Why Venous Leg Ulcers Have Difficulty Healing: Overview on Pathophysiology, Clinical Consequences, and Treatment


 ,
, 
Abstract
1. Introduction and Scope of the Problem
2. Pathophysiology
2.1. Definition and Etiology of Venous Leg Ulcers
2.2. Leg Ulcer Differential Diagnosis and Misdiagnosis
2.3. Clinical Manifestations, Healing, and Consequences
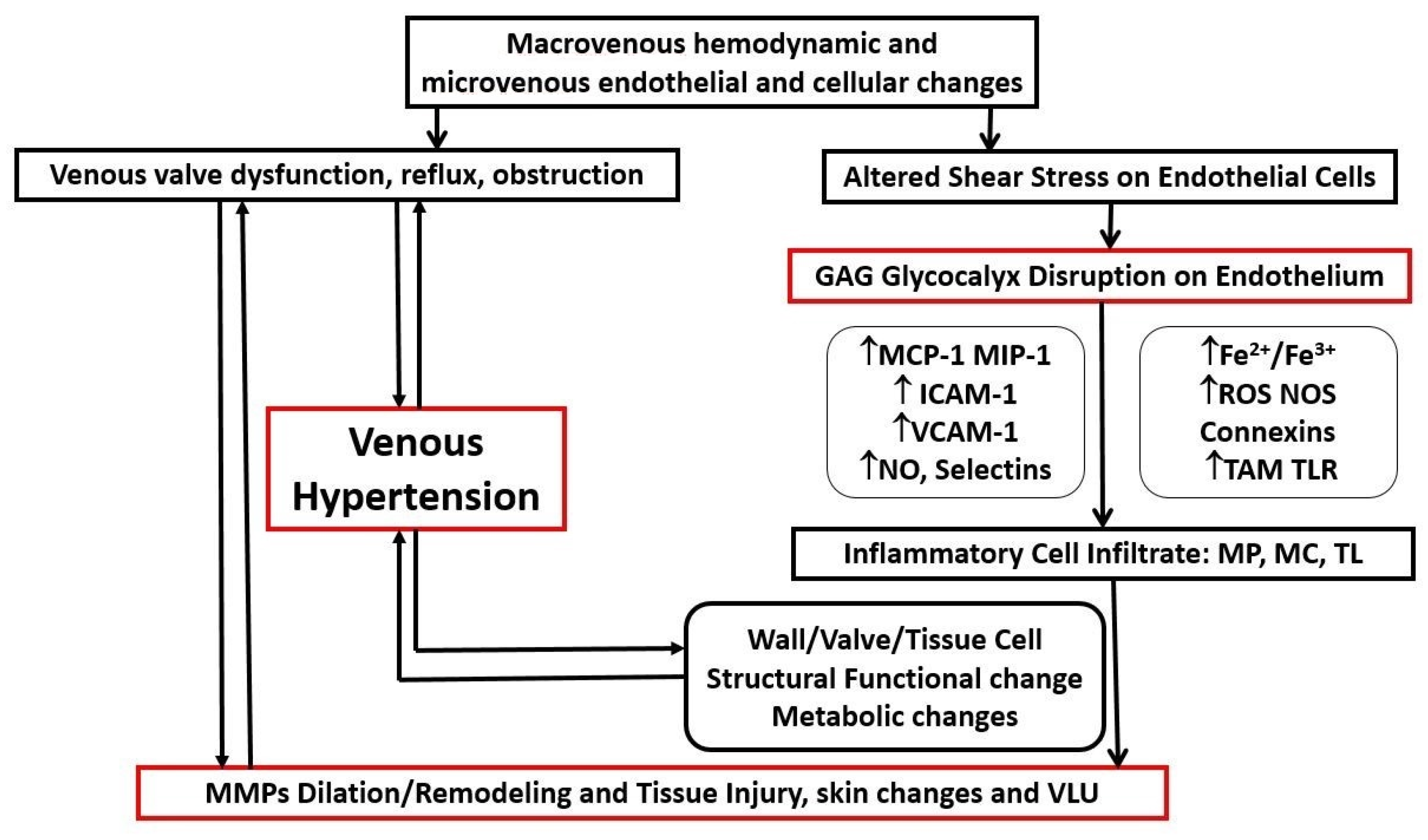
2.4. Pathophysiological Mechanisms
2.5. Biomarkers and Implications for Translational Research and Clinical Practice
3. Clinical Aspects
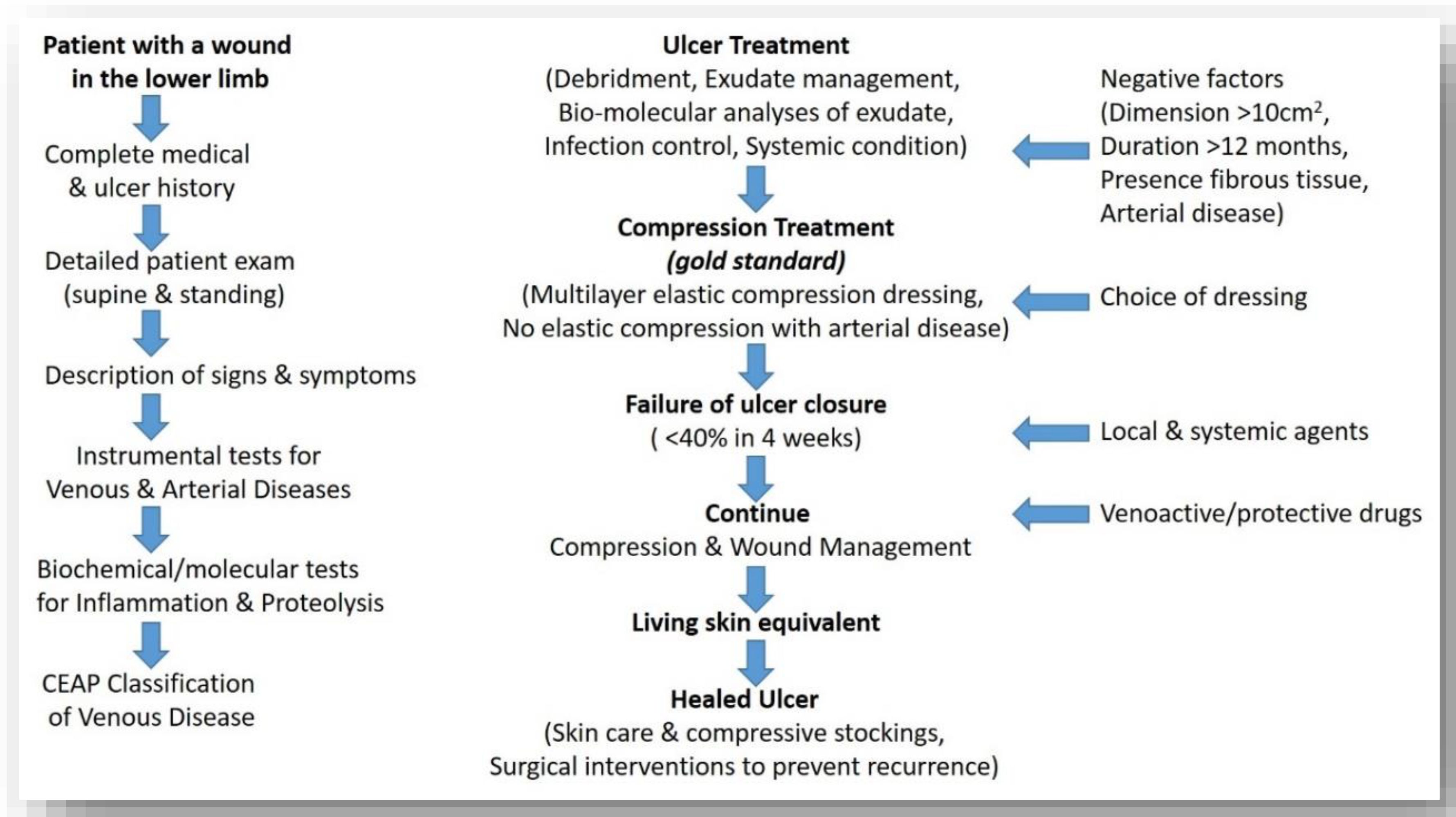
3.1. Recalcitrant Ulcers: Factors Prolonging Healing
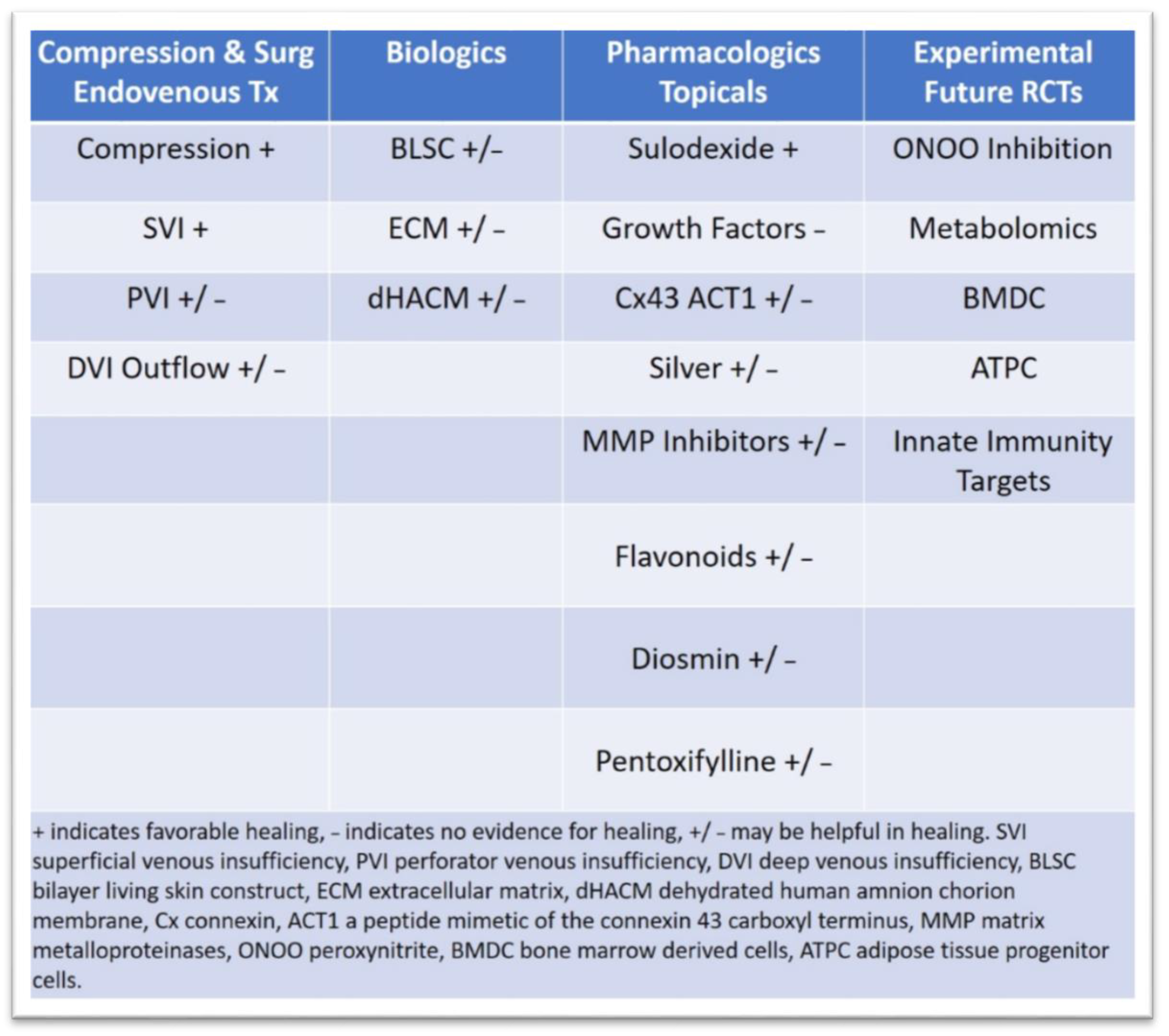
3.2. Latest Innovations in Surgical Treatment and Drug Therapies
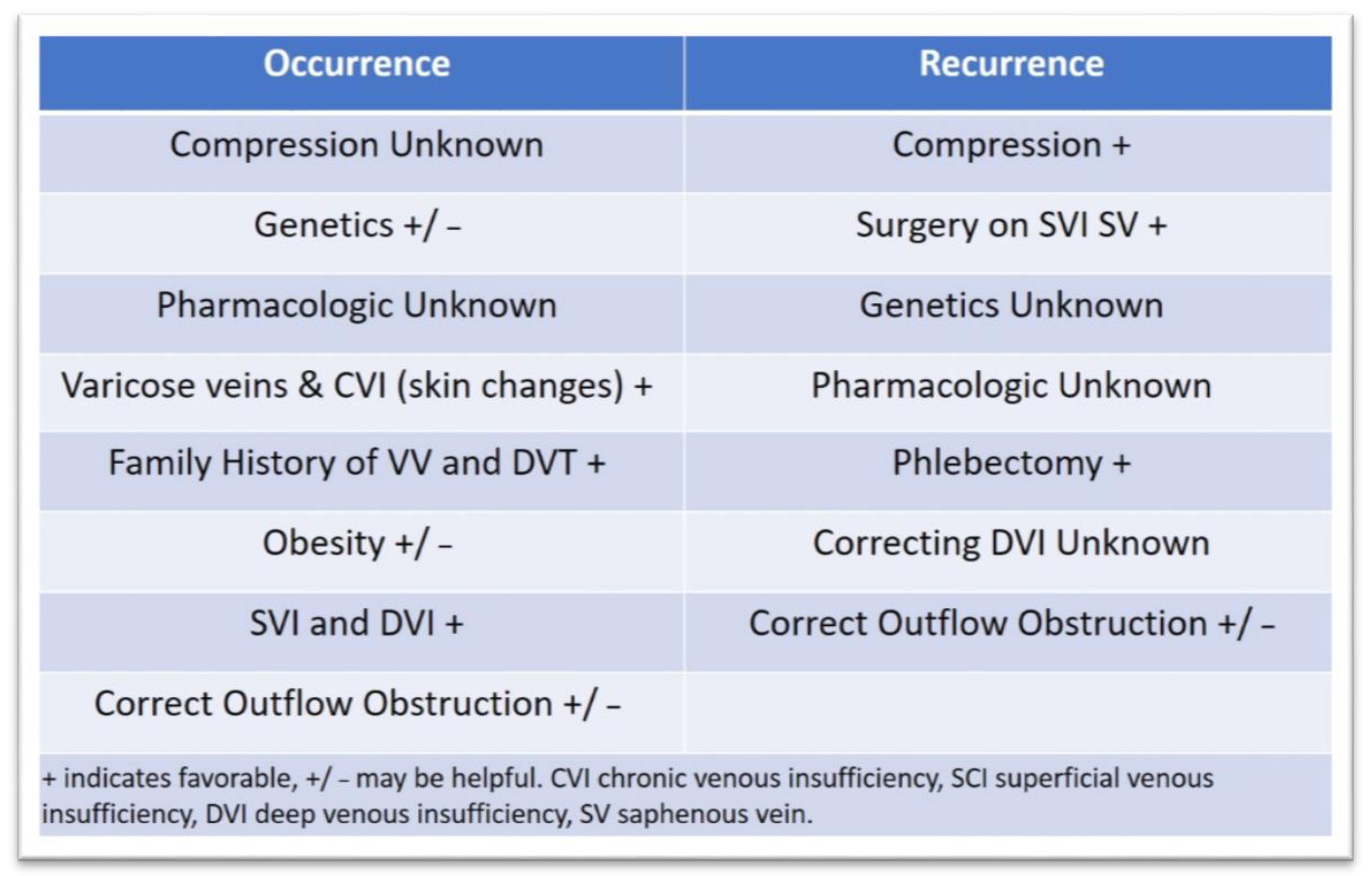
3.3. Approaches to Prevent Ulcer Occurrence and Recurrence
4. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- O’Donnell, T.F., Jr.; Passman, M.A.; Marston, W.A.; Ennis, W.J.; Dalsing, M.; Kistner, R.L.; Lurie, F.; Henke, P.K.; Gloviczki, M.L.; Eklöf, B.G.; et al. Management of venous leg ulcers: Clinical practice guidelines of the Society for Vascular Surgery® and the American Venous Forum. J. Vasc. Surg. 2014, 60, 3S–59S. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, R.T.; Raffetto, J.D. Chronic venous insufficiency. Circulation 2014, 130, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.W.; Raffetto, J.D. Venous leg ulceration pathophysiology and evidence based treatment. Vasc. Med. 2015, 20, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Broszczak, D.A.; Sydes, E.R.; Wallace, D.; Parker, T.J. Molecular Aspects of Wound Healing and the Rise of Venous Leg Ulceration: Omics Approaches to Enhance Knowledge and Aid Diagnostic Discovery. Clin. Biochem. Rev. 2017, 38, 35–55. [Google Scholar]
- Marston, W.A.; Carlin, R.E.; Passman, M.A.; Farber, M.A.; Keagy, B.A. Healing rates and cost efficacy of outpatient compression treatment for leg ulcers associated with venous insufficiency. J. Vasc. Surg. 1999, 30, 491–498. [Google Scholar] [CrossRef][Green Version]
- Ma, H.; O’Donnell, T.F., Jr.; Rosen, N.A.; Iafrati, M.D. The real cost of treating venous ulcers in a contemporary vascular practice. J. Vasc. Surg. Venous Lymphat. Disord. 2014, 2, 355–361. [Google Scholar] [CrossRef]
- Rice, J.B.; Desai, U.; Cummings, A.K.; Birnbaum, H.G.; Skornicki, M.; Parsons, N. Burden of venous leg ulcers in the United States. J. Med. Econ. 2014, 17, 347–356. [Google Scholar] [CrossRef]
- Eklof, B.; Rutherford, R.B.; Bergan, J.J.; Carpentier, P.H.; Gloviczki, P.; Kistner, R.L.; Meissner, M.H.; Moneta, G.L.; Myers, K.; Padberg, F.T.; et al. Revision of the CEAP classification for chronic venous disorders: Consensus statement. J. Vasc. Surg. 2004, 40, 1248–1252. [Google Scholar] [CrossRef]
- Meissner, M.H.; Moneta, G.; Burnand, K.; Gloviczki, P.; Lohr, J.M.; Lurie, F.; Mattos, M.A.; McLafferty, R.B.; Mozes, G.; Rutherford, R.B.; et al. The hemodynamics and diagnosis of venous disease. J. Vasc. Surg. 2007, 46, 4S–24S. [Google Scholar] [CrossRef]
- Labropoulos, N.; Waggoner, T.; Sammis, W.; Samali, S.; Pappas, P.J. The effect of venous thrombus location and extent on the development of post-thrombotic signs and symptoms. J. Vasc. Surg. 2008, 48, 407–412. [Google Scholar] [CrossRef]
- Raffetto, J.D.; Mannello, F. Pathophysiology of chronic venous disease. Int. Angiol. 2014, 33, 212–221. [Google Scholar] [PubMed]
- Mannello, F.; Ligi, D.; Canale, M.; Raffetto, J.D. Omics profiles in chronic venous ulcer wound fluid: Innovative applications for translational medicine. Expert Rev. Mol. Diagn. 2014, 14, 737–762. [Google Scholar] [CrossRef] [PubMed]
- Raffetto, J.D. Pathophysiology of Chronic Venous Disease and Venous Ulcers. Surg. Clin. N. Am. 2018, 98, 337–347. [Google Scholar] [CrossRef]
