagerstrom Test for ND
2.4.3. The Severity of Addiction
2.4.4. Alcohol Use Disorder Identification Test (AUDIT)
2.4.5. Collection of Saliva Samples
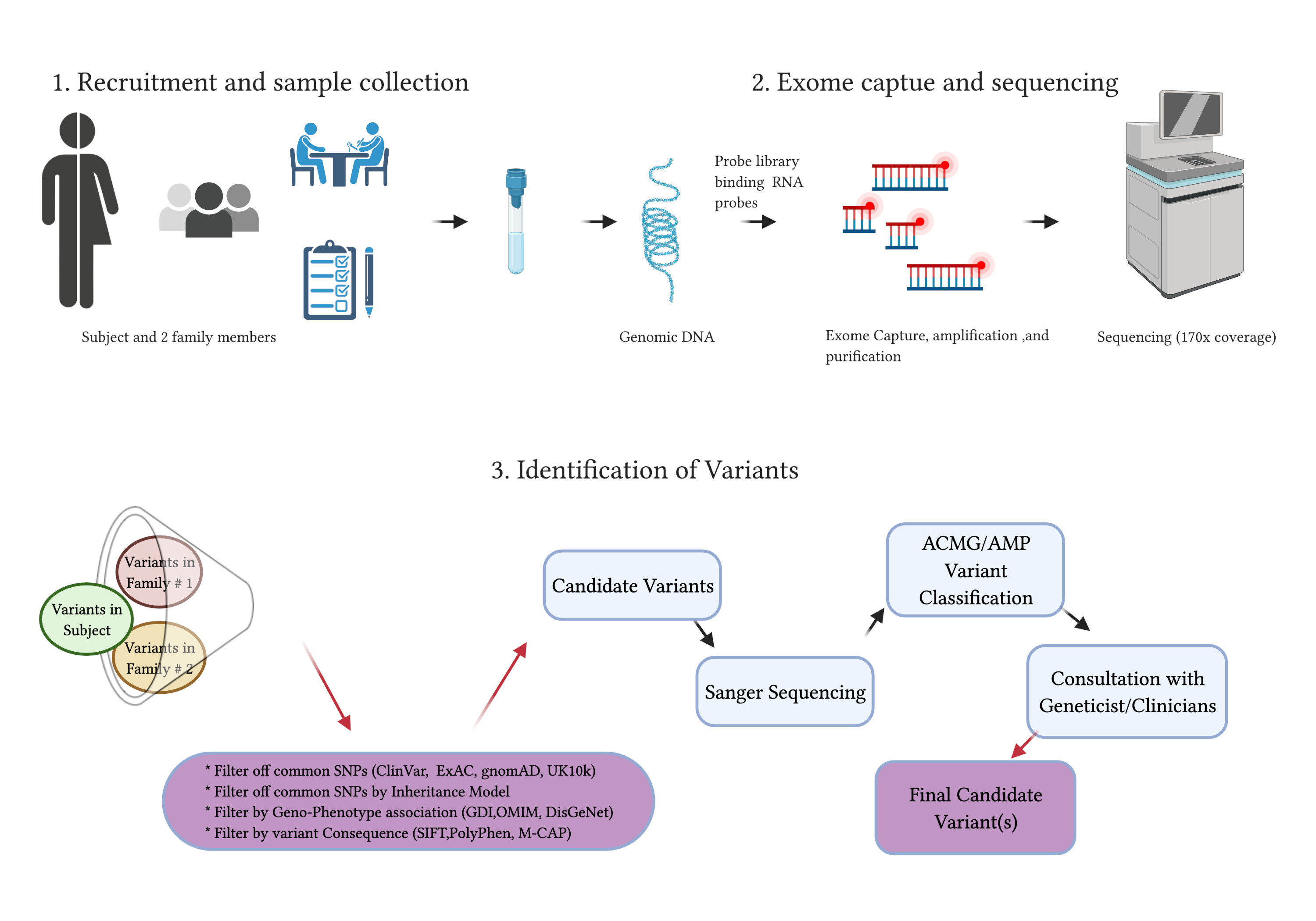
2.4.6. Whole Exome Sequencing
2.4.7. Exome Capture and Sequencing
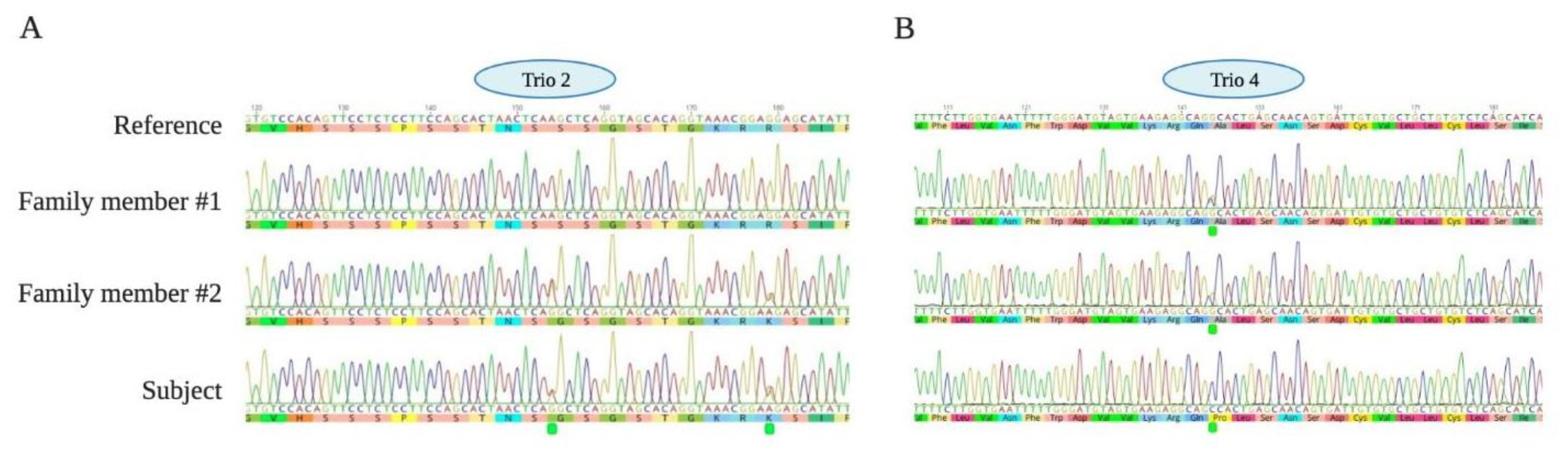
2.4.8. Identification of Variants and Filtering of Common and Family-Specific Variants
2.5. Data Analysis
3. Results
3.1. Demographics
3.2. Substance Use Characteristics
3.3. Genetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- United Nations Office on Drugs and Crime World Drug Report, United Nations. 2019. Available online: https://wdr.unodc.org/wdr2019/ (accessed on 5 April 2020).
- World Health Organization. Global Status Report on Alcohol and Health 2018; World Health Organization: Geneva, Switzerland, 2019; ISBN 9789241565639. [Google Scholar]
- Global Burden of Diseases 2015 Tobacco Collaborators Smoking Prevalence and Attributable Disease Burden in 195 Countries and Territories, 1990-2015: A Systematic Analysis from the Global Burden of Disease Study 2015. Lancet 2017, 389, 1885–1906. [CrossRef] [Green Version]
- Shahwan, S.; Abdin, E.; Shafie, S.; Chang, S.; Sambasivam, R.; Zhang, Y.; Vaingankar, J.A.; Teo, Y.Y.; Heng, D.; Chong, S.A.; et al. Prevalence and Correlates of Smoking and Nicotine Dependence: Results of a Nationwide Cross-Sectional Survey among Singapore Residents. BMJ Open 2019, 9, e032198. [Google Scholar] [CrossRef] [Green Version]
- MOH|Disease Burden. Available online: https://www.moh.gov.sg/resources-statistics/singapore-health-facts/disease-burden (accessed on 23 April 2020).
- Subramaniam, M.; Abdin, E.; Vaingankar, J.A.; Shafie, S.; Chua, B.Y.; Sambasivam, R.; Zhang, Y.J.; Shahwan, S.; Chang, S.; Chua, H.C.; et al. Tracking the Mental Health of a Nation: Prevalence and Correlates of Mental Disorders in the Second Singapore Mental Health Study. Epidemiol. Psychiatr. Sci. 2019, 29, e29. [Google Scholar] [CrossRef] [Green Version]
- Beydoun, M.A.; Beydoun, H.A.; Gamaldo, A.A.; Teel, A.; Zonderman, A.B.; Wang, Y. Epidemiologic Studies of Modifiable Factors Associated with Cognition and Dementia: Systematic Review and Meta-Analysis. BMC Public Health 2014, 14, 643. [Google Scholar] [CrossRef] [Green Version]
- Bassiony, M.; Seleem, D. Drug-Related Problems among Polysubstance and Monosubstance Users: A Cross-Sectional Study. J. Subst. Use 2020, 25, 1–6. [Google Scholar] [CrossRef]
- Hjemsæter, A.J.; Bramness, J.G.; Drake, R.; Skeie, I.; Monsbakken, B.; Benth, J.Š.; Landheim, A.S. Mortality, Cause of Death and Risk Factors in Patients with Alcohol Use Disorder Alone or Poly-Substance Use Disorders: A 19-Year Prospective Cohort Study. BMC Psychiatry 2019, 19, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pv, A.; Jun Wen, T.; Karuvetil, M.Z.; Cheong, A.; Cheok, C.; Kandasami, G. Unnatural Death among Treatment Seeking Substance Users in Singapore: A Retrospective Study. Int. J. Environ. Res. Public Health 2019, 16, 2743. [Google Scholar] [CrossRef] [Green Version]
- Shield, K.; Manthey, J.; Rylett, M.; Probst, C.; Wettlaufer, A.; Parry, C.D.H.; Rehm, J. National, Regional, and Global Burdens of Disease from 2000 to 2016 Attributable to Alcohol Use: A Comparative Risk Assessment Study. Lancet Public Health 2020, 5, e51–e61. [Google Scholar] [CrossRef] [Green Version]
- Richmond-Rakerd, L.S.; Slutske, W.S.; Deutsch, A.R.; Lynskey, M.T.; Agrawal, A.; Madden, P.A.F.; Bucholz, K.K.; Heath, A.C.; Martin, N.G. Progression in Substance Use Initiation: A Multilevel Discordant Monozygotic Twin Design. J. Abnorm. Psychol. 2015, 124, 596–605. [Google Scholar] [CrossRef]
- Bierut, L.J.; Dinwiddie, S.H.; Begleiter, H.; Crowe, R.R.; Hesselbrock, V.; Nurnberger, J.I.; Porjesz, B.; Schuckit, M.A.; Reich, T. Familial Transmission of Substance Dependence: Alcohol, Marijuana, Cocaine, and Habitual Smoking. Arch. Gen. Psychiatry 1998, 55, 982. [Google Scholar] [CrossRef]
- Hancock, D.B.; Markunas, C.A.; Bierut, L.J.; Johnson, E.O. Human Genetics of Addiction: New Insights and Future Directions. Curr. Psychiatry Rep. 2018, 20, 8. [Google Scholar] [CrossRef]
- Kendler, K.S.; PirouziFard, M.; Lönn, S.; Edwards, A.C.; Maes, H.H.; Lichtenstein, P.; Sundquist, J.; Sundquist, K. A National Swedish Twin-Sibling Study of Alcohol Use Disorders. Twin Res. Hum. Genet. 2016, 19, 430–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verweij, K.J.H.; Zietsch, B.P.; Lynskey, M.T.; Medland, S.E.; Neale, M.C.; Martin, N.G.; Boomsma, D.I.; Vink, J.M. Genetic and Environmental Influences on Cannabis Use Initiation and Problematic Use: A Meta-Analysis of Twin Studies. Addiction 2010, 105, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Tsuang, M.T.; Bar, J.L.; Harley, R.M.; Lyons, M.J. The Harvard Twin Study of Substance Abuse: What We Have Learned. Harv. Rev. Psychiatry 2001, 9, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Mathuru, A.S. A Little Rein on Addiction. Semin. Cell Dev. Biol. 2018, 78, 120–129. [Google Scholar] [CrossRef]
- Wang, S.; Yang, Z.; Ma, J.Z.; Payne, T.J.; Li, M.D. Introduction to Deep Sequencing and Its Application to Drug Addiction Research with a Focus on Rare Variants. Mol. Neurobiol. 2014, 49, 601–614. [Google Scholar] [CrossRef] [Green Version]
- Rabbani, B.; Tekin, M.; Mahdieh, N. The Promise of Whole-Exome Sequencing in Medical Genetics. J. Hum. Genet. 2014, 59, 5–15. [Google Scholar] [CrossRef]
- Singleton, A.B. Exome Sequencing: A Transformative Technology. Lancet Neurol. 2011, 10, 942–946. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.M.T.; Manning, V.; Teoh, H.C.; Winslow, M.; Lee, A.; Subramaniam, M.; Guo, S.; Wong, K.E. Stress-Coping Morbidity among Family Members of Addiction Patients in Singapore. Drug Alcohol. Rev. 2011, 30, 441–447. [Google Scholar] [CrossRef]
- McLellan, A.T.; Luborsky, L.; O’Brien, C.P.; Woody, G.E. An Improved Evaluation Instrument for Substance Abuse Patients: The Addiction Severity Index. Probl. Drug Depend. 1980, 168, 26–33. [Google Scholar] [CrossRef]
- Xue, S.; Maluenda, J.; Marguet, F.; Shboul, M.; Quevarec, L.; Bonnard, C.; Ng, A.Y.J.; Tohari, S.; Tan, T.T.; Kong, M.K.; et al. Loss-of-Function Mutations in LGI4, a Secreted Ligand Involved in Schwann Cell Myelination, Are Responsible for Arthrogryposis Multiplex Congenita. Am. J. Hum. Genet. 2017, 100, 659–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rackham, O.J.L.; Shihab, H.A.; Johnson, M.R.; Petretto, E. EvoTol: A Protein-Sequence Based Evolutionary Intolerance Framework for Disease-Gene Prioritization. Nucleic Acids Res. 2015, 43, e33. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Risso, D.S.; Mezzavilla, M.; Pagani, L.; Robino, A.; Morini, G.; Tofanelli, S.; Carrai, M.; Campa, D.; Barale, R.; Caradonna, F.; et al. Global Diversity in the TAS2R38 Bitter Taste Receptor: Revisiting a Classic Evolutionary PROPosal. Sci. Rep. 2016, 6, 25506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, J.E.; Behm, F.M.; Drgon, T.; Johnson, C.; Uhl, G. Personalized Smoking Cessation: Interactions between Nicotine Dose, Dependence and Quit-Success Genotype Score. Mol. Med. 2010, 16, 247–253. [Google Scholar] [CrossRef]
- Polimanti, R.; Zhao, H.; Farrer, L.A.; Kranzler, H.R.; Gelernter, J. Ancestry-Specific and Sex-Specific Risk Alleles Identified in a Genome-Wide Gene-by-Alcohol Dependence Interaction Study of Risky Sexual Behaviors. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.H.; Jensen, K.P.; Li, J.; Nunez, Y.; Farrer, L.A.; Hakonarson, H.; Cook-Sather, S.D.; Kranzler, H.R.; Gelernter, J. Genome-wide association study of therapeutic opioid dosing identifies a novel locus upstream of OPRM1. Mol. Psychiatry 2017, 22, 346–352. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.C.; Hinrichs, A.L.; Bertelsen, S.; Stock, H.; Budde, J.P.; Dick, D.M.; Bucholz, K.K.; Rice, J.; Saccone, N.; Edenberg, H.J.; et al. Functional Variants in TAS2R38 and TAS2R16 Influence Alcohol Consumption in High-Risk Families of African-American Origin. Alcohol Clin. Exp. Res. 2007, 31, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Lopez, O.; Roman, S.; Martinez-Lopez, E.; Gonzalez-Aldaco, K.; Ojeda-Granados, C.; Sepulveda-Villegas, M.; Panduro, A. Association of a novel TAS2R38 haplotype with alcohol intake among Mexican-Mestizo population. Ann. Hepatol. 2015, 14, 729–734. [Google Scholar] [CrossRef]
- Duffy, V.B.; Davidson, A.C.; Kidd, J.R.; Kidd, K.K.; Speed, W.C.; Pakstis, A.J.; Reed, D.R.; Snyder, D.J.; Bartoshuk, L.M. Bitter receptor gene (TAS2R38), 6-n-propylthiouracil (PROP) bitterness and alcohol intake. Alcohol Clin. Exp. Res. 2004, 28, 1629–1637. [Google Scholar] [CrossRef] [Green Version]
- Keller, M.; Liu, X.; Wohland, T.; Rohde, K.; Gast, M.-T.; Stumvoll, M.; Kovacs, P.; Tönjes, A.; Böttcher, Y. TAS2R38 and its influence on smoking behavior and glucose homeostasis in the German Sorbs. PLoS ONE. 2013, 8, e80512. [Google Scholar] [CrossRef] [Green Version]
- Wain, L.V.; Shrine, N.; Miller, S.; Jackson, V.E.; Ntalla, I.; Soler Artigas, M.; Billington, C.K.; Kheirallah, A.K.; Allen, R.; Cook, J.P.; et al. Novel insights into the genetics of smoking behaviour, lung function, and chronic obstructive pulmonary disease (UK BiLEVE): A genetic association study in UK Biobank. Lancet. Respir. Med. 2015, 3, 769–781. [Google Scholar] [CrossRef]
- Chambers, R.A.; McClintick, J.N.; Sentir, A.M.; Berg, S.A.; Runyan, M.; Choi, K.H.; Edenberg, H.J. Cortical-striatal gene expression in neonatal hippocampal lesion (NVHL)-amplified cocaine sensitization. Genes Brain Behav. 2013, 12, 564–575. [Google Scholar] [CrossRef]
- Available online: https://varsome.com/variant/hg19/PLEKHM3%20Leu637Val?annotation-mode=germline (accessed on 5 April 2020).
- Available online: https://varsome.com/variant/hg19/PLEKHM3%20His182Arg?annotation-mode=germline (accessed on 5 April 2020).
- Available online: https://varsome.com/variant/hg19/CCSER1%3AS52G?annotation-mode=germline (accessed on 5 April 2020).
- Available online: https://varsome.com/variant/hg19/NM_001145065.2(CCSER1)%3AR60K?annotation-mode=germline (accessed on 5 April 2020).
- Available online: https://varsome.com/variant/hg19/LIMCH1%20Tyr430Cys?annotation-mode=germline (accessed on 5 April 2020).
- Available online: https://varsome.com/variant/hg19/TAS2R38%20Ile296Val?annotation-mode=germline (accessed on 5 April 2020).
- Available online: https://varsome.com/variant/hg19/TAS2R38%20Ala49Pro?annotation-mode=germline (accessed on 5 April 2020).
- Available online: https://varsome.com/variant/hg19/DNAH8%20Leu3810Arg?annotation-mode=germline (accessed on 5 April 2020).
- Nathan, F.M.; Kibat, C.; Goel, T.; Stewart, J.; Claridge-Chang, A.; Mathuru, A.S. Contingent-Behavior Assay to Study the Neurogenetics of Addiction Shows Zebrafish Preference for Alcohol Is Biphasic. bioRxiv 2021. [Google Scholar] [CrossRef]
- Subramaniam, M.; Abdin, E.; Picco, L.; Pang, S.; Shafie, S.; Vaingankar, J.A.; Kwok, K.W.; Verma, K.; Chong, S.A. Stigma towards People with Mental Disorders and Its Components—a Perspective from Multi-Ethnic Singapore. Epidemiol. Psychiatr. Sci. 2017, 26, 371–382. [Google Scholar]
- Millum, J.; Campbell, M.; Luna, F.; Malekzadeh, A.; Karim, Q.A. Ethical Challenges in Global Health-Related Stigma Research. Bmc Med. 2019, 17, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, J.L.; Dick, D.M. Genetic and Environmental Risk Factors for Adolescent-Onset Substance Use Disorders. Child. Adolesc. Psychiatr. Clin. N. Am. 2010, 19, 465–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, V.; Gomez, B.; Koh, P.K.; Ng, A.; Guo, S.; Kandasami, G.; Wong, K.E. Treatment Outcome and Its Predictors among Asian Problem Drinkers. Drug Alcohol Rev. 2013, 32, 178–186. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Subject ID | Diagnosis | Age | Gender | Marital Status | Ethnicity | Employment Status | Education |
|---|---|---|---|---|---|---|---|
| Trio 1 | OUD | 32 | M | Single | Malay | Unemployed | Diploma/Pre-U/Higher Diploma |
| - | 60 | F | Married | Malay | Unemployed | Secondary School | |
| - | 62 | M | Married | Malay | Unemployed | Secondary School | |
| Trio 2 | AUD | 44 | F | Married | Indian | Employed | Secondary school |
| - | 68 | F | Married | Indian | Employed | Primary school | |
| - | 49 | M | Married | Indian | Employed | Secondary | |
| Trio 3 | AUD | 22 | M | Single | Indian | Unemployed | Secondary school |
| - | 53 | F | Married | Indian | Employed | Diploma/Pre-U/Higher Diploma | |
| - | 56 | M | Married | Indian | Employed | Degree/above | |
| Trio 4 | AUD | 21 | F | Single | Chinese | Unemployed | Degree/above |
| - | 54 | F | Married | Indian | Unemployed | Secondary school | |
| - | 67 | M | Married | Chinese | Unemployed | Primary school | |
| Trio 5 | OUD | 38 | M | Married | Chinese | Unemployed | Primary school |
| - | 65 | F | Widowed | Chinese | Employed | Secondary | |
| - | 35 | F | Married | Chinese | Unemployed | Degree/above |
| Subject ID/Diagnosis | Medical | Employment | Alcohol | Drugs | Legal Status | Family/Social Relationships | Psychiatric Status |
|---|---|---|---|---|---|---|---|
| Trio 1 (OUD) | 0.70 * | 0.50 | 0.00 | 0.23 | 0.100 | 0.37 | 0.56 |
| 0.00 | 1.00 | 0.00 | 0.00 | 0.000 | 0.10 | 0.00 | |
| 0.00 | 0.75 | 0.00 | 0.00 | 0.000 | 0.10 | 0.00 | |
| Trio 2 (AUD) | 0.74 | 0.37 | 0.85 | 0.00 | 0.20 | 0.57 | 0.41 |
| 0.00 | 0.55 | 0.15 | 0.00 | 0.00 | 0.15 | 0.30 | |
| 0.00 | 0.76 | 0.45 | 0.00 | 0.00 | 0.00 | 0.00 | |
| Trio 3 (AUD) | 0.71 | 0.50 | 0.37 | 0.00 | 0.00 | 0.75 | 0.74 |
| 0.00 | 0.75 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| 0.00 | 0.25 | 0.00 | 0.00 | 0.00 | 0.20 | 0.00 | |
| Trio 4 (AUD) | 0.00 | 1.00 | 0.35 | 0.15 | 0.30 | 0.29 | 0.30 |
| 0.03 | 0.50 | 0.00 | 0.00 | 0.00 | 0.20 | 0.00 | |
| 0.00 | 0.50 | 0.00 | 0.00 | 0.00 | 0.20 | 0.00 | |
| Trio 5 (OUD) | 0.00 | 0.63 | 0.00 | 0.21 | 0.00 | 0.20 | 0.00 |
| 0.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.20 | 0.00 | |
| 0.00 | 0.75 | 0.01 | 0.00 | 0.00 | 0.20 | 0.00 |
| Diagnosis of the Subject | Gene Symbol | Common Name | Known Function | HGNC ID | Disease (Based on DisGeNET) | References Linking This Gene to SUD in Human Genetic Studies |
|---|---|---|---|---|---|---|
| OUD + ND (Subject 1) | PLEKHM3 | Pleckstrin homology domain-containing family M member 3 | Muscle differentiation (scaffold protein) | HGNC:34006 | Tobacco use disorder | [28] |
| AUD + ND (Subject 2) | CCSER1 | Coiled-coil serine-rich Protein 1 | Cell division | HGNC:29349 | Cocaine-related disorders | [29,30] |
| AUD + ND (Subject 3) | LIMCH1 | LIM and calponin homology domains-containing protein 1 | Cell spreading and migration | HGNC:29191 | Substance-related disorders | [28] |
| AUD + ND (Subject 4) | TAS2R38 | Taste 2 Receptor Member 38 | Sensory perception (bitterness) | HGNC:9584 | Alcoholism | [31,32,33,34] |
| OUD + ND (Subject 5) | DNAH8 and TAS2R38 | Dynein axonemal heavy chain 8 | Force generating protein for cilia | HGNC:2952 | Cocaine-related disorders | [35,36] |
| Subject ID | Genotype | Gene Name | Polyphen Score | Exon | Protein | Coding | GnomAD Frequency | CADD Score PHRED | ACMG Classification |
|---|---|---|---|---|---|---|---|---|---|
| Subject 1 | G/C | PLEKHM3 | 0.97 | 6 | p. Leu637Val | c.1909C> G | 0.000456 | 17.08 | Uncertain Significance [37] |
| T/C | PLEKHM3 | 0 | 2 | p. His182Arg | c.545A>G | 0.0014517 | 2.805 | Likely Benign [38] | |
| Subject 2 | A/G | CCSER1 | 0.998 | 2 | p. Ser52Gly | c.154A>G | 0.0000284733 | 21.8 | Uncertain Significance [39] |
| G/A | CCSER1 | 0.998 | 2 | p. Arg60Lys | c.179G>A | 0.0000284866 | 33 | Uncertain Significance [40] | |
| Subject 3 | A/G | LIMCH1 | 1 | 12 | p. Tyr430Cys | c.1289A>G | 0.000012188 | 20.9 | Uncertain Significance [41] |
| Subject 4 | C/C | TAS2R38 | 0.75 | 1 | p. Ile296Val | c.886A>G | 0.485108 | 8.017 | Benign [42] |
| G/G | TAS2R38 | 0 | 1 | p. Ala49Pro | c.145G>C | 0.456251 | 0.978 | Benign [43] | |
| Subject 5 | T/G | DNAH8 | 1 | 76 | p. Leu3810Arg | c.11429T>G | 0.0000508 | 24.8 | Uncertain significance [44] |
| C/C | TAS2R38 | 0.75 | 1 | p. Ile296Val | c.886A>G | 0.485108 | 8.017 | Benign [42] | |
| G/G | TAS2R38 | 0 | 1 | p. Ala49Pro | c.145G>C | 0.456251 | 0.978 | Benign [43] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
AshaRani, P.V.; Amron, S.; Zainuldin, N.A.B.; Tohari, S.; Ng, A.Y.J.; Song, G.; Venkatesh, B.; Mathuru, A.S. Whole-Exome Sequencing to Identify Potential Genetic Risk in Substance Use Disorders: A Pilot Feasibility Study. J. Clin. Med. 2021, 10, 2810. https://doi.org/10.3390/jcm10132810
AshaRani PV, Amron S, Zainuldin NAB, Tohari S, Ng AYJ, Song G, Venkatesh B, Mathuru AS. Whole-Exome Sequencing to Identify Potential Genetic Risk in Substance Use Disorders: A Pilot Feasibility Study. Journal of Clinical Medicine. 2021; 10(13):2810. https://doi.org/10.3390/jcm10132810
Chicago/Turabian StyleAshaRani, P. V., Syidda Amron, Noor Azizah Bte Zainuldin, Sumanty Tohari, Alvin Y. J. Ng, Guo Song, Byrappa Venkatesh, and Ajay S. Mathuru. 2021. "Whole-Exome Sequencing to Identify Potential Genetic Risk in Substance Use Disorders: A Pilot Feasibility Study" Journal of Clinical Medicine 10, no. 13: 2810. https://doi.org/10.3390/jcm10132810
APA StyleAshaRani, P. V., Amron, S., Zainuldin, N. A. B., Tohari, S., Ng, A. Y. J., Song, G., Venkatesh, B., & Mathuru, A. S. (2021). Whole-Exome Sequencing to Identify Potential Genetic Risk in Substance Use Disorders: A Pilot Feasibility Study. Journal of Clinical Medicine, 10(13), 2810. https://doi.org/10.3390/jcm10132810