
Genotype-Phenotype Correlations in RP1-Associated Retinal Dystrophies: A Multi-Center Cohort Study in JAPAN

 , , , ,
, , , ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Molecular Genetic Study
2.2.1. Next-Generation Sequencing
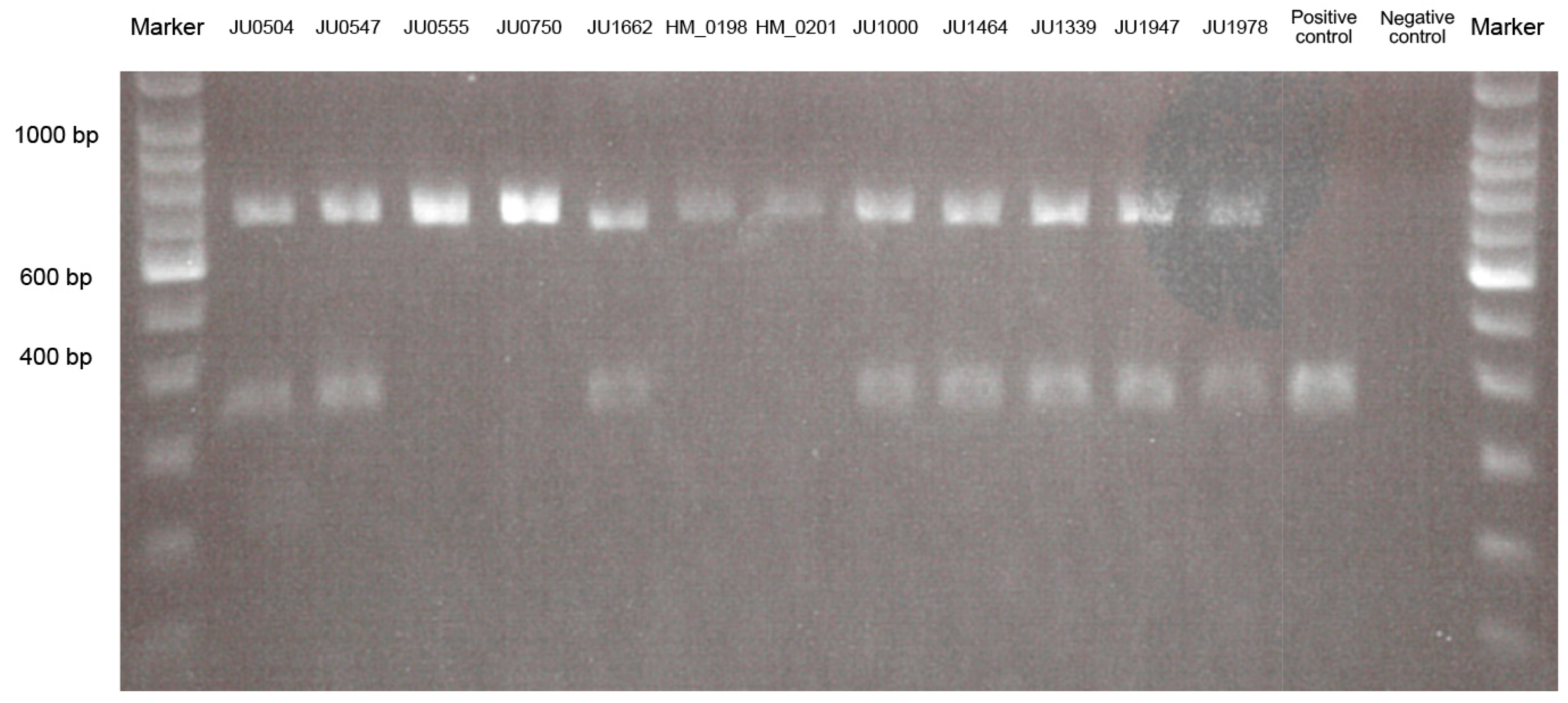
2.2.2. Screening for Alu Element Insertion
2.3. Clinical Examinations
2.4. Statistical Analysis
3. Results
3.1. Molecular Genetic Findings
3.1.1. Autosomal-Dominant Retinitis Pigmentosa
3.1.2. Autosomal-Recessive Retinitis Pigmentosa
3.1.3. Autosomal-Recessive Cone Dystrophy/Cone-Rod Dystrophy
3.1.4. Alu Element Insertion Analysis
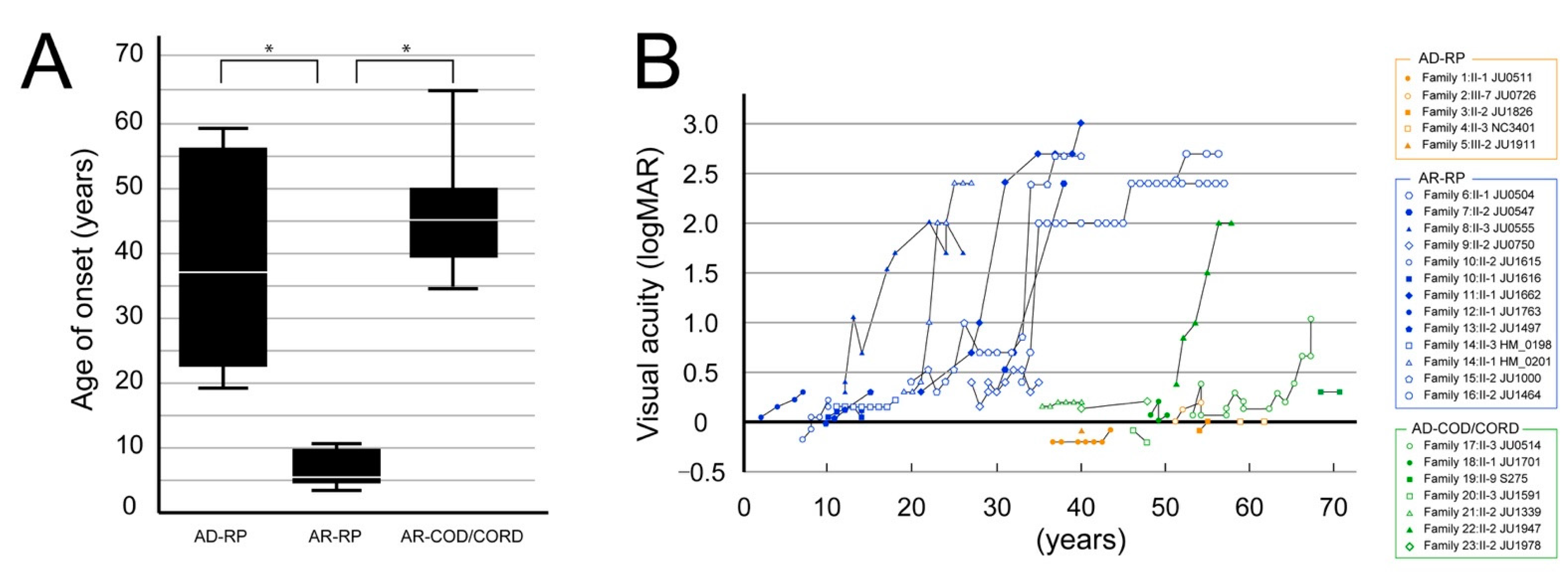
3.2. Clinical Findings
3.2.1. Visual Acuity Assessment
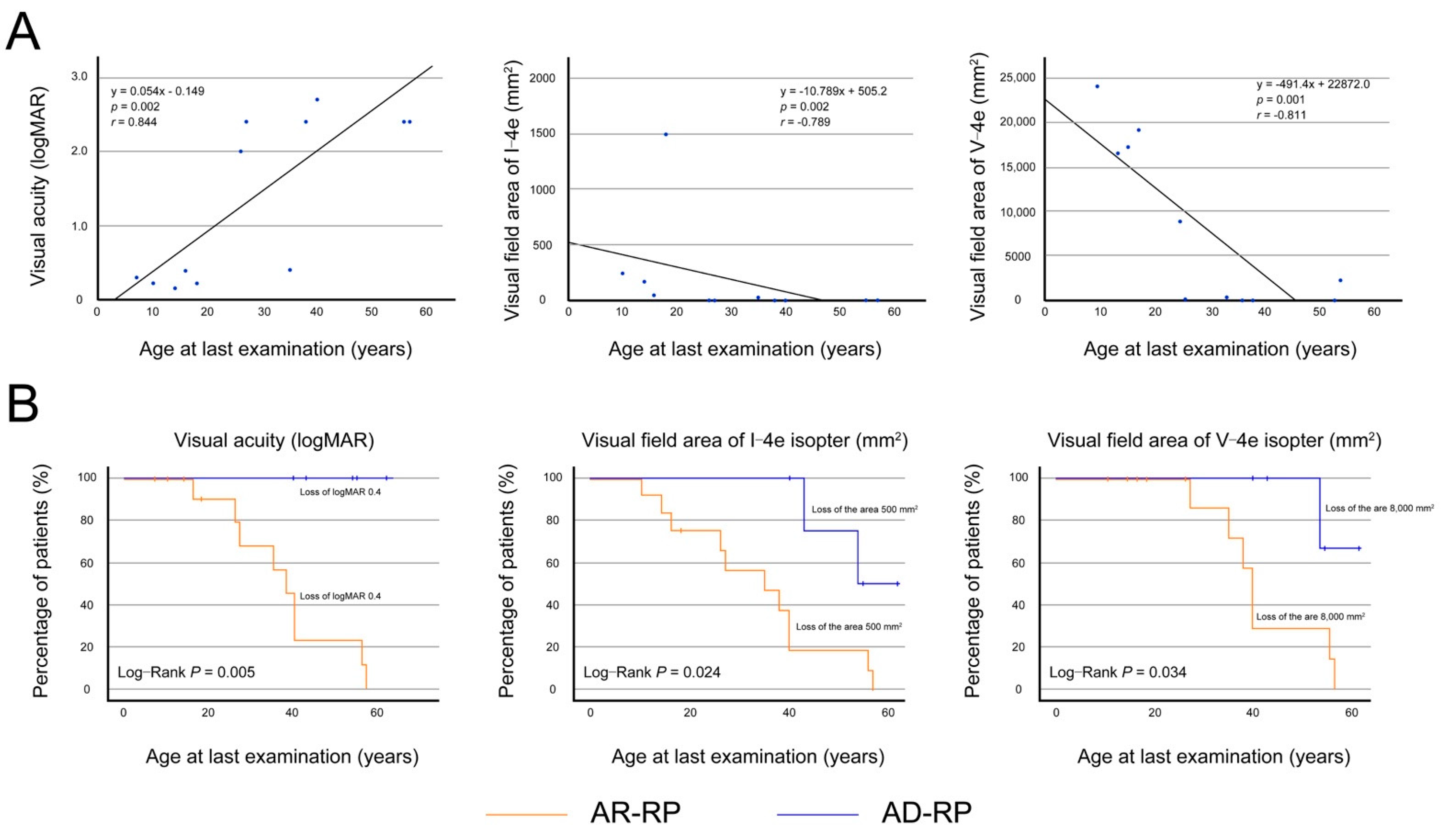
3.2.2. Visual Acuity and Visual Fields in Patients with Autosomal Recessive Retinitis Pigmentosa
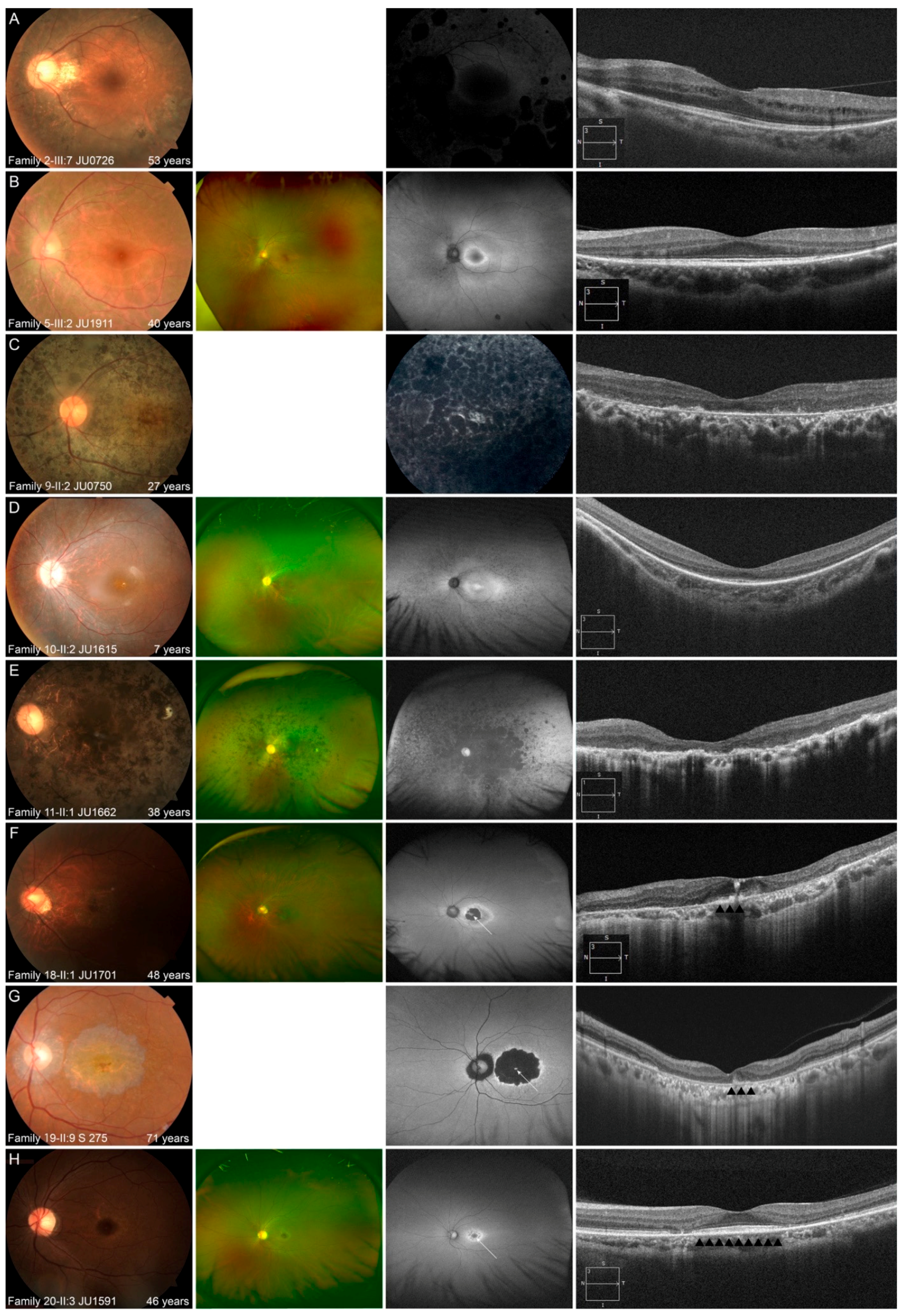
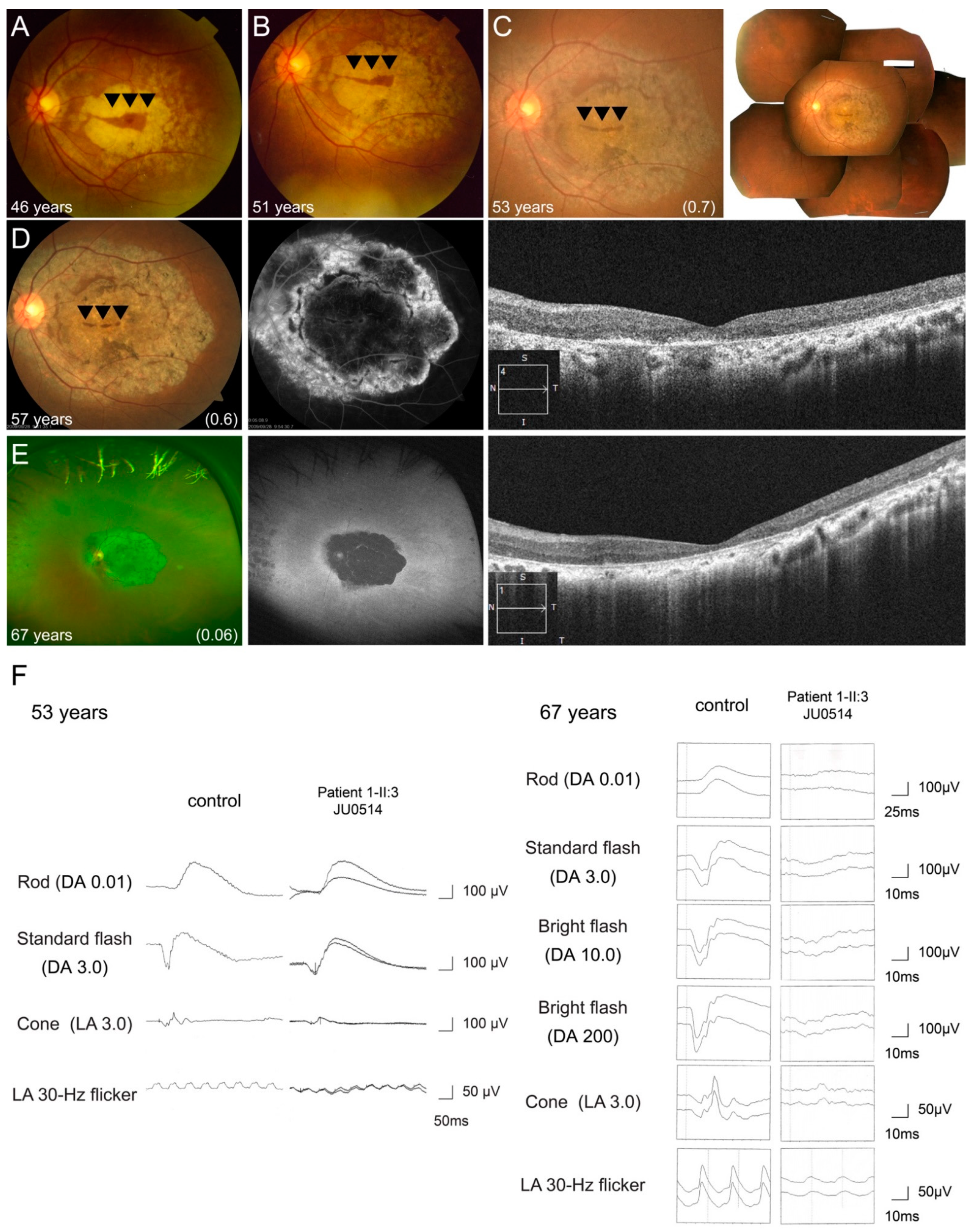
3.2.3. Multimodal Retinal Imaging in Each Phenotype
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bunker, C.H.; Berson, E.L.; Bromley, W.C.; Hayes, R.P.; Roderick, T.H. Prevalence of retinitis pigmentosa in Maine. Am. J. Ophthalmol. 1984, 97, 357–365. [Google Scholar] [CrossRef]
- Guillonneau, X.; Piriev, N.I.; Danciger, M.; Kozak, C.A.; Cideciyan, A.V.; Jacobson, S.G.; Farber, D.B. A nonsense mutation in a novel gene is associated with retinitis pigmentosa in a family linked to the RP1 locus. Hum. Mol. Genet. 1999, 8, 1541–1546. [Google Scholar] [CrossRef] [PubMed]
- Mansergh, F.C.; Millington-Ward, S.; Kennan, A.; Kiang, A.S.; Humphries, M.; Farrar, G.J.; Humphries, P.; Kenna, P.F. Retinitis pigmentosa and progressive sensorineural hearing loss caused by a C12258A mutation in the mitochondrial MTTS2 gene. Am. J. Hum. Genet. 1999, 64, 971–985. [Google Scholar] [CrossRef] [PubMed]
- Rivolta, C.; Sharon, D.; DeAngelis, M.M.; Dryja, T.P. Retinitis pigmentosa and allied diseases: Numerous diseases, genes, and inheritance patterns. Hum. Mol. Genet. 2002, 11, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Pierce, E.A.; Quinn, T.; Meehan, T.; McGee, T.L.; Berson, E.L.; Dryja, T.P. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat. Genet. 1999, 22, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.S.; Heckenlively, J.R.; Bowne, S.J.; Zuo, J.; Hide, W.A.; Gal, A.; Denton, M.; Inglehearn, C.F.; Blanton, S.H.; Daiger, S.P. Mutations in a novel retina-specific gene cause autosomal dominant retinitis pigmentosa. Nat. Genet. 1999, 22, 255–259. [Google Scholar] [CrossRef]
- Liu, Q.; Zuo, J.; Pierce, E.A. The retinitis pigmentosa 1 protein is a photoreceptor microtubule-associated protein. J. Neurosci. 2004, 24, 6427–6436. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, J.; Daiger, S.P.; Farber, D.B.; Heckenlively, J.R.; Smith, J.E.; Sullivan, L.S.; Zuo, J.; Milam, A.H.; Pierce, E.A. Identification and subcellular localization of the RP1 protein in human and mouse photoreceptors. Invest. Ophthalmol. Vis. Sci. 2002, 43, 22–32. [Google Scholar] [PubMed]
- Yamashita, T.; Liu, J.; Gao, J.; LeNoue, S.; Wang, C.; Kaminoh, J.; Bowne, S.J.; Sullivan, L.S.; Daiger, S.P.; Zhang, K.; et al. Essential and synergistic roles of RP1 and RP1L1 in rod photoreceptor axoneme and retinitis pigmentosa. J. Neurosci. 2009, 29, 9748–9760. [Google Scholar] [CrossRef]
- Kaplan, M.W.; Iwata, R.T.; Sears, R.C. Lengths of immunolabeled ciliary microtubules in frog photoreceptor outer segments. Exp. Eye Res. 1987, 44, 623–632. [Google Scholar] [CrossRef]
- Ran, X.; Cai, W.J.; Huang, X.F.; Liu, Q.; Lu, F.; Qu, J.; Wu, J.; Jin, Z.B. ‘RetinoGenetics’: A comprehensive mutation database for genes related to inherited retinal degeneration. Database 2014, 2014. [Google Scholar] [CrossRef]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Avila-Fernandez, A.; Corton, M.; Nishiguchi, K.M.; Munoz-Sanz, N.; Benavides-Mori, B.; Blanco-Kelly, F.; Riveiro-Alvarez, R.; Garcia-Sandoval, B.; Rivolta, C.; Ayuso, C. Identification of an RP1 prevalent founder mutation and related phenotype in Spanish patients with early-onset autosomal recessive retinitis. Ophthalmology 2012, 119, 2616–2621. [Google Scholar] [CrossRef] [PubMed]
- El Shamieh, S.; Boulanger-Scemama, E.; Lancelot, M.E.; Antonio, A.; Demontant, V.; Condroyer, C.; Letexier, M.; Saraiva, J.P.; Mohand-Said, S.; Sahel, J.A.; et al. Targeted next generation sequencing identifies novel mutations in RP1 as a relatively common cause of autosomal recessive rod-cone dystrophy. Biomed. Res. Int. 2015, 2015, 485624. [Google Scholar] [CrossRef] [PubMed]
- Berson, E.L.; Grimsby, J.L.; Adams, S.M.; McGee, T.L.; Sweklo, E.; Pierce, E.A.; Sandberg, M.A.; Dryja, T.P. Clinical features and mutations in patients with dominant retinitis pigmentosa-1 (RP1). Investig. Ophthalmol. Vis. Sci. 2001, 42, 2217–2224. [Google Scholar]
- Liu, Q.; Collin, R.W.; Cremers, F.P.; den Hollander, A.I.; van den Born, L.I.; Pierce, E.A. Expression of wild-type Rp1 protein in Rp1 knock-in mice rescues the retinal degeneration phenotype. PLoS ONE 2012, 7, e43251. [Google Scholar] [CrossRef]
- Siemiatkowska, A.M.; Astuti, G.D.; Arimadyo, K.; den Hollander, A.I.; Faradz, S.M.; Cremers, F.P.; Collin, R.W. Identification of a novel nonsense mutation in RP1 that causes autosomal recessive retinitis pigmentosa in an Indonesian family. Mol. Vis. 2012, 18, 2411–2419. [Google Scholar]
- Lafont, E.L.; Manes, G.L.; Sénéchal, A.; Bocquet, B.A.; Coustès-Chazalette, D.; Baudoin, C.; Ksantini, M.; Bourien, J.R.M.; Devos, A.; Dollfus, H.L.N.; et al. Patients with Retinitis Pigmentosa due to RP1 Mutations Show Greater Severity in Recessive than in Dominant Cases. J. Clin. Exp. Ophthalmol. 2011. [Google Scholar] [CrossRef]
- Nikopoulos, K.; Cisarova, K.; Quinodoz, M.; Koskiniemi-Kuendig, H.; Miyake, N.; Farinelli, P.; Rehman, A.U.; Khan, M.I.; Prunotto, A.; Akiyama, M.; et al. A frequent variant in the Japanese population determines quasi-Mendelian inheritance of rare retinal ciliopathy. Nat. Commun. 2019, 10, 2884. [Google Scholar] [CrossRef]
- Nishiguchi, K.M.; Fujita, K.; Ikeda, Y.; Kunikata, H.; Koyanagi, Y.; Akiyama, M.; Abe, T.; Wada, Y.; Sonoda, K.H.; Nakazawa, T. A founder Alu insertion in RP1 gene in Japanese patients with retinitis pigmentosa. Jpn. J. Ophthalmol. 2020, 64, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Ueno, S.; Koyanagi, Y.; Kominami, T.; Ito, Y.; Kawano, K.; Nishiguchi, K.M.; Rivolta, C.; Nakazawa, T.; Sonoda, K.H.; Terasaki, H. Clinical characteristics and high resolution retinal imaging of retinitis pigmentosa caused by RP1 gene variants. Jpn. J. Ophthalmol. 2020, 64, 485–496. [Google Scholar] [CrossRef]
- Liu, X.; Xiao, J.; Huang, H.; Guan, L.; Zhao, K.; Xu, Q.; Zhang, X.; Pan, X.; Gu, S.; Chen, Y.; et al. Molecular genetic testing in clinical diagnostic assessments that demonstrate correlations in patients with autosomal recessive inherited retinal dystrophy. JAMA Ophthalmol. 2015, 133, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Ellingford, J.M.; Barton, S.; Bhaskar, S.; O’Sullivan, J.; Williams, S.G.; Lamb, J.A.; Panda, B.; Sergouniotis, P.I.; Gillespie, R.L.; Daiger, S.P.; et al. Molecular findings from 537 individuals with inherited retinal disease. J. Med. Genet. 2016, 53, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Verbakel, S.K.; van Huet, R.A.C.; den Hollander, A.I.; Geerlings, M.J.; Kersten, E.; Klevering, B.J.; Klaver, C.C.W.; Plomp, A.S.; Wesseling, N.L.; Bergen, A.A.B.; et al. Macular Dystrophy and Cone-Rod Dystrophy Caused by Mutations in the RP1 Gene: Extending the RP1 Disease Spectrum. Invest. Ophthalmol. Vis. Sci. 2019, 60, 1192–1203. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, S.; Yoshitake, K.; Akahori, M.; Hayashi, T.; Furuno, M.; Nishino, J.; Ikeo, K.; Tsuneoka, H.; Iwata, T. Whole-exome sequencing identifies a novel ALMS1 mutation (p.Q2051X) in two Japanese brothers with Alstrom syndrome. Mol. Vis. 2013, 19, 2393–2406. [Google Scholar]
- Katagiri, S.; Akahori, M.; Sergeev, Y.; Yoshitake, K.; Ikeo, K.; Furuno, M.; Hayashi, T.; Kondo, M.; Ueno, S.; Tsunoda, K.; et al. Whole exome analysis identifies frequent CNGA1 mutations in Japanese population with autosomal recessive retinitis pigmentosa. PLoS ONE 2014, 9, e108721. [Google Scholar] [CrossRef] [PubMed]
- Kubota, D.; Gocho, K.; Kikuchi, S.; Akeo, K.; Miura, M.; Yamaki, K.; Takahashi, H.; Kameya, S. CEP250 mutations associated with mild cone-rod dystrophy and sensorineural hearing loss in a Japanese family. Ophthalmic Genet. 2018, 1–8. [Google Scholar] [CrossRef]
- Katagiri, S.; Hayashi, T.; Nakamura, M.; Mizobuchi, K.; Gekka, T.; Komori, S.; Ueno, S.; Terasaki, H.; Sakuramoto, H.; Kuniyoshi, K.; et al. RDH5-Related Fundus Albipunctatus in a Large Japanese Cohort. Investig. Ophthalmol. Vis. Sci. 2020, 61, 53. [Google Scholar] [CrossRef]
- Mizobuchi, K.; Hayashi, T.; Yoshitake, K.; Fujinami, K.; Tachibana, T.; Tsunoda, K.; Iwata, T.; Nakano, T. Novel homozygous CLN3 missense variant in isolated retinal dystrophy: A case report and electron microscopic findings. Mol. Genet. Genom. Med. 2020, e1308. [Google Scholar] [CrossRef]
- Hayashi, T.; Kameya, S.; Mizobuchi, K.; Kubota, D.; Kikuchi, S.; Yoshitake, K.; Mizota, A.; Murakami, A.; Iwata, T.; Nakano, T. Genetic defects of CHM and visual acuity outcome in 24 choroideremia patients from 16 Japanese families. Sci. Rep. 2020, 10, 15883. [Google Scholar] [CrossRef]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. Adv. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Hayashi, T.; Bedell, M.; Zhang, K.; Yamada, H.; Tsuneoka, H. A novel haplotype with the R345W mutation in the EFEMP1 gene associated with autosomal dominant drusen in a Japanese family. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1643–1650. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, S.; Hosono, K.; Hayashi, T.; Kurata, K.; Mizobuchi, K.; Matsuura, T.; Yoshitake, K.; Iwata, T.; Nakano, T.; Hotta, Y. Early onset flecked retinal dystrophy associated with new compound heterozygous RPE65 variants. Mol. Vis. 2018, 24, 286–296. [Google Scholar] [PubMed]
- Kutsuma, T.; Katagiri, S.; Hayashi, T.; Yoshitake, K.; Iejima, D.; Gekka, T.; Kohzaki, K.; Mizobuchi, K.; Baba, Y.; Terauchi, R.; et al. Novel biallelic loss-of-function KCNV2 variants in cone dystrophy with supernormal rod responses. Doc. Ophthalmol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, W.; Mizobuchi, K.; Hayashi, T.; Okude, S.; Katagiri, S.; Kubo, A.; Masuhara, N.; Nakano, T. Electroretinographic abnormalities associated with pregabalin: A case report. Doc. Ophthalmol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kubota, D.; Matsumoto, K.; Hayashi, M.; Oishi, N.; Gocho, K.; Yamaki, K.; Kobayakawa, S.; Igarashi, T.; Takahashi, H.; Kameya, S. High-resolution photoreceptor imaging analysis of patients with autosomal dominant retinitis pigmentosa (adRP) caused by HK1 mutation. Ophthalmic. Genet. 2020, 41, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Kameya, S.; Fujinami, K.; Ueno, S.; Hayashi, T.; Kuniyoshi, K.; Ideta, R.; Kikuchi, S.; Kubota, D.; Yoshitake, K.; Katagiri, S.; et al. Phenotypical Characteristics of POC1B-Associated Retinopathy in Japanese Cohort: Cone Dystrophy with Normal Funduscopic Appearance. Invest. Ophthalmol. Vis. Sci. 2019, 60, 3432–3446. [Google Scholar] [CrossRef]
- Grover, S.; Fishman, G.A.; Alexander, K.R.; Anderson, R.J.; Derlacki, D.J. Visual acuity impairment in patients with retinitis pigmentosa. Ophthalmology 1996, 103, 1593–1600. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, V.W.; Feng, Y.; Tian, X.; Li, F.Y.; Truong, C.; Wang, G.; Chiang, P.W.; Lewis, R.A.; Wong, L.J. Dependable and efficient clinical utility of target capture-based deep sequencing in molecular diagnosis of retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 2014, 55, 6213–6223. [Google Scholar] [CrossRef] [PubMed]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Kurata, K.; Hosono, K.; Hotta, Y. Clinical and genetic findings of a Japanese patient with RP1-related autosomal recessive retinitis pigmentosa. Doc. Ophthalmol. 2018, 137, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, S.; Sun, W.; Xiao, X.; Jia, X.; Liu, M.; Xu, L.; Long, Y.; Zhang, Q. An Ophthalmic Targeted Exome Sequencing Panel as a Powerful Tool to Identify Causative Mutations in Patients Suspected of Hereditary Eye Diseases. Transl. Vis. Sci. Technol. 2019, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.J.; Lai, T.Y.; Tam, P.O.; Chiang, S.W.; Zhang, X.; Lam, S.; Lai, R.Y.; Lam, D.S.; Pang, C.P. Compound heterozygosity of two novel truncation mutations in RP1 causing autosomal recessive retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2236–2242. [Google Scholar] [CrossRef] [PubMed]
- Audo, I.; Mohand-Saïd, S.; Dhaenens, C.M.; Germain, A.; Orhan, E.; Antonio, A.; Hamel, C.; Sahel, J.A.; Bhattacharya, S.S.; Zeitz, C. RP1 and autosomal dominant rod-cone dystrophy: Novel mutations, a review of published variants, and genotype-phenotype correlation. Hum. Mutat. 2012, 33, 73–80. [Google Scholar] [CrossRef]
- Singh, H.P.; Jalali, S.; Narayanan, R.; Kannabiran, C. Genetic analysis of Indian families with autosomal recessive retinitis pigmentosa by homozygosity screening. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4065–4071. [Google Scholar] [CrossRef]
- Perez-Carro, R.; Corton, M.; Sanchez-Navarro, I.; Zurita, O.; Sanchez-Bolivar, N.; Sanchez-Alcudia, R.; Lelieveld, S.H.; Aller, E.; Lopez-Martinez, M.A.; Lopez-Molina, M.I.; et al. Panel-based NGS Reveals Novel Pathogenic Mutations in Autosomal Recessive Retinitis Pigmentosa. Sci. Rep. 2016, 6, 19531. [Google Scholar] [CrossRef]
- Li, S.; Yang, M.; Liu, W.; Liu, Y.; Zhang, L.; Yang, Y.; Sundaresan, P.; Yang, Z.; Zhu, X. Targeted Next-Generation Sequencing Reveals Novel RP1 Mutations in Autosomal Recessive Retinitis Pigmentosa. Genet. Test. Mol. Biomark. 2018, 22, 109–114. [Google Scholar] [CrossRef]
- Riazuddin, S.A.; Zulfiqar, F.; Zhang, Q.; Sergeev, Y.V.; Qazi, Z.A.; Husnain, T.; Caruso, R.; Riazuddin, S.; Sieving, P.A.; Hejtmancik, J.F. Autosomal recessive retinitis pigmentosa is associated with mutations in RP1 in three consanguineous Pakistani families. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2264–2270. [Google Scholar] [CrossRef]
- Aldahmesh, M.A.; Safieh, L.A.; Alkuraya, H.; Al-Rajhi, A.; Shamseldin, H.; Hashem, M.; Alzahrani, F.; Khan, A.O.; Alqahtani, F.; Rahbeeni, Z.; et al. Molecular characterization of retinitis pigmentosa in Saudi Arabia. Mol. Vis. 2009, 15, 2464–2469. [Google Scholar]
- Zhang, X.; Chen, L.J.; Law, J.P.; Lai, T.Y.; Chiang, S.W.; Tam, P.O.; Chu, K.Y.; Wang, N.; Zhang, M.; Pang, C.P. Differential pattern of RP1 mutations in retinitis pigmentosa. Mol. Vis. 2010, 16, 1353–1360. [Google Scholar]
- Eisenberger, T.; Neuhaus, C.; Khan, A.O.; Decker, C.; Preising, M.N.; Friedburg, C.; Bieg, A.; Gliem, M.; Charbel Issa, P.; Holz, F.G.; et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: The example of retinal dystrophies. PLoS ONE 2013, 8, e78496. [Google Scholar] [CrossRef]
- Xu, Y.; Guan, L.; Shen, T.; Zhang, J.; Xiao, X.; Jiang, H.; Li, S.; Yang, J.; Jia, X.; Yin, Y.; et al. Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Hum. Genet. 2014, 133, 1255–1271. [Google Scholar] [CrossRef] [PubMed]
- Beheshtian, M.; Saee Rad, S.; Babanejad, M.; Mohseni, M.; Hashemi, H.; Eshghabadi, A.; Hajizadeh, F.; Akbari, M.R.; Kahrizi, K.; Riazi Esfahani, M.; et al. Impact of whole exome sequencing among Iranian patients with autosomal recessive retinitis pigmentosa. Arch. Iran. Med. 2015, 18, 776–785. [Google Scholar] [PubMed]
- Zhao, L.; Wang, F.; Wang, H.; Li, Y.; Alexander, S.; Wang, K.; Willoughby, C.E.; Zaneveld, J.E.; Jiang, L.; Soens, Z.T.; et al. Next-generation sequencing-based molecular diagnosis of 82 retinitis pigmentosa probands from Northern Ireland. Hum. Genet. 2015, 134, 217–230. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, Q.; Huang, X.; Qu, C.; Ma, S.; Mao, Y.; Yang, J.; Li, Y.; Li, Y.; Tan, C.; et al. Mutation screening in genes known to be responsible for Retinitis Pigmentosa in 98 Small Han Chinese Families. Sci. Rep. 2017, 7, 1948. [Google Scholar] [CrossRef] [PubMed]
- Ezquerra-Inchausti, M.; Anasagasti, A.; Barandika, O.; Garay-Aramburu, G.; Galdós, M.; López de Munain, A.; Irigoyen, C.; Ruiz-Ederra, J. A new approach based on targeted pooled DNA sequencing identifies novel mutations in patients with Inherited Retinal Dystrophies. Sci. Rep. 2018, 8, 15457. [Google Scholar] [CrossRef]
- Neveling, K.; Collin, R.W.; Gilissen, C.; van Huet, R.A.; Visser, L.; Kwint, M.P.; Gijsen, S.J.; Zonneveld, M.N.; Wieskamp, N.; de Ligt, J.; et al. Next-generation genetic testing for retinitis pigmentosa. Hum. Mutat. 2012, 33, 963–972. [Google Scholar] [CrossRef]
- Mendez-Vidal, C.; Bravo-Gil, N.; Gonzalez-Del Pozo, M.; Vela-Boza, A.; Dopazo, J.; Borrego, S.; Antinolo, G. Novel RP1 mutations and a recurrent BBS1 variant explain the co-existence of two distinct retinal phenotypes in the same pedigree. BMC Genet. 2014, 15, 143. [Google Scholar] [CrossRef]
- Leitch, C.C.; Zaghloul, N.A.; Davis, E.E.; Stoetzel, C.; Diaz-Font, A.; Rix, S.; Alfadhel, M.; Lewis, R.A.; Eyaid, W.; Banin, E.; et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 2008, 40, 443–448. [Google Scholar] [CrossRef]
- Badano, J.L.; Katsanis, N. Beyond Mendel: An evolving view of human genetic disease transmission. Nat. Rev. Genet. 2002, 3, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Alfano, G.; Kruczek, P.M.; Shah, A.Z.; Kramarz, B.; Jeffery, G.; Zelhof, A.C.; Bhattacharya, S.S. EYS Is a Protein Associated with the Ciliary Axoneme in Rods and Cones. PLoS ONE 2016, 11, e0166397. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family ID Patient ID/Affeceted Number | Nucleotide Change Protein Change | Zygosity | Classification | Phenotype | dP SNP ID | Frequency in Database (%) | ACMG | Known Novel | ||
|---|---|---|---|---|---|---|---|---|---|---|
| HGVD | ToMMo | gnomAD | Classification Criteria | |||||||
| Family 1 JU0511/1 | c.2029C > T (p.Arg677Ter) | Hetero | Exon 4 Class II | AD-RP | rs104894082 | NR | NR | NR | Likely pathogenic (PM, PM2, PM4, PP3, PP4, PP5) | Known |
| Family 2 JU0726/1 | c.2599A > T (p.Lys867Ter) | Hetero | Exon 4 Class II | AD-RP | NR | NR | NR | NR | Likely pathogenic (PM, PM2, PM4, PP3, PP4, PP5) | Novel |
| Family 3 JU1826/1 | c.2557A > T (p.Lys853Ter) | Hetero | Exon 4 Class II | AD-RP | NR | NR | NR | NR | Likely pathogenic (PM1, PM2, PM4, PP4) | Known |
| Family 4 NC3401/1 | c.2613dupA (p.Arg872ThrfsTer2) | Hetero | Exon 4 Class II | AD-RP | rs1449723475 | NR | NR | NR | Likely pathogenic (PM1, PM2, PM4, PP3, PP4, PP5) | Known |
| Family 5 JU1911/1 | c.2032C > T (p.Gln678Ter) | Hetero | Exon 4 Class II | AD-RP | rs878853328 | NR | NR | 0.000 | Likely pathogenic (PM1,PM2, PM4, PP3, PP4, PP5) | Novel |
| Family 6 JU0504/1 | c.473T > G (p.Val158Gly) | Compound hetero | Exon 2 Class I | AR-RP | NR | NR | NR | NR | Likely pathogenic (PM1, PM2, PM3, PP3) | Known |
| c.4052_4053ins328 (p.Tyr1352AlafsTer9) | Exon 4 Class III | rs775253277 | NR | NR | NR | Pathogenic (PVS1, PM2, PM4, PP3) | Novel | |||
| Family 7-JU0547/1 | c.2020dupA (p.Ser676IlefsTer22) | Compound hetero | Exon 4 Class II | AR-RP | NR | NR | NR | NR | Pathogenic (PVS1, PM2, PM3, PM4) | Known |
| c.4052_4053ins328 (p.Tyr1352AlafsTer9) | Exon 4 Class III | rs775253277 | NR | NR | NR | Pathogenic (PVS1, PM2, PM4, PP3) | Known | |||
| Family 8 JU0555/1 | c.4052_4053ins328 (p.Tyr1352AlafsTer9) | Homo | Exon 4 Class III | AR-RP | rs775253277 | NR | NR | NR | Pathogenic (PVS1, PM2, PM4, PP3) | Known |
| Family 9 JU0750/1 | c.4052_4053ins328 (p.Tyr1352AlafsTer9) | Homo | Exon 4 Class III | AR-RP | rs775253277 | NR | NR | NR | Pathogenic (PVS1, PM2, PM4, PP3) | Known |
| Family 10 JU1615, JU1616/2 | c.3669C > A (p.Cys1223Ter) | Compound hetero | Exon 4 Class II | AR-RP | rs765129639 | NR | 0.000 | 0.000 | Pathogenic (PVS1, PM2, PM4, PP1, PP3, PP5) | Novel |
| c.4400delC (p.Ser1467PhefsTer5) | Exon 4 Class III | NR | NR | NR | NR | Pathogenic (PVS1, PM2, PM3, PM4, PP1) | Known | |||
| Family 11 JU1662/1 | c.4052_4053ins328 (p.Tyr1352AlafsTer9) | Compound hetero | Exon 4 Class III | AR-RP | rs775253277 | NR | NR | NR | Pathogenic (PVS1, PM2, PM4, PP3) | Known |
| c.4196delG (p.Cys1399LeufsTer5) | Exon 4 Class III | rs762951570 | NR | 0.000 | 0.000 | Pathogenic (PVS1, PM2, PM3, PM4, PP5) | Known | |||
| Family 12-JU1763/1 | c.4196delG (p.Cys1399LeufsTer5) | Homo | Exon 4 Class III | AR-RP | rs762951570 | NR | 0.000 | 0.000 | Pathogenic (PVS1, PM2, PM4, PP5) | Known |
| Family 13- JU1497/1 | c.4196delG (p.Cys1399LeufsTer5) | Compound hetero | Exon 4 Class III | AR-RP | rs762951570 | NR | 0.000 | 0.000 | Pathogenic (PVS1, PM2, PM4, PP5) | Novel |
| c.4591_4592delAG (p.Arg1531AlafsTer12) | Exon 4 Class III | NR | NR | NR | NR | Pathogenic (PVS1, PM2, PM3, PM4) | Known | |||
| Family 14 HM_0198, HM_0201/2 | c.4052_4053ins328 (p.Tyr1352AlafsTer9) | Homo | Exon 4 Class III | AR-RP | rs775253277 | NR | NR | NR | Pathogenic (PVS1, PM2, PM4, PP3) | Novel |
| Family 15 JU1000/1 | c.3843dupT (p.Pro1282SerfsTer2) | Compound hetero | Exon 4 Class III | AR-RP | NR | NR | NR | NR | Pathogenic (PVS1, PM2, PM3, PM4) | Known |
| c.4052_4053ins328 (p.Tyr1352AlafsTer9) | Exon 4 Class III | rs775253277 | NR | NR | NR | Pathogenic (PVS1, PM2, PM4, PP3) | Known | |||
| Family 16 JU1464/1 | c.1498_1499delAT (p.Met500ValfsTer7) | Compound hetero | Exon 4 Class II | AR-RP | rs765129639 | NR | 0.000 | 0.000 | Pathogenic (PVS1, PM2, PM3, PM4, PP3, PP5) | Known |
| c.4052_4053ins328 (p.Tyr1352AlafsTer9) | Exon 4 Class III | rs775253277 | NR | NR | NR | Pathogenic (PVS1, PM2, PM4, PP3) | Novel | |||
| Family 17 JU0514/1 | c.392G>A (p.Arg131Gln) | Compound hetero | Exon 2 Class I | AR-COD/ CORD | rs752150870 | 0.002 | 0.003 | 0.000 | Uncertain Significance (PM2, PM3, PP3) | Known |
| c.2116G > C (p.Gly706Arg) | Exon 4 Class II | rs199879316 | 0.000 | 0.000 | 0.000 | Uncertain Significance (PM2, PP5) | Known | |||
| Family 18 JU1701/1 | c.1498_1499delAT (p.Met500ValfsTer7) | Compound hetero | Exon 4 Class II | AR-COD/ CORD | rs765129639 | NR | 0.000 | 0.000 | Pathogenic (PVS1, PM2, PM3, PM4, PP3, PP5) | Known |
| c.5797C > T (p.Arg1933Ter) | Exon 4 Class IV | rs118031911 | 0.003 | 0.003 | 0.000 | Pathogenic (PVS1, PM2, PM3, PM4) | Known | |||
| Family 19 S275/1 | c.1498_1499delAT (p.Met500ValfsTer7) | Compound hetero | Exon 4 Class II | AR-COD/ CORD | rs765129639 | NR | 0.000 | 0.000 | Pathogenic (PVS1, PM2, PM3, PM4, PP3, PP5) | Known |
| c.5797C > T (p.Arg1933Ter) | Exon 4 Class IV | rs118031911 | 0.003 | 0.003 | 0.000 | Pathogenic (PVS1, PM2, PM3, PM4) | Known | |||
| Family 20 JU1591/1 | c.2377delA (p.Arg793GlufsTer55) | Compound hetero | Exon 4 Class II | AR-COD/ CORD | NR | NR | NR | NR | Pathogenic (PVS1, PM2, PM3, PM4) | Known |
| c.5797C > T (p.Arg1933Ter) | Exon 4 Class IV | rs118031911 | 0.003 | 0.003 | 0.000 | Pathogenic (PVS1, PM2, PM3, PM4) | Known | |||
| Family 21 JU1339/1 | c.4052_4053ins328 (p.Tyr1352AlafsTer9) | Compound hetero | Exon 4 Class III | AR-COD/ CORD | rs775253277 | NR | NR | NR | Pathogenic (PVS1, PM2, PM3, PM4, PP3) | Known |
| c.5797C > T (p.Arg1933Ter) | Exon 4 Class IV | rs118031911 | 0.003 | 0.003 | 0.000 | Pathogenic (PVS1, PM2, PM3, PM4) | Known | |||
| Family 22 JU1947/1 | c.4052_4053ins328 (p.Tyr1352AlafsTer9) | Compound hetero | Exon 4 Class III | AR-COD/ CORD | rs775253277 | NR | NR | NR | Pathogenic (PVS1, PM2, PM3, PM4, PP3) | Known |
| c.5797C>T (p.Arg1933Ter) | Exon 4 Class IV | rs118031911 | 0.003 | 0.003 | 0.000 | Pathogenic (PVS1, PM2, PM3, PM4) | Known | |||
| Family 23 JU1978/1 | c.4052_4053ins328 (p.Tyr1352AlafsTer9) | Compound hetero | Exon 4 Class III | AR-COD/ CORD | rs775253277 | NR | NR | NR | Pathogenic (PVS1, PM2, PM3, PM4, PP3) | Known |
| c.5797C > T (p.Arg1933Ter) | Exon 4 Class IV | rs118031911 | 0.003 | 0.003 | 0.000 | Pathogenic (PVS1, PM2, PM3, PM4) | Known | |||
| Family 24 JU1672/1 | c.5913C > A (p.Asn1971Lys) | Hetero | Exon 4 Class IV | AD-RP | rs754290174 | 0.005 | 0.005 | 0.000 | Likely benign (PM2, BP1, BP4) | Novel |
| Family 25 JU0518/1 | c.5913C > A (p.Asn1971Lys) | Hetero | Exon 4 Class IV | AD-RP | rs754290174 | 0.005 | 0.005 | 0.000 | Likely benign (PM2, BP1, BP4) | Novel |
| Family 26 JU0616/1 | c.3188A > G (p.Gln1063Arg) | Hetero | Exon 4 Class III | AD-RP | rs199550930 | NR | NR | 0.000 | Likely benign (PM2, BP1, BP4) | Novel |
| Family 27 JU0523/1 | c.5797C > T (p.Arg1933Ter) | Hetero | Exon 4 Class IV | AR-RP | rs118031911 | 0.003 | 0.003 | 0.000 | Pathogenic (PVS1, PM2, PM3, PM4) | Known |
| Family 28 JU0525/1 | c.2400A > T (p.Lys800Asn) | Hetero | Exon 4 Class II | AR-RP | NR | NR | NR | NR | Uncertain Significance (PM2, BP1) | Novel |
| Family 29 JU0553/1 | c.2951A > G (p.Asp984Gly) | Hetero | Exon 4 Class II | AR-RP | rs200135800 | 0.003 | 0.005 | 0.000 | Likely benign (PM2, PP5, BP1, BP4) | Known |
| Family 30 JU1791/1 | c.2400A > T (p.Lys800Asn) | Hetero | Exon 4 Class II | AR-RP | NR | NR | NR | NR | Uncertain Significance (PM2, BP1) | Novel |
| Family 31 JU1942/1 | c.5797C > T (p.Arg1933Ter) | Hetero | Exon 4 Class IV | AR-RP | rs118031911 | 0.003 | 0.003 | 0.000 | Pathogenic (PVS1, PM2, PM3, PM4) | Known |
| Family 32 JU0565/1 | c.5797C > T (p.Arg1933Ter) | Hetero | Exon 4 Class IV | AR-COD/ CORD | rs118031911 | 0.003 | 0.003 | 0.000 | Pathogenic (PVS1, PM2, PM3, PM4) | Known |
| Family 33 JU0632/1 | c.5797C > T (p.Arg1933Ter) | Hetero | Exon 4 Class IV | AR-COD/ CORD | rs118031911 | 0.003 | 0.003 | 0.000 | Pathogenic (PVS1, PM2, PM3, PM4) | Known |
| Family 34 JU0830/1 | c.2960G > A (p.Cys987Tyr) | Hetero | Exon 4 Class II | AR-COD/ CORD | rs747536867 | 0.000 | 0.001 | 0.000 | Likely benign (PM2, BP1, BP4) | Novel |
| Family 35 JU1191/1 | c.2951A > G (p.Asp984Gly) | Hetero | Exon 4 Class II | AR-COD/ CORD | rs200135800 | 0.003 | 0.005 | 0.000 | Likely benign (PM2, PP5, BP1, BP4) | Known |
| Family 36 JU1955/1 | c.2894G > T (p.Ser965Ile) | Hetero | Exon 4 Class II | AR-COD/ CORD | rs201110322 | 0.042 | 0.040 | 0.005 | Likely benign (PM2, BP1, BP4, BP6) | Novel |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizobuchi, K.; Hayashi, T.; Oishi, N.; Kubota, D.; Kameya, S.; Higasa, K.; Futami, T.; Kondo, H.; Hosono, K.; Kurata, K.; et al. Genotype-Phenotype Correlations in RP1-Associated Retinal Dystrophies: A Multi-Center Cohort Study in JAPAN. J. Clin. Med. 2021, 10, 2265. https://doi.org/10.3390/jcm10112265
Mizobuchi K, Hayashi T, Oishi N, Kubota D, Kameya S, Higasa K, Futami T, Kondo H, Hosono K, Kurata K, et al. Genotype-Phenotype Correlations in RP1-Associated Retinal Dystrophies: A Multi-Center Cohort Study in JAPAN. Journal of Clinical Medicine. 2021; 10(11):2265. https://doi.org/10.3390/jcm10112265
Chicago/Turabian StyleMizobuchi, Kei, Takaaki Hayashi, Noriko Oishi, Daiki Kubota, Shuhei Kameya, Koichiro Higasa, Takuma Futami, Hiroyuki Kondo, Katsuhiro Hosono, Kentaro Kurata, and et al. 2021. "Genotype-Phenotype Correlations in RP1-Associated Retinal Dystrophies: A Multi-Center Cohort Study in JAPAN" Journal of Clinical Medicine 10, no. 11: 2265. https://doi.org/10.3390/jcm10112265
APA StyleMizobuchi, K., Hayashi, T., Oishi, N., Kubota, D., Kameya, S., Higasa, K., Futami, T., Kondo, H., Hosono, K., Kurata, K., Hotta, Y., Yoshitake, K., Iwata, T., Matsuura, T., & Nakano, T. (2021). Genotype-Phenotype Correlations in RP1-Associated Retinal Dystrophies: A Multi-Center Cohort Study in JAPAN. Journal of Clinical Medicine, 10(11), 2265. https://doi.org/10.3390/jcm10112265