- Onida, S.; Tan, M.K.H.; Kafeza, M.; Bergner, R.T.; Shalhoub, J.; Holmes, E.; Davies, A.H. Metabolic Phenotyping in Venous Disease: The Need for Standardization. J. Proteome Res. 2019, 18, 3809–3820. [Google Scholar] [CrossRef]
- Pannier, F.; Rabe, E. Differential diagnosis of leg ulcers. Phlebology 2013, 28 (Suppl. 1), 55–60. [Google Scholar] [CrossRef]
- Makrantonaki, E.; Wlaschek, M.; Scharffetter-Kochanek, K. Pathogenesis of wound healing disorders in the elderly. J. Dtsch. Dermatol. Ges. 2017, 15, 255–275. [Google Scholar] [CrossRef] [PubMed]
- Alavi, A.; Sibbald, R.G.; Phillips, T.J.; Miller, O.F.; Margolis, D.J.; Marston, W.; Woo, K.; Romanelli, M.; Kirsner, R.S. What’s new: Management of venous leg ulcers: Approach to venous leg ulcers. J. Am. Acad. Dermatol. 2016, 74, 627–642. [Google Scholar] [CrossRef] [PubMed]
- Morton, L.M.; Phillips, T.J. Wound healing and treating wounds: Differential diagnosis and evaluation of chronic wounds. J. Am. Acad. Dermatol. 2016, 74, 589–606. [Google Scholar] [CrossRef] [PubMed]
- Santler, B.; Goerge, T. Chronic venous insufficiency—A review of pathophysiology, diagnosis, and treatment. J. Dtsch. Dermatol. Ges. 2017, 15, 538–556. [Google Scholar] [CrossRef] [PubMed]
- Ligi, D.; Mosti, G.; Croce, L.; Raffetto, J.D.; Mannello, F. Chronic venous disease—Part I: Inflammatory biomarkers in wound healing. Biochim. Biophys. Acta 2016, 1862, 1964–1974. [Google Scholar] [CrossRef]
- Abbade, L.P.F.; Lastória, S. Venous ulcer: Epidemiology, physiopathology, diagnosis and treatment. Int. J. Dermatol. 2005, 44, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Mansilha, A.; Sousa, J. Pathophysiological Mechanisms of Chronic Venous Disease and Implications for Venoactive Drug Therapy. Int. J. Mol. Sci. 2018, 19, 1669. [Google Scholar] [CrossRef] [PubMed]
- Comerota, A.; Lurie, F. Pathogenesis of venous ulcer. Semin. Vasc. Surg. 2015, 28, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Raffetto, J.D.; Khalil, R.A. Mechanisms of varicose vein formation: Valve dysfunction and wall dilation. Phlebology 2008, 23, 85–98. [Google Scholar] [CrossRef]
- Labropoulos, N.; Leon, M.; Nicolaides, A.N.; Giannoukas, A.D.; Volteas, N.; Chan, P. Superficial venous insufficiency: Correlation of anatomic extent of reflux with clinical symptoms and signs. J. Vasc. Surg. 1994, 20, 953–958. [Google Scholar] [CrossRef]
- Labropoulos, N.; Gasparis, A.P.; Tassiopoulos, A.K. Prospective evaluation of the clinical deterioration in post-thrombotic limbs. J. Vasc. Surg. 2009, 50, 826–830. [Google Scholar] [CrossRef]
- Labropoulos, N.; Leon, M.; Nicolaides, A.N.; Sowade, O.; Volteas, N.; Ortega, F.; Chan, P. Venous reflux in patients with previous deep venous thrombosis: Correlation with ulceration and other symptoms. J. Vasc. Surg. 1994, 20, 20–26. [Google Scholar] [CrossRef][Green Version]
- Raju, S.; Knepper, J.; May, C.; Knight, A.; Pace, N.; Jayaraj, A. Ambulatory venous pressure, air plethysmography, and the role of calf venous pump in chronic venous disease. J. Vasc. Surg. Venous Lymphat. Disord. 2019, 7, 428–440. [Google Scholar] [CrossRef]
- Raffetto, J.D. Inflammation in chronic venous ulcers. Phlebology 2013, 28, 61–67. [Google Scholar] [CrossRef]
- Bergan, J. Molecular mechanisms in chronic venous insufficiency. Ann. Vasc. Surg. 2007, 21, 260–266. [Google Scholar] [CrossRef]
- Pocock, E.S.; Alsaigh, T.; Mazor, R.; Schmid-Schönbein, G.W. Cellular and molecular basis of Venous insufficiency. Vasc. Cell 2014, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Huang, Z.; Yin, H.; Lin, Y.; Wang, S. In vitro differences between smooth muscle cells derived from varicose veins and normal veins. J. Vasc. Surg. 2009, 50, 1149–1154. [Google Scholar] [CrossRef]
- Badier-Commander, C.; Couvelard, A.; Henin, D.; Verbeuren, T.; Michel, J.B.; Jacob, M.P. Smooth muscle cell modulation and cytokine overproduction in varicose veins. An in situ study. J. Pathol. 2001, 193, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.M.; Lal, B.K.; Durán, W.N.; Pappas, P.J. Pathophysiology of venous ulceration. J. Vasc. Surg. Venous Lymphat. Disord. 2017, 5, 596–605. [Google Scholar] [CrossRef]
- Travers, J.P.; Brookes, C.E.; Evans, J.; Baker, D.M.; Kent, C.; Makin, G.S.; Mayhew, T.M. Assessment of wall structure and composition of varicose veins with reference to collagen, elastin and smooth muscle content. Eur. J. Vasc. Endovasc. Surg. 1996, 11, 230–237. [Google Scholar] [CrossRef]
- Wali, M.A.; Eid, R.A. Changes of elastic and collagen fibers in varicose veins. Int. Angiol. 2002, 21, 337–343. [Google Scholar] [PubMed]
- Sansilvestri-Morel, P.; Rupin, A.; Badier-Commander, C.; Kern, P.; Fabiani, J.N.; Verbeuren, T.J.; Vanhoutte, P.M. Imbalance in the synthesis of collagen type I and collagen type III in smooth muscle cells derived from human varicose veins. J. Vasc. Res. 2001, 38, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Sansilvestri-Morel, P.; Rupin, A.; Jullien, N.D.; Lembrez, N.; Mestries-Dubois, P.; Fabiani, J.N.; Verbeuren, T.J. Decreased production of collagen Type III in cultured smooth muscle cells from varicose vein patients is due to a degradation by MMPs: Possible implication of MMP-3. J. Vasc. Res. 2005, 42, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Wlaschek, M.; Singh, K.; Sindrilaru, A.; Crisan, D.; Scharffetter-Kochanek, K. Iron and iron-dependent reactive oxygen species in the regulation of macrophages and fibroblasts in non-healing chronic wounds. Free Radic. Biol. Med. 2019, 133, 262–275. [Google Scholar] [CrossRef]
- Caggiati, A.; Rosi, C.; Casini, A.; Cirenza, M.; Petrozza, V.; Acconcia, M.C.; Zamboni, P. Skin iron deposition characterises lipodermatosclerosis and leg ulcer. Eur. J. Vasc. Endovasc. Surg. 2010, 40, 777–782. [Google Scholar] [CrossRef]
- Pappas, P.J.; DeFouw, D.O.; Venezio, L.M.; Gorti, R.; Padberg, F.T., Jr.; Silva, M.B., Jr.; Goldberg, M.C.; Durán, W.N.; Hobson, R.W., 2nd. Morphometric assessment of the dermal microcirculation in patients with chronic venous insufficiency. J. Vasc. Surg. 1997, 26, 784–795. [Google Scholar] [CrossRef]
- Wilkinson, L.S.; Bunker, C.; Edwards, J.C.; Scurr, J.H.; Smith, P.D. Leukocytes: Their role in the etiopathogenesis of skin damage in venous disease. J. Vasc. Surg. 1993, 17, 669–675. [Google Scholar] [CrossRef]
- Thomas, P.R.; Dormandy, J.A. White cell and platelet trapping in patients with chronic venous insufficiency. Phlebologie 1988, 41, 771–776. [Google Scholar] [PubMed]
- Tisato, V.; Zauli, G.; Gianesini, S.; Menegatti, E.; Brunelli, L.; Manfredini, R.; Zamboni, P.; Secchiero, P. Modulation of circulating cytokine-chemokine profile in patients affected by chronic venous insufficiency undergoing surgical hemodynamic correction. J. Immunol. Res. 2014, 2014, 473765. [Google Scholar] [CrossRef]
- Poredos, P.; Spirkoska, A.; Rucigaj, T.; Fareed, J.; Jezovnik, M.K. Do blood constituents in varicose veins differ from the systemic blood constituents? Eur. J. Vasc. Endovasc. Surg. 2015, 50, 250–256. [Google Scholar] [CrossRef]
- Solá, L.d.R.; Aceves, M.; Dueñas, A.I.; González-Fajardo, J.A.; Vaquero, C.; Crespo, M.S.; García-Rodríguez, C. Varicose veins show enhanced chemokine expression. Eur. J. Vasc. Endovasc. Surg. 2009, 38, 635–641. [Google Scholar] [CrossRef]
- Lattimer, C.R.; Kalodiki, E.; Geroulakos, G.; Hoppensteadt, D.; Fareed, J. Are Inflammatory Biomarkers Increased in Varicose Vein Blood? Clin. Appl. Thromb. Hemost. 2016, 22, 656–664. [Google Scholar] [CrossRef]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef]
- Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar] [CrossRef]
- Lyons, O.T.; Saha, P.; Smith, A. Redox dysregulation in the pathogenesis of chronic venous ulceration. Free Radic. Biol. Med. 2019. [Google Scholar] [CrossRef]
- Stacey, M.C. Biomarker directed chronic wound therapy—A new treatment paradigm. J. Tissue Viability 2019. [Google Scholar] [CrossRef] [PubMed]
- Edwards, H.E.; Parker, C.N.; Miller, C.; Gibb, M.; Kapp, S.; Ogrin, R.; Anderson, J.; Coleman, K.; Smith, D.; Finlayson, K.J. Predicting delayed healing: The diagnostic accuracy of a venous leg ulcer risk assessment tool. Int. Wound J. 2018, 15, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Qing, C. The molecular biology in wound healing & non-healing wound. Chin. J. Traumatol. Zhonghua Chuang Shang Za Zhi 2017, 20, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Diegelmann, R.F.; Evans, M.C. Wound healing: An overview of acute, fibrotic and delayed healing. Front. Biosci. J. Virtual Libr. 2004, 9, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Ceilley, R. Chronic Wound Healing: A Review of Current Management and Treatments. Adv. Ther. 2017, 34, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Lindley, L.E.; Stojadinovic, O.; Pastar, I.; Tomic-Canic, M. Biology and Biomarkers for Wound Healing. Plast. Reconstr. Surg. 2016, 138, 18S–28S. [Google Scholar] [CrossRef]
- Pastar, I.; Wong, L.L.; Egger, A.N.; Tomic-Canic, M. Descriptive vs mechanistic scientific approach to study wound healing and its inhibition: Is there a value of translational research involving human subjects? Exp. Dermatol. 2018, 27, 551–562. [Google Scholar] [CrossRef]
- Parnell, L.K.S.; Volk, S.W. The Evolution of Animal Models in Wound Healing Research: 1993–2017. Adv. Wound Care 2019, 8, 692–702. [Google Scholar] [CrossRef]
- Moore, K.; Huddleston, E.; Stacey, M.C.; Harding, K.G. Venous leg ulcers—The search for a prognostic indicator. Int. Wound J. 2007, 4, 163–172. [Google Scholar] [CrossRef]
- Simka, M. Cellular and molecular mechanisms of venous leg ulcers development—The “puzzle” theory. Int. Angiol. 2010, 29, 1–19. [Google Scholar]
- Claudy, A.L.; Mirshahi, M.; Soria, C.; Soria, J. Detection of undegraded fibrin and tumor necrosis factor-alpha in venous leg ulcers. J. Am. Acad. Dermatol. 1991, 25, 623–627. [Google Scholar] [CrossRef]
- Higley, H.R.; Ksander, G.A.; Gerhardt, C.O.; Falanga, V. Extravasation of macromolecules and possible trapping of transforming growth factor-beta in venous ulceration. Br. J. Dermatol. 1995, 132, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.R.; Yee, K.C.; Walters, C.E.; Cunliffe, W.J.; Kearney, J.N.; Wood, E.J.; Ingham, E. Cytokine and protease levels in healing and non-healing chronic venous leg ulcers. Exp. Dermatol. 1995, 4, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Pappas, P.J.; Fallek, S.R.; Garcia, A.; Araki, C.T.; Back, T.L.; Durán, W.N.; Hobson, R.W., 2nd. Role of leukocyte activation in patients with venous stasis ulcers. J. Surg. Res. 1995, 59, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.; Murata, H.; Falabella, A.; Ochoa, S.; Zhou, L.; Badiavas, E.; Falanga, V. Dermal fibroblasts from venous ulcers are unresponsive to the action of transforming growth factor-beta 1. J. Dermatol. Sci. 1997, 16, 59–66. [Google Scholar] [CrossRef]
- He, C.F.; Cherry, G.W.; Arnold, F. Postural vasoregulation and mediators of reperfusion injury in venous ulceration. J. Vasc. Surg. 1997, 25, 647–653. [Google Scholar] [CrossRef][Green Version]
- Fivenson, D.P.; Faria, D.T.; Nickoloff, B.J.; Poverini, P.J.; Kunkel, S.; Burdick, M.; Strieter, R.M. Chemokine and inflammatory cytokine changes during chronic wound healing. Wound Repair Regen. 1997, 5, 310–322. [Google Scholar] [CrossRef]
- