
Emerging Immunogenicity and Genotoxicity Considerations of Adeno-Associated Virus Vector Gene Therapy for Hemophilia


 and
and
Abstract
1. Introduction
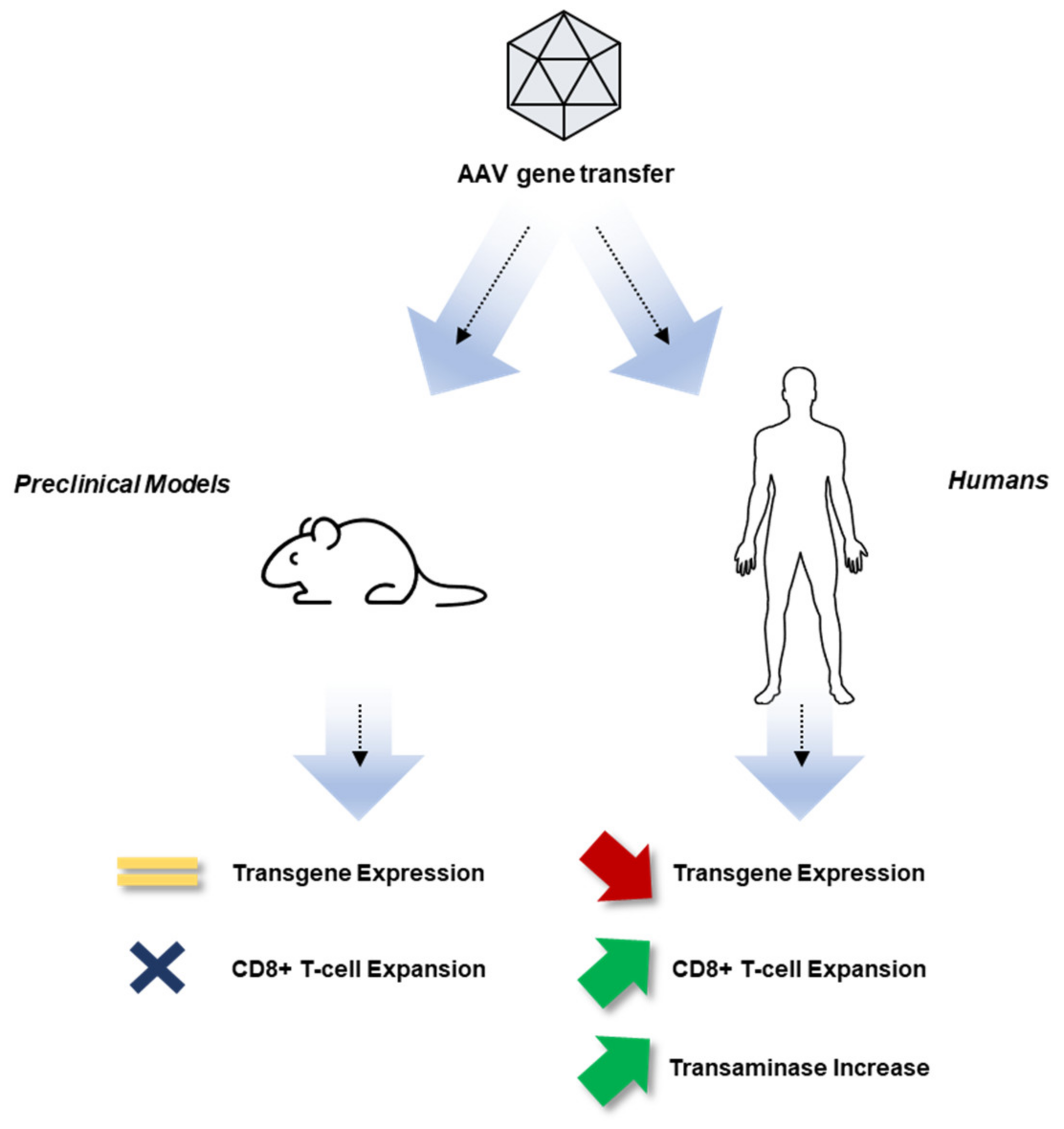
2. Experience with Gene Therapy for Hemophilia to Date
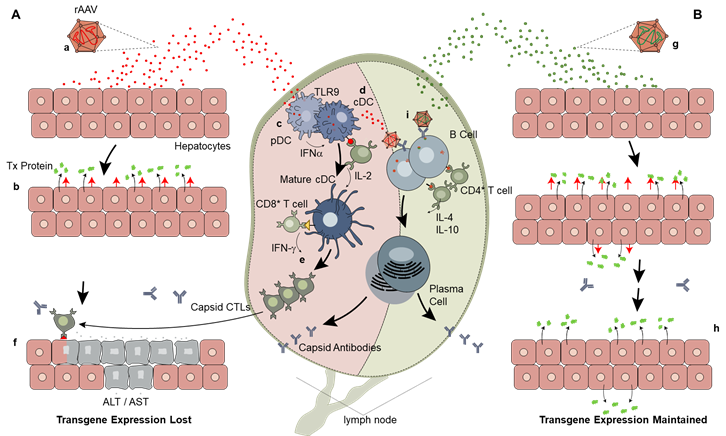
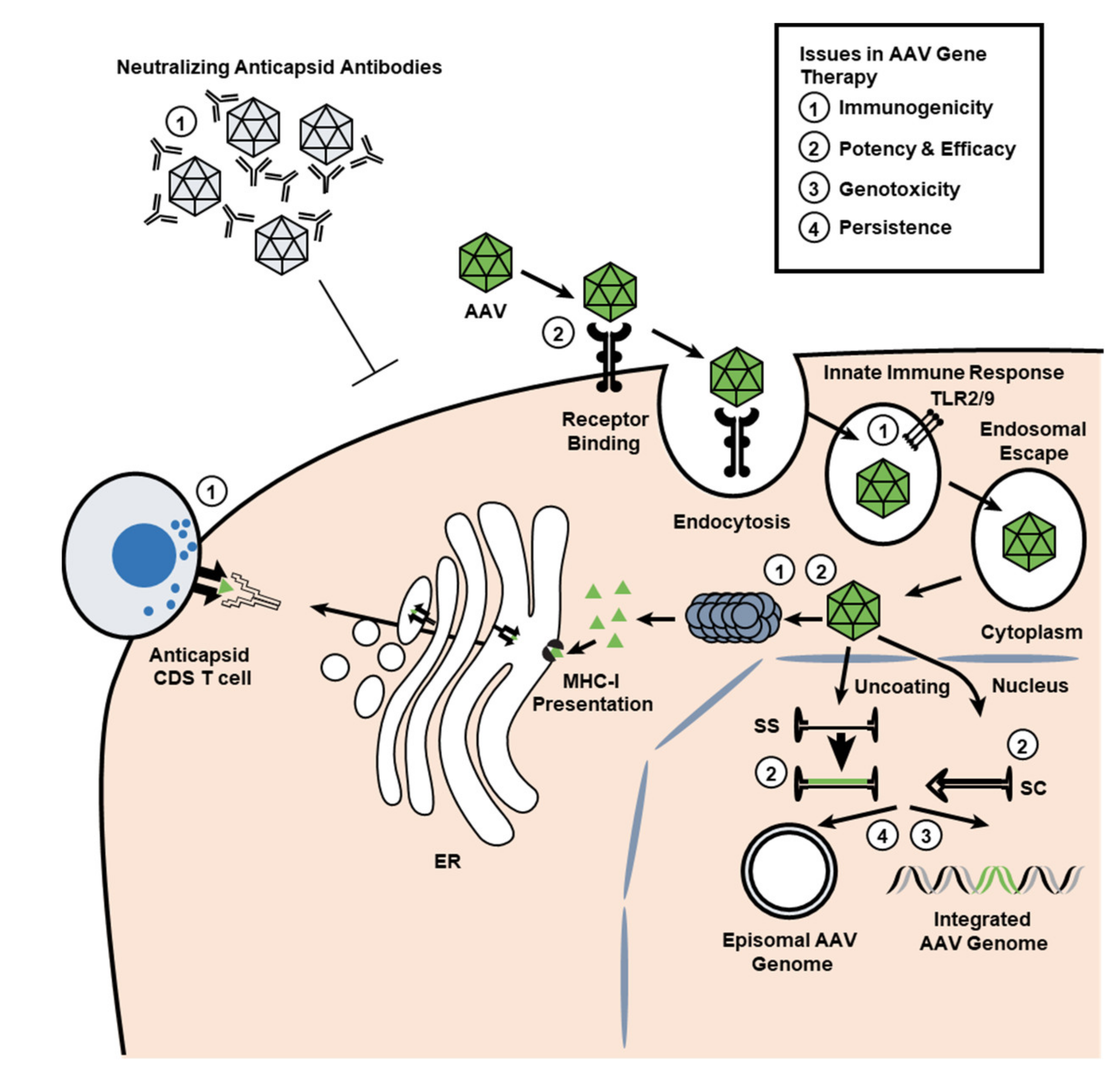
3. Overview of the Immune System
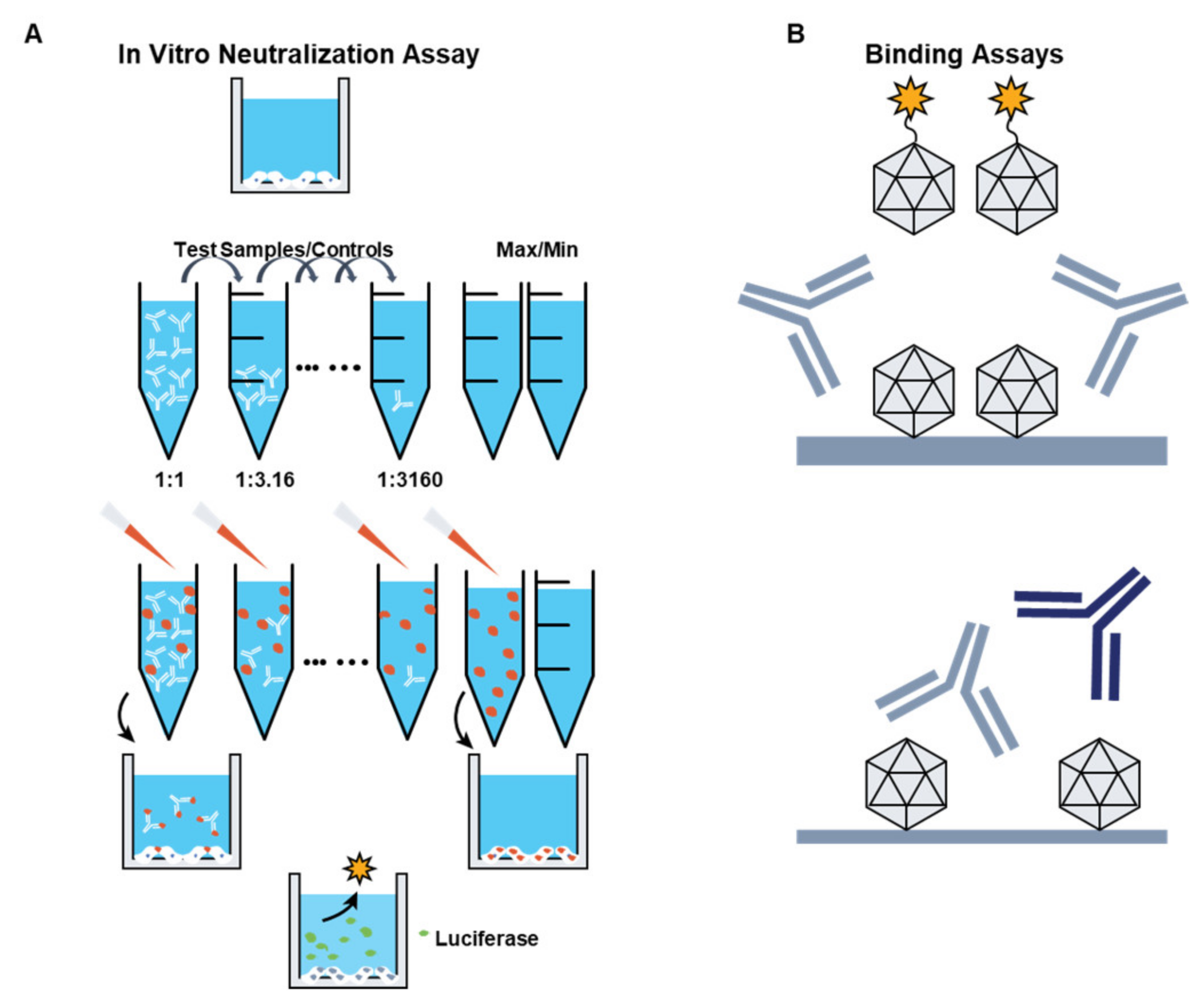
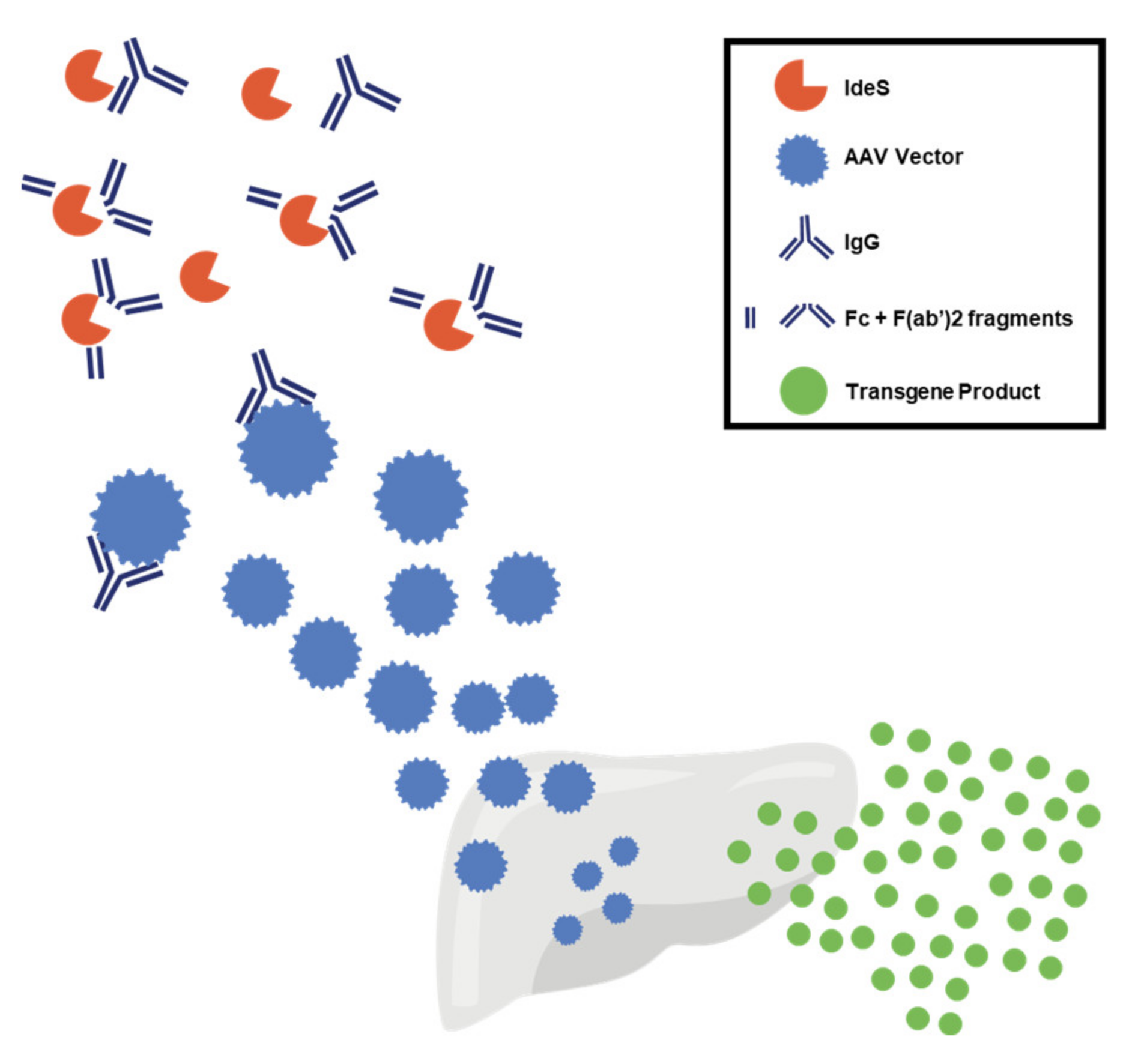
3.1. Preexisting Immunity to AAV Vectors
3.2. Innate Immunity
3.3. Adaptive Immunity
- T-cell reactivity to AAV has, in most trials, been measured with an IFN-γ enzyme- linked immune absorbent spot (ELISpot) assay, which has a limited readout, being focused on a single cytokine, and does not distinguish between T-cell subsets. The use of multicytokine ELISpot-based assays or more complex immunophenotyping of T-cells may help address this limitation.
- In all hemophilia gene transfer trials, immune responses have been monitored using circulating PBMCs, which may not represent the subset of T-cells residing in tissues transduced by AAV vectors. Recent progress toward characterizing and phenotyping of CD8+ T-cells that home to tissues, which may present specific surface markers detectable by flow cytometry [90], may help address this limitation of current immune-monitoring technologies.
- Importantly, the use of immunomodulatory regimens in gene transfer, although essential to maintain long-term expression following gene transfer, may limit or confound the ability to correlate immunology readouts with outcomes.
4. Mitigation Strategies to Overcome Vector Immunogenicity
5. Inhibitors: Transgene-Related Immune Responses
6. Immune Tolerance Induction
Regulatory T-Cells and Liver Tolerance
7. AAV Vector Integration into the Host Genome
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nathwani, A.C.; Davidoff, A.M.; Tuddenham, E.G.D. Advances in Gene Therapy for Hemophilia. Hum. Gene Ther. 2017, 28, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Doshi, B.S.; Arruda, V.R. Gene therapy for hemophilia: What does the future hold? Ther. Adv. Hematol. 2018, 9, 273–293. [Google Scholar] [CrossRef]
- Srivastava, A.; Santagostino, E.; Dougall, A.; Kitchen, S.; Sutherland, M.; Pipe, S.W.; Carcao, M.; Mahlangu, J.; Ragni, M.V.; Windyga, J.; et al. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia 2020, 26, 1–158. [Google Scholar] [CrossRef]
- Verdera, H.C.; Kuranda, K.; Mingozzi, F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. 2020, 28, 723–746. [Google Scholar] [CrossRef] [PubMed]
- Konkle, B.A.; Walsh, C.E.; Escobar, M.A.; Josephson, N.C.; Young, G.; von Drygalski, A.; McPhee, S.W.J.; Samulski, R.J.; Bilic, I.; de la Rosa, M.; et al. BAX 335 hemophilia B gene therapy clinical trial results: Potential impact of CpG sequences on gene expression. Blood 2021, 137, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, A.C.; Davidoff, A.M.; Tuddenham, E.G. Gene Therapy for Hemophilia. Hematol. Clin. N. Am. 2017, 31, 853–868. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, F.; Garagiola, I. Clinical advances in gene therapy updates on clinical trials of gene therapy in haemophilia. Haemophilia 2019, 25, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Pasi, K.J.; Rangarajan, S.; Mitchell, N.; Lester, W.; Symington, E.; Madan, B.; Laffan, M.; Russell, C.B.; Li, M.; Pierce, G.F.; et al. Multiyear Follow-up of AAV5-hFVIII-SQ Gene Therapy for Hemophilia A. N. Engl. J. Med. 2020, 382, 29–40. [Google Scholar] [CrossRef]
- Miesbach, W.; Meijer, K.; Coppens, M.; Kampmann, P.; Klamroth, R.; Schutgens, R.; Tangelder, M.; Castaman, G.; Schwäble, J.; Bonig, H.; et al. Gene therapy with adeno-associated virus vector 5–human factor IX in adults with hemophilia B. Blood 2018, 131, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, A.C.; Reiss, U.M.; Tuddenham, E.G.; Rosales, C.; Chowdary, P.; McIntosh, J.; Della Peruta, M.; Lheriteau, E.; Patel, N.; Raj, D.; et al. Long-Term Safety and Efficacy of Factor IX Gene Therapy in Hemophilia B. N. Engl. J. Med. 2014, 371, 1994–2004. [Google Scholar] [CrossRef]
- George, L.A.; Sullivan, S.K.; Giermasz, A.; Rasko, J.E.; Samelson-Jones, B.J.; Ducore, J.; Cuker, A.; Sullivan, L.M.; Majumdar, S.; Teitel, J.; et al. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N. Engl. J. Med. 2017, 377, 2215–2227. [Google Scholar] [CrossRef]
- Manno, C.S.; Pierce, G.F.; Arruda, V.R.; Glader, B.; Ragni, M.; Rasko, J.J.; Ozelo, M.C.; Hoots, K.; Blatt, P.; Konkle, B.; et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006, 12, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Manno, C.S.; Chew, A.J.; Hutchison, S.; Larson, P.J.; Herzog, R.W.; Arruda, V.R.; Tai, S.J.; Ragni, M.V.; Thompson, A.; Ozelo, M.; et al. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood 2003, 101, 2963–2972. [Google Scholar] [CrossRef] [PubMed]
- Simioni, P.; Tormene, D.; Tognin, G.; Gavasso, S.; Bulato, C.; Iacobelli, N.P.; Finn, J.D.; Spiezia, L.; Radu, C.; Arruda, V.R. X-Linked Thrombophilia with a Mutant Factor IX (Factor IX Padua). N. Engl. J. Med. 2009, 361, 1671–1675. [Google Scholar] [CrossRef] [PubMed]
- Monahan, P.E.; Sun, J.; Gui, T.; Hu, G.; Hannah, W.B.; Wichlan, D.G.; Wu, Z.; Grieger, J.C.; Li, C.; Suwanmanee, T.; et al. Employing a Gain-of-Function Factor IX Variant R338L to Advance the Efficacy and Safety of Hemophilia B Human Gene Therapy: Preclinical Evaluation Supporting an Ongoing Adeno-Associated Virus Clinical Trial. Hum. Gene Ther. 2015, 26, 69–81. [Google Scholar] [CrossRef]
- Mingozzi, F.; Maus, M.V.; Hui, D.J.; Sabatino, D.E.; Murphy, S.L.; Rasko, J.E.J.; Ragni, M.V.; Manno, C.S.; Sommer, J.; Jiang, H.; et al. CD8+ T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 2007, 13, 419–422. [Google Scholar] [CrossRef] [PubMed]
- George, L.A.; Ragni, M.V.; Rasko, J.E.; Raffini, L.J.; Samelson-Jones, B.J.; Ozelo, M.; Hazbon, M.; Runowski, A.R.; Wellman, J.A.; Wachtel, K.; et al. Long-Term Follow-Up of the First in Human Intravascular Delivery of AAV for Gene Transfer: AAV2-hFIX16 for Severe Hemophilia B. Mol. Ther. 2020, 28, 2073–2082. [Google Scholar] [CrossRef]
- Gao, G.-P.; Alvira, M.R.; Wang, L.; Calcedo, R.; Johnston, J.; Wilson, J.M. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA 2002, 99, 11854–11859. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Reiss, U.; Tuddenham, E.; Chowdary, P.; McIntosh, J.; Riddell, A.; Pie, J.; Mahlangu, J.N.; Recht, M.; Shen, Y.-M.; et al. Adeno-Associated Mediated Gene Transfer for Hemophilia B:8 Year Follow up and Impact of Removing "Empty Viral Particles" on Safety and Efficacy of Gene Transfer. Blood 2018, 132, 491. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Tuddenham, E.G.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-Associated Virus Vector–Mediated Gene Transfer in Hemophilia B. N. Engl. J. Med. 2011, 365, 2357–2365. [Google Scholar] [CrossRef]
- Pipe, S.; Stine, K.; Rajasekhar, A.; Everington, T.; Poma, A.; Crombez, E.; Hay, C.R. 101HEMB01 is a phase 1/2 open-label, single ascending dose-finding trial of DTX101 (AAVrh10FIX) in patients with moderate/severe hemophilia B that demonstrated meaningful but transient expression of human Factor IX (hFIX). Blood 2017, 130, 3331. [Google Scholar]
- High, K.A.; Anguela, X. Modified Factor IX, and Compositions, Methods and Uses for Gene Transfer to Cells, Organs, and Tissues. U.S. Patent 10,799,566, 29 December 2016. [Google Scholar]
- Samelson-Jones, B.J.; Finn, J.D.; George, L.A.; Camire, R.M.; Arruda, V.R. Hyperactivity of factor IX Padua (R338L) depends on factor VIIIa cofactor activity. JCI Insight 2019, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Martino, A.T.; Suzuki, M.; Markusic, D.M.; Zolotukhin, I.; Ryals, R.C.; Moghimi, B.; Ertl, H.C.J.; Muruve, D.A.; Lee, B.; Herzog, R.W. The genome of self-complementary adeno-associated viral vectors increases Toll-like receptor 9–dependent innate immune responses in the liver. Blood 2011, 117, 6459–6468. [Google Scholar] [CrossRef]
- Rogers, G.L.; Shirley, J.L.; Zolotukhin, I.; Kumar, S.R.P.; Sherman, A.; Perrin, G.Q.; Hoffman, B.E.; Srivastava, A.; Basner-Tschakarjan, E.; Wallet, M.A.; et al. Plasmacytoid and conventional dendritic cells cooperate in crosspriming AAV capsid-specific CD8+ T cells. Blood 2017, 129, 3184–3195. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Huang, X.; Yang, Y. The TLR9-MyD88 pathway is critical for adaptive immune responses to adeno-associated virus gene therapy vectors in mice. J. Clin. Investig. 2009, 119, 2388–2398. [Google Scholar] [CrossRef]
- Shirley, J.L.; Keeler, G.D.; Sherman, A.; Zolotukhin, I.; Markusic, D.M.; Hoffman, B.E.; Morel, L.M.; Wallet, M.A.; Terhorst, C.; Herzog, R.W. Type I IFN Sensing by cDCs and CD4+ T Cell Help Are Both Requisite for Cross-Priming of AAV Capsid-Specific CD8+ T Cells. Mol. Ther. 2020, 28, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Kuranda, K.; Jean-Alphonse, P.; Leborgne, C.; Hardet, R.; Collaud, F.; Marmier, S.; Verdera, H.C.; Ronzitti, G.; Veron, P.; Mingozzi, F. Exposure to wild-type AAV drives distinct capsid immunity profiles in humans. J. Clin. Investig. 2018, 128, 5267–5279. [Google Scholar] [CrossRef]
- Xiang, Z.; Kurupati, R.K.; Li, Y.; Kuranda, K.; Zhou, X.; Mingozzi, F.; High, K.A.; Ertl, H.C. The Effect of CpG Sequences on Capsid-Specific CD8+ T Cell Responses to AAV Vector Gene Transfer. Mol. Ther. 2020, 28, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.F. Quantification of CpG Motifs in rAAV Genomes: Avoiding the Toll. Mol. Ther. 2020, 28, 1756–1758. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, A.C. Gene therapy for hemophilia. Hematology 2019, 2019, 1–8. [Google Scholar] [CrossRef]
- Leebeek, F.W.; Meijer, K.; Coppens, M.; Kampmann, P.; Klamroth, R.; Schutgens, M.R.; Castaman, G.; Seifried, E.; Schwaeble, J.; Bönig, H.; et al. AMT-060 Gene Therapy in Adults with Severe or Moderate-Severe Hemophilia B Confirm Stable FIX Expression and Durable Reductions in Bleeding and Factor IX Consumption for up to 5 Years. Blood 2020, 136, 26. [Google Scholar] [CrossRef]
- Von Drygalski, A.; Giermasz, A.; Castaman, G.; Key, N.S.; Lattimore, S.; Leebeek, F.W.G.; Miesbach, W.; Recht, M.; Long, A.; Gut, R.; et al. Etranacogene dezaparvovec (AMT-061 phase 2b): Normal/near normal FIX activity and bleed cessation in hemophilia B. Blood Adv. 2019, 3, 3241–3247. [Google Scholar] [CrossRef] [PubMed]
- Pipe, S.W.; Recht, M.; Key, N.S.; Leebeek, F.W.; Castaman, G.; Lattimore, S.U.; Van Der Valk, P.; Peerlinck, K.; Coppens, M.; O’Connell, N.; et al. First Data from the Phase 3 HOPE-B Gene Therapy Trial: Efficacy and Safety of Etranacogene Dezaparvovec (AAV5-Padua hFIX variant; AMT-061) in Adults with Severe or Moderate-Severe Hemophilia B Treated Irrespective of Pre-Existing Anti-Capsid Neutralizing Antibodies. Blood 2020, 136, 6. [Google Scholar] [CrossRef]
- George, L.E.E.; Ragni, M.; Sullivan, S.; Samelson-Jones, B.; Evans, M.; MacDougall, A.; Curran, M.; Tompkins, S.; Wachtel, K.; Takefman, D.; et al. Phase I/II Trial of SPK-8011: Stable and Durable FVIII Expression for >2 Years with Significant ABR Improvements in Initial Dose Cohorts Following AAV-Mediated FVIII Gene Transfer for Hemophilia A [abstract]. Res. Pract. Thromb. Haemost. 2020, 4 (Suppl 1). Available online: https://abstracts.isth.org/abstract/phase-i-ii-trial-of-spk-8011-stable-and-durable-fviii-expression-for-2-years-with-significant-abr-improvements-in-initial-dose-cohorts-following-aav-mediated-fviii-gene-transfer-for-hemophilia-a/ (accessed on 2 March 2021).
- Sullivan, S.K. SPK-8016: Preliminary Results From a Phase 1/2 Clinical Trial of Gene Therapy For Hemophilia A. in European Association for Haemophilia and Allied Disorders (EAHAD). Virtual Haemoph. 2021, 27 (Suppl. 2), 18–181. [Google Scholar]
- Nathwani, A.C.; Tuddenham, E.; Chowdary, P.; McIntosh, J.; Lee, D.; Rosales, C.; Phillips, M.; Pie, J.; Junfang, Z.; Meagher, M.M.; et al. GO-8: Preliminary Results of a Phase I/II Dose Escalation Trial of Gene Therapy for Haemophilia a Using a Novel Human Factor VIII Variant. Blood 2018, 132, 489. [Google Scholar] [CrossRef]
- Leavitt, A.D.; Konkle, B.A.; Stine, K.; Visweshwar, N.; Harrington, T.J.; Giermasz, A.; Arkin, S.; Fang, A.; Plonski, F.; Smith, L.; et al. Updated Follow-up of the Alta Study, a Phase 1/2 Study of Giroctocogene Fitelparvovec (SB-525) Gene Therapy in Adults with Severe Hemophilia a. Blood 2020, 136 (Suppl. 1), 12. [Google Scholar] [CrossRef]
- Chapin, J. Results from a Phase 1/2 Safety and Dose Escalation Study of TAK-754, an AAV8 Vector With a Codon-Optimized B-Domain-Deleted Factor VIII Transgene in Severe Hemophilia A. in European Association for Haemophilia and Allied Disorders (EAHAD). Virtual Haemoph. 2021, 27 (Suppl. 2), 18–181. [Google Scholar]
- Pipe, S.W.; Ferrante, F.; Reis, M.; Wiegmann, S.; Lange, C.; Braun, M.; Michaels, L.A. First-in-Human Gene Therapy Study of AAVhu37 Capsid Vector Technology in Severe Hemophilia A-BAY 2599023 has Broad Patient Eligibility and Stable and Sustained Long-Term Expression of FVIII. Blood 2020, 136, 44–45. [Google Scholar] [CrossRef]
- Lind, P.; Larsson, K.; Spira, J.; Sydow-Bäckman, M.; Almstedt, A.; Gray, E.; Sandberg, H. Novel Forms of B-Domain-Deleted Recombinant Factor VIII Molecules. Construction and Biochemical Characterization. JBIC J. Biol. Inorg. Chem. 1995, 232, 19–27. [Google Scholar] [CrossRef]
- Samelson-Jones, B.J.; Arruda, V.R. Translational Potential of Immune Tolerance Induction by AAV Liver-Directed Factor VIII Gene Therapy for Hemophilia A. Front. Immunol. 2020, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.; Lenting, P.J.; Rosales, C.; Lee, D.; Rabbanian, S.; Raj, D.; Patel, N.; Tuddenham, E.G.D.; Christophe, O.D.; McVey, J.H.; et al. Therapeutic levels of FVIII following a single peripheral vein administration of rAAV vector encoding a novel human factor VIII variant. Blood 2013, 121, 3335–3344. [Google Scholar] [CrossRef]
- Ward, N.N.; Buckley, S.M.K.S.; Waddington, S.S.; VandenDriessche, T.; Chuah, M.K.L.; Nathwani, A.A.; McIntosh, J.J.; Tuddenham, E.G.D.E.; Kinnon, C.C.; Thrasher, A.; et al. Codon optimization of human factor VIII cDNAs leads to high-level expression. Blood 2011, 117, 798–807. [Google Scholar] [CrossRef]
- Long, B.R.; Veron, P.; Kuranda, K.; Hardet, R.; Mitchell, N.; Hayes, G.M.; Wong, W.Y.; Lau, K.; Li, M.; Hock, M.B.; et al. Early Phase Clinical Immunogenicity of Valoctocogene Roxaparvovec, an AAV5-Mediated Gene Therapy for Hemophilia A. Mol. Ther. 2021, 29, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, S.; Walsh, L.; Lester, W.; Perry, D.; Madan, B.; Laffan, M.; Yu, H.; Vettermann, C.; Pierce, G.F.; Wong, W.Y.; et al. AAV5–Factor VIII Gene Transfer in Severe Hemophilia A. N. Engl. J. Med. 2017, 377, 2519–2530. [Google Scholar] [CrossRef]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; Hasbrouck, N.C.; Basner-Tschakarjan, E.; Edmonson, S.A.; Hui, D.J.; Sabatino, D.E.; Zhou, S.; Wright, J.F.; Jiang, H.; Pierce, G.F.; et al. Modulation of tolerance to the transgene product in a nonhuman primate model of AAV-mediated gene transfer to liver. Blood 2007, 110, 2334–2341. [Google Scholar] [CrossRef]
- Hösel, M.; Broxtermann, M.; Janicki, H.; Esser, K.; Arzberger, S.; Hartmann, P.; Gillen, S.; Kleeff, J.; Stabenow, D.; Odenthal, M.; et al. Toll-like receptor 2-mediated innate immune response in human nonparenchymal liver cells toward adeno-associated viral vectors. Hepatology 2011, 55, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Fields, P.A.; Arruda, V.R.; Armstrong, E.; Chu, K.; Mingozzi, F.; Hagstrom, J.; Herzog, R.W.; High, K.A. Risk and Prevention of Anti-factor IX Formation in AAV-Mediated Gene Transfer in the Context of a Large Deletion of F9. Mol. Ther. 2001, 4, 201–210. [Google Scholar] [CrossRef]
- National Hemophilia Foundation. HIV/AIDS. 2020. Available online: https://www.hemophilia.org/Bleeding-Disorders/Blood-Safety/HIVAIDS (accessed on 1 May 2021).
- Riley, L.; Womack, W. Hepatitis and Hemophilia; National Hemophilia Foundation: New York, NY, USA, 2012; Chapter 10; pp. 1–20. Available online: https://www.hemophilia.org/sites/default/files/document/files/nurses-guide-chapter-10-hepatitis-and-hemophilia.pdf (accessed on 2 March 2021).
- Mazepa, M.A.; Monahan, P.E.; Baker, J.R.; Riske, B.K.; Soucie, J.M. Men with severe hemophilia in the United States: Birth cohort analysis of a large national database. Blood 2016, 127, 3073–3081. [Google Scholar] [CrossRef] [PubMed]
- Hösel, M.; Lucifora, J.; Michler, T.; Holz, G.; Gruffaz, M.; Stahnke, S.; Zoulim, F.; Durantel, D.; Heikenwalder, M.; Nierhoff, D.; et al. Hepatitis B virus infection enhances susceptibility toward adeno-associated viral vector transduction in vitro and in vivo. Hepatology 2014, 59, 2110–2120. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A. Adeno-Associated Virus: The Naturally Occurring Virus Versus the Recombinant Vector. Hum. Gene Ther. 2016, 27, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Montus, M.F.; Masurier, C. Prevalence of Serum IgG and Neutralizing Factors Against Adeno-Associated Virus (AAV) Types 1, 2, 5, 6, 8, and 9 in the Healthy Population: Implications for Gene Therapy Using AAV Vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef]
- Kruzik, A.; Fetahagic, D.; Hartlieb, B.; Dorn, S.; Koppensteiner, H.; Horling, F.M.; Scheiflinger, F.; Reipert, B.M.; de la Rosa, M. Prevalence of Anti-Adeno-Associated Virus Immune Responses in International Cohorts of Healthy Donors. Mol. Ther. Methods Clin. Dev. 2019, 14, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Narkbunnam, N.; Samulski, R.J.; Asokan, A.; Hu, G.; Jacobson, L.J.; Manco-Johnson, M.J.; Monahan, P.E. Neutralizing antibodies against adeno-associated virus examined prospectively in pediatric patients with hemophilia. Gene Ther. 2011, 19, 288–294. [Google Scholar] [CrossRef]
- Gao, G.; Vandenberghe, L.H.; Alvira, M.R.; Lu, Y.; Calcedo, R.; Zhou, X.; Wilson, J.M. Clades of Adeno-Associated Viruses Are Widely Disseminated in Human Tissues. J. Virol. 2004, 78, 6381–6388. [Google Scholar] [CrossRef]
- Fitzpatrick, Z.; Leborgne, C.; Barbon, E.; Masat, E.; Ronzitti, G.; van Wittenberghe, L.; Vignaud, A.; Collaud, F.; Charles, S.; Sola, M.S.; et al. Influence of Pre-existing Anti-capsid Neutralizing and Binding Antibodies on AAV Vector Transduction. Mol. Ther. Methods Clin. Dev. 2018, 9, 119–129. [Google Scholar] [CrossRef]
- Mimuro, J.; Mizukami, H.; Shima, M.; Matsushita, T.; Taki, M.; Muto, S.; Higasa, S.; Sakai, M.; Ohmori, T.; Madoiwa, S.; et al. The prevalence of neutralizing antibodies against adeno-associated virus capsids is reduced in young Japanese individuals. J. Med. Virol. 2014, 86, 1990–1997. [Google Scholar] [CrossRef]
- Leborgne, C.; Latournerie, V.; Boutin, S.; Desgue, D.; Quéré, A.; Pignot, E.; Collaud, F.; Charles, S.; Sola, M.S.; Masat, E.; et al. Prevalence and long-term monitoring of humoral immunity against adeno-associated virus in Duchenne Muscular Dystrophy patients. Cell. Immunol. 2019, 342, 103780. [Google Scholar] [CrossRef] [PubMed]
- Calcedo, R.; Vandenberghe, L.H.; Gao, G.; Lin, J.; Wilson, J.M. Worldwide Epidemiology of Neutralizing Antibodies to Adeno-Associated Viruses. J. Infect. Dis. 2009, 199, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Huang, W.; Zhang, H.; Wang, Y.; Zhao, J.; Song, A.; Xie, H.; Zhao, C.; Gao, D. Neutralizing antibodies against AAV2, AAV5 and AAV8 in healthy and HIV-1-infected subjects in China: Implications for gene therapy using AAV vectors. Gene Ther. 2014, 21, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, B.H.; Butler, J.A.; Felker, G.M.; Ponikowski, P.; Voors, A.A.; Pogoda, J.M.; Provost, R.; Guerrero, J.L.; Hajjar, R.J.; Zsebo, K.M. Prevalence of AAV1 neutralizing antibodies and consequences for a clinical trial of gene transfer for advanced heart failure. Gene Ther. 2016, 23, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Couto, L.B.; Patarroyo-White, S.; Liu, T.; Nagy, D.; Vargas, J.A.; Zhou, S.; Scallan, C.D.; Sommer, J.; Vijay, S.; et al. Effects of transient immunosuppression on adenoassociated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy. Blood 2006, 108, 3321–3328. [Google Scholar] [CrossRef] [PubMed]
- Falese, L.; Sandza, K.; Yates, B.; Triffault, S.; Gangar, S.; Long, B.; Tsuruda, L.; Carter, B.; Vettermann, C.; Zoog, S.J.; et al. Strategy to detect pre-existing immunity to AAV gene therapy. Gene Ther. 2017, 24, 768–778. [Google Scholar] [CrossRef]
- Meliani, A.; Leborgne, C.; Triffault, S.; Jeanson-Leh, L.; Veron, P.; Mingozzi, F. Determination of Anti-Adeno-Associated Virus Vector Neutralizing Antibody Titer with an In Vitro Reporter System. Hum. Gene Ther. Methods 2015, 26, 45–53. [Google Scholar] [CrossRef]
- Vandamme, C.; Xicluna, R.; Hesnard, L.; Devaux, M.; Jaulin, N.; Guilbaud, M.; Le Duff, J.; Couzinié, C.; Moullier, P.; Saulquin, X.; et al. Tetramer-Based Enrichment of Preexisting Anti-AAV8 CD8+ T Cells in Human Donors Allows the Detection of a TEMRA Subpopulation. Front. Immunol. 2020, 10, 3110. [Google Scholar] [CrossRef]
- Hui, D.J.; Edmonson, S.C.; Podsakoff, G.M.; Pien, G.C.; Ivanciu, L.; Camire, R.M.; Ertl, H.; Mingozzi, F.; High, K.A.; Basner-Tschakarjan, E. AAV capsid CD8+ T-cell epitopes are highly conserved across AAV serotypes. Mol. Ther. Methods Clin. Dev. 2015, 2, 15029. [Google Scholar] [CrossRef]
- Veron, P.; Leborgne, C.; Monteilhet, V.; Boutin, S.; Martin, S.; Moullier, P.; Masurier, C. Humoral and Cellular Capsid-Specific Immune Responses to Adeno-Associated Virus Type 1 in Randomized Healthy Donors. J. Immunol. 2012, 188, 6418–6424. [Google Scholar] [CrossRef]
- Trinchieri, G.; Sher, A. Cooperation of Toll-like receptor signals in innate immune defence. Nat. Rev. Immunol. 2007, 7, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Herzog, R.W.; Cooper, M.; Perrin, G.Q.; Biswas, M.; Martino, A.T.; Morel, L.; Terhorst, C.; Hoffman, B.E. Regulatory T cells and TLR9 activation shape antibody formation to a secreted transgene product in AAV muscle gene transfer. Cell. Immunol. 2019, 342, 103682. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.L.; Suzuki, M.; Zolotukhin, I.; Markusic, D.M.; Morel, L.M.; Lee, B.; Ertl, H.C.; Herzog, R.W. Unique Roles of TLR9- and MyD88-Dependent and -Independent Pathways in Adaptive Immune Responses to AAV-Mediated Gene Transfer. J. Innate Immun. 2015, 7, 302–314. [Google Scholar] [CrossRef]
- Shao, W.; Earley, L.F.; Chai, Z.; Chen, X.; Sun, J.; He, T.; Deng, M.; Hirsch, M.L.; Ting, J.; Samulski, R.J.; et al. Double-stranded RNA innate immune response activation from long-term adeno-associated virus vector transduction. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, A.K.; Cotter, M.J.; White, L.R.; Clark, S.A.; Wong, N.C.W.; Holers, V.M.; Bartlett, J.S.; Muruve, D.A. Complement Is an Essential Component of the Immune Response to Adeno-Associated Virus Vectors. J. Virol. 2008, 82, 2727–2740. [Google Scholar] [CrossRef] [PubMed]
- Denard, J.; Marolleau, B.; Jenny, C.; Rao, T.N.; Fehling, H.J.; Voit, T.; Svinartchouk, F. C-Reactive Protein (CRP) Is Essential for Efficient Systemic Transduction of Recombinant Adeno-Associated Virus Vector 1 (rAAV-1) and rAAV-6 in Mice. J. Virol. 2013, 87, 10784–10791. [Google Scholar] [CrossRef] [PubMed]
- Solid Biosciences Provides SGT-001 Program Update. Available online: https://www.solidbio.com/about/media/press-releases/solid-biosciences-provides-sgt-001-program-update (accessed on 7 October 2020).
- Duan, D. Systemic AAV Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2018, 26, 2337–2356. [Google Scholar] [CrossRef] [PubMed]
- Solid Biosciences Announces FDA Removes Clinical Hold on SGT-001. 2018. Available online: https://investors.solidbio.com/news-releases/news-release-details/solid-biosciences-announces-fda-removes-clinical-hold-sgt-001 (accessed on 1 May 2021).
- Muhuri, M.; Maeda, Y.; Ma, H.; Ram, S.; Fitzgerald, K.A.; Tai, P.W.; Gao, G. Overcoming innate immune barriers that impede AAV gene therapy vectors. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Calcedo, R.; Bell, P.; Lin, J.; Grant, R.L.; Siegel, D.L.; Wilson, J.M. Impact of Pre-Existing Immunity on Gene Transfer to Nonhuman Primate Liver with Adeno-Associated Virus 8 Vectors. Hum. Gene Ther. 2011, 22, 1389–1401. [Google Scholar] [CrossRef]
- Calcedo, R.; Morizono, H.; Wang, L.; McCarter, R.; He, J.; Jones, D.; Batshaw, M.L.; Wilson, J.M. Adeno-Associated Virus Antibody Profiles in Newborns, Children, and Adolescents. Clin. Vaccine Immunol. 2011, 18, 1586–1588. [Google Scholar] [CrossRef]
- Tseng, Y.-S.; Gurda, B.L.; Chipman, P.; McKenna, R.; Afione, S.; Chiorini, J.A.; Muzyczka, N.; Olson, N.H.; Baker, T.S.; Kleinschmidt, J.; et al. Adeno-Associated Virus Serotype 1 (AAV1)- and AAV5-Antibody Complex Structures Reveal Evolutionary Commonalities in Parvovirus Antigenic Reactivity. J. Virol. 2014, 89, 1794–1808. [Google Scholar] [CrossRef]
- Solid Biosciences. Investor Presentation. In Proceedings of the 39th Annual J.P. Healthcare Conference, San Francisco, CA, USA, 11–14 January 2021. [Google Scholar]
- Li, C.; Hirsch, M.; Asokan, A.; Zeithaml, B.; Ma, H.; Kafri, T.; Samulski, R.J. Adeno-Associated Virus Type 2 (AAV2) Capsid-Specific Cytotoxic T Lymphocytes Eliminate Only Vector-Transduced Cells Coexpressing the AAV2 Capsid In Vivo. J. Virol. 2007, 81, 7540–7547. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Murphy, S.L.; Giles-Davis, W.; Edmonson, S.; Xiang, Z.; Li, Y.; Lasaro, M.O.; High, K.A.; Ertl, H.C. Pre-existing AAV Capsid-specific CD8+ T Cells are Unable to Eliminate AAV-transduced Hepatocytes. Mol. Ther. 2007, 15, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Wang, Q.; Dai, Z.; Calcedo, R.; Sun, X.; Li, G.; Wilson, J.M. Adenovirus based vaccines generate cytotoxic t lymphocytes to epitopes of ns1 from dengue virus that are present in all major serotypes. Hum. Gene Ther. 2008, 19, 927–936. [Google Scholar] [CrossRef]
- Buggert, M.; Vella, L.A.; Nguyen, S.; Wu, V.; Chen, Z.; Sekine, T.; Perez-Potti, A.; Maldini, C.R.; Manne, S.; Darko, S.; et al. The Identity of Human Tissue-Emigrant CD8+ T Cells. Cell 2020, 183, 1946–1961.e15. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, A.C. Data update from the B-AMAZE Phase 1/2 Study–Verbrinacogene setparvovec (FLT180a). Freeline Corporate Presentation. Available online: https://www.freeline.life/media/1349/presentation-freeline-company-update-14-dec-2020.pdf (accessed on 26 May 2021).
- Sherman, A.; Biswas, M.; Herzog, R.W. Innovative Approaches for Immune Tolerance to Factor VIII in the Treatment of Hemophilia A. Front. Immunol. 2017, 8, 1604. [Google Scholar] [CrossRef] [PubMed]
- Cao, O.; Dobrzynski, E.; Wang, L.; Nayak, S.; Mingle, B.; Terhorst, C.; Herzog, R.W. Induction and role of regulatory CD4+CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer. Blood 2007, 110, 1132–1140. [Google Scholar] [CrossRef]
- Samelson-Jones, B.J.; Finn, J.D.; Favaro, P.; Wright, J.F.; Arruda, V.R. Timing of Intensive Immunosuppression Impacts Risk of Transgene Antibodies after AAV Gene Therapy in Nonhuman Primates. Mol. Ther. Methods Clin. Dev. 2020, 17, 1129–1138. [Google Scholar] [CrossRef]
- Tse, L.V.; Klinc, K.A.; Madigan, V.J.; Rivera, R.M.C.; Wells, L.F.; Havlik, L.P.; Smith, J.K.; Agbandje-McKenna, M.; Asokan, A. Structure-guided evolution of antigenically distinct adeno-associated virus variants for immune evasion. Proc. Natl. Acad. Sci. USA 2017, 114, E4812–E4821. [Google Scholar] [CrossRef] [PubMed]
- Lisowski, L.; Dane, A.P.; Chu, K.; Zhang, Y.; Cunningham, S.C.; Wilson, E.M.; Nygaard, S.; Grompe, M.; Alexander, I.E.; Kay, M.A. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nat. Cell Biol. 2014, 506, 382–386. [Google Scholar] [CrossRef]
- Ogden, P.J.; Kelsic, E.D.; Sinai, S.; Church, G.M. Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science 2019, 366, 1139–1143. [Google Scholar] [CrossRef]
- Li Ca Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar]
- Kimchi-Sarfaty, C.; Schiller, T.; Hamasaki-Katagiri, N.; Khan, M.A.; Yanover, C.; Sauna, Z.E. Building better drugs: Developing and regulating engineered therapeutic proteins. Trends Pharmacol. Sci. 2013, 34, 534–548. [Google Scholar] [CrossRef]
- Bali, V.; Bebok, Z. Decoding mechanisms by which silent codon changes influence protein biogenesis and function. Int. J. Biochem. Cell Biol. 2015, 64, 58–74. [Google Scholar] [CrossRef]
- Simhadri, V.L.; Hamasaki-Katagiri, N.; Lin, B.; Hunt, R.; Jha, S.; Tseng, S.C.; Wu, A.; Bentley, A.A.; Zichel, R.; Lu, Q.; et al. Single synonymous mutation in factor IX alters protein properties and underlies haemophilia B. J. Med. Genet. 2017, 54, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Knobe, K.E.; Sjrin, E.; Ljung, R.C.R.; Sjörin, E. Why does the mutationG17736AVal107Val (silent) in theF9gene cause mild haemophilia B in five Swedish families? Haemophilia 2008, 14, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Mccarty, D.M.; Monahan, P.E.; Samulski, R.J. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001, 8, 1248–1254. [Google Scholar] [CrossRef]
- Wu, Z.; Sun, J.; Zhang, T.; Yin, C.; Yin, F.; Van Dyke, T.; Samulski, R.J.; Monahan, P.E. Optimization of Self-complementary AAV Vectors for Liver-directed Expression Results in Sustained Correction of Hemophilia B at Low Vector Dose. Mol. Ther. 2008, 16, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Mauro, V.P.; Chappell, S.A. A critical analysis of codon optimization in human therapeutics. Trends Mol. Med. 2014, 20, 604–613. [Google Scholar] [CrossRef]
- Alexaki, A.; Hettiarachchi, G.K.; Athey, J.C.; Katneni, U.; Simhadri, V.; Hamasaki-Katagiri, N.; Nanavaty, P.; Lin, B.; Takeda, K.; Freedberg, D.; et al. Effects of codon optimization on coagulation factor IX translation and structure: Implications for protein and gene therapies. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Mingozzi, F.; Anguela, X.M.; Pavani, G.; Chen, Y.; Davidson, R.J.; Hui, D.J.; Yazicioglu, M.; Elkouby, L.; Hinderer, C.J.; Faella, A.; et al. Overcoming Preexisting Humoral Immunity to AAV Using Capsid Decoys. Sci. Transl. Med. 2013, 5, 194ra92. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; High, K.A. Immune responses to AAV vectors: Overcoming barriers to successful gene therapy. Blood 2013, 122, 23–36. [Google Scholar] [CrossRef]
- Meliani, A.; Boisgerault, F.; Hardet, R.; Marmier, S.; Collaud, F.; Ronzitti, G.; Leborgne, C.; Verdera, H.C.; Sola, M.S.; Charles, S.; et al. Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration. Nat. Commun. 2018, 9, 4098. [Google Scholar] [CrossRef]
- Corti, M.; Cleaver, B.; Clément, N.; Conlon, T.J.; Faris, K.J.; Wang, G.; Benson, J.; Tarantal, A.F.; Fuller, D.; Herzog, R.W.; et al. Evaluation of Readministration of a Recombinant Adeno-Associated Virus Vector Expressing Acid Alpha-Glucosidase in Pompe Disease: Preclinical to Clinical Planning. Hum. Gene Ther. Clin. Dev. 2015, 26, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Unzu, C.; Hervás-Stubbs, S.; Sampedro, A.; Mauleón, I.; Mancheño, U.; Alfaro, C.; De Salamanca, R.E.; Benito, A.; Beattie, S.G.; Petry, H.; et al. Transient and intensive pharmacological immunosuppression fails to improve AAV-based liver gene transfer in non-human primates. J. Transl. Med. 2012, 10, 122. [Google Scholar] [CrossRef] [PubMed]
- Corti, M.; Elder, M.; Falk, D.; Lawson, L.; Smith, B.; Nayak, S.; Conlon, T.; Clément, N.; Erger, K.; Lavassani, E.; et al. B-cell depletion is protective against anti-AAV capsid immune response: A human subject case study. Mol. Ther. Methods Clin. Dev. 2014, 1, 14033. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; Chen, Y.; Edmonson, S.; Zhou, S.; Thurlings, R.M.; Tak, P.P.; High, K.A.; Vervoordeldonk, M.J. Prevalence and pharmacological modulation of humoral immunity to AAV vectors in gene transfer to synovial tissue. Gene Ther. 2012, 20, 417–424. [Google Scholar] [CrossRef]
- Salas, D.; Kwikkers, K.L.; Zabaleta, N.; Bazo, A.; Petry, H.; Van Deventer, S.J.; Aseguinolaza, G.G.; Ferreira, V. Immunoadsorption enables successful rAAV5-mediated repeated hepatic gene delivery in nonhuman primates. Blood Adv. 2019, 3, 2632–2641. [Google Scholar] [CrossRef]
- Monteilhet, V.; Saheb, S.; Boutin, S.; Leborgne, C.; Veron, P.; Montus, M.-F.; Moullier, P.; Benveniste, O.; Masurier, C. A 10 Patient Case Report on the Impact of Plasmapheresis Upon Neutralizing Factors Against Adeno-associated Virus (AAV) Types 1, 2, 6, and 8. Mol. Ther. 2011, 19, 2084–2091. [Google Scholar] [CrossRef]
- Bertin, B.; Veron, P.; Leborgne, C.; Deschamps, J.-Y.; Moullec, S.; Fromes, Y.; Collaud, F.; Boutin, S.; Latournerie, V.; Van Wittenberghe, L.; et al. Capsid-specific removal of circulating antibodies to adeno-associated virus vectors. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, A.; Katz, M.G.; Gubara, S.M.; Fargnoli, A.S.; Fish, K.M.; Weber, T. Successful Transduction with AAV Vectors after Selective Depletion of Anti-AAV Antibodies by Immunoadsorption. Mol. Ther. Methods Clin. Dev. 2020, 16, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Hurlbut, G.D.; Ziegler, R.J.; Nietupski, J.B.; Foley, J.W.; Woodworth, L.A.; Meyers, E.; Bercury, S.D.; Pande, N.N.; Souza, D.W.; Bree, M.P.; et al. Preexisting Immunity and Low Expression in Primates Highlight Translational Challenges for Liver-directed AAV8-mediated Gene Therapy. Mol. Ther. 2010, 18, 1983–1994. [Google Scholar] [CrossRef]
- Chicoine, L.; Montgomery, C.; Bremer, W.; Shontz, K.; Griffin, D.; Heller, K.; Lewis, S.; Malik, V.; Grose, W.; Shilling, C.; et al. Plasmapheresis Eliminates the Negative Impact of AAV Antibodies on Microdystrophin Gene Expression Following Vascular Delivery. Mol. Ther. 2014, 22, 338–347. [Google Scholar] [CrossRef]
- Leborgne, C.; Barbon, E.; Alexander, J.M.; Hanby, H.; Delignat, S.; Cohen, D.M.; Collaud, F.; Muraleetharan, S.; Lupo, D.; Silverberg, J.; et al. IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat. Med. 2020, 26, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.C.; Lorant, T.; Choi, J.; Kjellman, C.; Winstedt, L.; Bengtsson, M.; Zhang, X.; Eich, T.; Toyoda, M.; Eriksson, B.-M.; et al. IgG Endopeptidase in Highly Sensitized Patients Undergoing Transplantation. N. Engl. J. Med. 2017, 377, 442–453. [Google Scholar] [CrossRef]
- Elmore, Z.C.; Oh, D.K.; Simon, K.E.; Fanous, M.M.; Asokan, A. Rescuing AAV gene transfer from neutralizing antibodies with an IgG-degrading enzyme. JCI Insight 2020, 5, e139881. [Google Scholar] [CrossRef]
- Kempton, C.L.; White, G.C. How we treat a hemophilia A patient with a factor VIII inhibitor. Blood 2009, 113, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Witmer, C.; Young, G. Factor VIII inhibitors in hemophilia A: Rationale and latest evidence. Ther. Adv. Hematol. 2012, 4, 59–72. [Google Scholar] [CrossRef]
- Castaman, G.; Matino, D. Hemophilia A and B: Molecular and clinical similarities and differences. Haematology 2019, 104, 1702–1709. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, J.; Pavlova, A. Genetic risk factors for inhibitors to factors VIII and IX. Haemophilia 2006, 12, 15–22. [Google Scholar] [CrossRef]
- Tiegs, G.; Lohse, A.W. Immune tolerance: What is unique about the liver. J. Autoimmunity 2010, 34, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Calne, R.Y.; Sells, R.A.; Pena, J.R.; Davis, D.R.; Millard, P.R.; Herbertson, B.M.; Binns, R.M.; Davies, D.A.L. Induction of Immunological Tolerance by Porcine Liver Allografts. Nat. Cell Biol. 1969, 223, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Grakoui, A.; Crispe, I.N. Presentation of hepatocellular antigens. Cell. Mol. Immunol. 2016, 13, 293–300. [Google Scholar] [CrossRef]
- Mingozzi, F.; Liu, Y.-L.; Dobrzynski, E.; Kaufhold, A.; Liu, J.H.; Wang, Y.; Arruda, V.R.; High, K.A.; Herzog, R.W. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J. Clin. Investig. 2003, 111, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Markusic, D.M.; Hoffman, B.E.; Perrin, G.Q.; Nayak, S.; Wang, X.; Loduca, P.A.; High, K.A.; Herzog, R.W. Effective gene therapy for haemophilic mice with pathogenic factor IX antibodies. EMBO Mol. Med. 2013, 5, 1698–1709. [Google Scholar] [CrossRef]
- Dobrzynski, E.; Mingozzi, F.; Liu, Y.-L.; Bendo, E.; Cao, O.; Wang, L.; Herzog, R.W. Induction of antigen-specific CD4+ T-cell anergy and deletion by in vivo viral gene transfer. Blood 2004, 104, 969–977. [Google Scholar] [CrossRef]
- Ashrani, A.A.; Reding, M.T.; Shet, A.; Osip, J.; Humar, A.; Lake, J.R.; Key, N.S. Successful liver transplantation in a patient with severe haemophilia A and a high-titre factor VIII inhibitor. Haemophilia 2004, 10, 735–737. [Google Scholar] [CrossRef] [PubMed]
- Finn, J.D.; Ozelo, M.C.; Sabatino, D.E.; Franck, H.W.G.; Merricks, E.P.; Crudele, J.M.; Zhou, S.; Kazazian, H.H.; Lillicrap, D.; Nichols, T.C.; et al. Eradication of neutralizing antibodies to factor VIII in canine hemophilia A after liver gene therapy. Blood 2010, 116, 5842–5848. [Google Scholar] [CrossRef] [PubMed]
- Poupiot, J.; Verdera, H.C.; Hardet, R.; Colella, P.; Collaud, F.; Bartolo, L.; Davoust, J.; Sanatine, P.; Mingozzi, F.; Richard, I.; et al. Role of Regulatory T Cell and Effector T Cell Exhaustion in Liver-Mediated Transgene Tolerance in Muscle. Mol. Ther. Methods Clin. Dev. 2019, 15, 83–100. [Google Scholar] [CrossRef]
- Le Guen, V.; Judor, J.-P.; Boeffard, F.; Gauttier, V.; Ferry, N.; Soulillou, J.-P.; Brouard, S.; Conchon, S. Alloantigen gene transfer to hepatocytes promotes tolerance to pancreatic islet graft by inducing CD8 + regulatory T cells. J. Hepatol. 2017, 66, 765–777. [Google Scholar] [CrossRef]
- Breous, E.; Somanathan, S.; Wilson, J.M. BALB/c Mice Show Impaired Hepatic Tolerogenic Response Following AAV Gene Transfer to the Liver. Mol. Ther. 2010, 18, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Keeler, G.D.; Kumar, S.; Palaschak, B.; Silverberg, E.L.; Markusic, D.M.; Jones, N.T.; Hoffman, B.E. Gene Therapy-Induced Antigen-Specific Tregs Inhibit Neuro-inflammation and Reverse Disease in a Mouse Model of Multiple Sclerosis. Mol. Ther. 2018, 26, 173–183. [Google Scholar] [CrossRef]
- Battaglia, M.; Stabilini, A.; Migliavacca, B.; Horejs-Hoeck, J.; Kaupper, T.; Roncarolo, M.-G. Rapamycin Promotes Expansion of Functional CD4+CD25+FOXP3+ Regulatory T Cells of Both Healthy Subjects and Type 1 Diabetic Patients. J. Immunol. 2006, 177, 8338–8347. [Google Scholar] [CrossRef] [PubMed]
- Crudele, J.M.; Finn, J.D.; Siner, J.I.; Martin, N.B.; Niemeyer, G.P.; Zhou, S.; Mingozzi, F.; Lothrop, J.C.D.; Arruda, V.R. AAV liver expression of FIX-Padua prevents and eradicates FIX inhibitor without increasing thrombogenicity in hemophilia B dogs and mice. Blood 2015, 125, 1553–1561. [Google Scholar] [CrossRef]
- Smith, R.H. Adeno-associated virus integration: Virus versus vector. Gene Ther. 2008, 15, 817–822. [Google Scholar] [CrossRef] [PubMed]
- McCarty, D.M.; Young, S.M., Jr.; Samulski, R.J. Integration of adeno-associated virus (AAV) and recombinant AAV vectors. Annu. Rev. Genet. 2004, 38, 819–845. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.-C.; Datta, S.; Imbeaud, S.; Franconi, A.; Mallet, M.; Couchy, G.; Letouzé, E.; Pilati, C.; Verret, B.; Blanc, J.-F.; et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet. 2015, 47, 1187–1193. [Google Scholar] [CrossRef]
- La Bella, T.; Imbeaud, S.; Peneau, C.; Mami, I.; Datta, S.; Bayard, Q.; Caruso, S.; Hirsch, T.Z.; Calderaro, J.; Morcrette, G.; et al. Adeno-associated virus in the liver: Natural history and consequences in tumour development. Gut 2020, 69, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.-C.; Mami, I.; La Bella, T.; Datta, S.; Imbeaud, S.; Franconi, A.; Mallet, M.; Couchy, G.; Letouzé, E.; Pilati, C.; et al. Wild-type AAV Insertions in Hepatocellular Carcinoma Do Not Inform Debate Over Genotoxicity Risk of Vectorized AAV. Mol. Ther. 2016, 24, 660–661. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.N.; Everett, J.K.; Kafle, S.; Roche, A.M.; Raymond, H.E.; Leiby, J.; Wood, C.; Assenmacher, C.-A.; Merricks, E.P.; Long, C.T.; et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021, 39, 47–55. [Google Scholar] [CrossRef]
- Chandler, R.J.; LaFave, M.C.; Varshney, G.K.; Trivedi, N.S.; Carrillo-Carrasco, N.; Senac, J.S.; Wu, W.; Hoffmann, V.; Elkahloun, A.G.; Burgess, S.M.; et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J. Clin. Investig. 2015, 125, 870–880. [Google Scholar] [CrossRef]
- Donsante, A.; Miller, D.; Li, Y.; Vogler, C.; Brunt, E.M.; Russell, D.W.; Sands, M.S. AAV Vector Integration Sites in Mouse Hepatocellular Carcinoma. Science 2007, 317, 477. [Google Scholar] [CrossRef] [PubMed]
- Chandler, R.J.; LaFave, M.C.; Varshney, G.K.; Burgess, S.M.; Venditti, C.P. Genotoxicity in Mice Following AAV Gene Delivery: A Safety Concern for Human Gene Therapy? Mol. Ther. 2016, 24, 198–201. [Google Scholar] [CrossRef]
- Li, H.; Malani, N.; Hamilton, S.R.; Schlachterman, A.; Bussadori, G.; Edmonson, S.E.; Shah, R.; Arruda, V.R.; Mingozzi, F.; Wright, J.F.; et al. Assessing the potential for AAV vector genotoxicity in a murine model. Blood 2011, 117, 3311–3319. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, G.P.; Herzog, R.W.; Mount, J.; Arruda, V.R.; Tillson, D.M.; Hathcock, J.; Van Ginkel, F.W.; High, K.A.; Lothrop, C.D. Long-term correction of inhibitor-prone hemophilia B dogs treated with liver-directed AAV2-mediated factor IX gene therapy. Blood 2009, 113, 797–806. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Rosales, C.; McIntosh, J.; Rastegarlari, G.; Nathwani, D.; Raj, D.; Nawathe, S.; Waddington, S.N.; Bronson, R.; Jackson, S.; et al. Long-term Safety and Efficacy Following Systemic Administration of a Self-complementary AAV Vector Encoding Human FIX Pseudotyped With Serotype 5 and 8 Capsid Proteins. Mol. Ther. 2011, 19, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Venditti, C.P. Safety questions for AAV gene therapy. Nat. Biotechnol. 2021, 39, 24–26. [Google Scholar] [CrossRef] [PubMed]
- uniQure. uniQure Announces FDA Removes Clinical Hold on Hemophilia B Gene Therapy Program. 2021. Available online: https://tools.eurolandir.com/tools/Pressreleases/GetPressRelease/?ID=3902770&lang=en-GB&companycode=nl-qure&v= (accessed on 20 May 2021).
- Dalwadi, D.A.; Torrens, L.; Abril-Fornaguera, J.; Pinyol, R.; Willoughby, C.; Posey, J.; Llovet, J.M.; Lanciault, C.; Russell, D.W.; Grompe, M.; et al. Liver Injury Increases the Incidence of HCC following AAV Gene Therapy in Mice. Mol. Ther. 2021, 29, 680–690. [Google Scholar] [CrossRef]
- Konkle, B.A.; Recht, M.; Hilger, A.; Marks, P. The critical need for postmarketing surveillance in gene therapy for haemophilia. Haemophilia 2021, 27, 126–131. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sponsor | Vector Serotype | Transgene | Manufacturing | Phase | FVIII Range, % of Normal | ClinicalTrials.gov Identifier |
|---|---|---|---|---|---|---|---|
| Valoctocogene roxaparvovec(BMN-270) [8] | Biomarin | AAV5 | FVIII-SQ | Baculovirus/insect cells | 3 | 4–100 a | NCT03370913 NCT03392974 |
| SPK-8011 [35] | Spark | LK03 | FVIII-SQ | Plasmid/mammalian cells | 1/2 | 5.2–19.8 b | NCT03003533 |
| SPK-8016 [36] | Spark | NA | FVIII-SQ | Plasmid/mammalian cells | 1/2 | 6–22 | NCT03734588 |
| AAV2/8-HLP-FVIII-V3 [37] | UCL | AAV8 | FVIII-V3 | Plasmid/mammalian cells | 1/2 | 6–69 | NCT03001830 |
| SB-525 [38] | Sangamo/Pfizer | AAV6 | FVIII-SQ | Baculovirus/insect cells | 1/2 | 56.5–80.1 | NCT03061201 |
| TAK754 (BAX888) [39] | Takeda (Shire) | AAV8 | FVIII-SQ | Plasmid/mammalian cells | 1/2 | NA | NCT03370172 |
| BAY 2,599,023 (DTX201) [40] | Bayer | AAVhu37 | FVIII-SQ | Plasmid/mammalian cells | 1/2 | 5–17 | NCT03588299 |
| Sponsor | Serotype/ Configuration | Number of CpG in ORF | Immune Suppression a | CTL b | Peak FIX | Duration |
|---|---|---|---|---|---|---|
| CHOP, Stanford Avigen [12,17] | AAV2-FIX/ss | 19 (Wild type) | − | ++ | 12% (n = 1) | <3 months |
| UCL, St. Jude [31] | AAV8-FIX/sc | 0 | + | + | 2–11% (n = 10) | >7 years |
| Takeda (Shire) (BAX335) [5] | AAV8-FIX Padua/sc | 99 | ++ | ++ | 4–58% (n = 8) | <3 months for 7/8 subjects >4 years for 1/8 |
| CHOP | AAV8-FIX 19/ss | 94 [22] | ++ | ++ [22] | ND | ND |
| Pfizer (SPK-9001) [11] | AAVSPK-FIX Padua/ss | 0 | + | + | 34% (n = 10) | >1 year |
| UniQure (AMT060) [32] | AAV5-FIX/sc | 0 | + | + | 7% (n = 10) | >4 year |
| Dimension (DTX101) [21] | AAVrh10-FIX/ss | 96 | ++ | ++ | 3–8% (n = 6) | <3 months |
| UniQure (AMT061) [34] | AAV5-FIX Padua/sc | 0 | − | + | 36–51% (n = 3) | >2 years |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monahan, P.E.; Négrier, C.; Tarantino, M.; Valentino, L.A.; Mingozzi, F. Emerging Immunogenicity and Genotoxicity Considerations of Adeno-Associated Virus Vector Gene Therapy for Hemophilia. J. Clin. Med. 2021, 10, 2471. https://doi.org/10.3390/jcm10112471
Monahan PE, Négrier C, Tarantino M, Valentino LA, Mingozzi F. Emerging Immunogenicity and Genotoxicity Considerations of Adeno-Associated Virus Vector Gene Therapy for Hemophilia. Journal of Clinical Medicine. 2021; 10(11):2471. https://doi.org/10.3390/jcm10112471
Chicago/Turabian StyleMonahan, Paul E., Claude Négrier, Michael Tarantino, Leonard A. Valentino, and Federico Mingozzi. 2021. "Emerging Immunogenicity and Genotoxicity Considerations of Adeno-Associated Virus Vector Gene Therapy for Hemophilia" Journal of Clinical Medicine 10, no. 11: 2471. https://doi.org/10.3390/jcm10112471
APA StyleMonahan, P. E., Négrier, C., Tarantino, M., Valentino, L. A., & Mingozzi, F. (2021). Emerging Immunogenicity and Genotoxicity Considerations of Adeno-Associated Virus Vector Gene Therapy for Hemophilia. Journal of Clinical Medicine, 10(11), 2471. https://doi.org/10.3390/jcm10112471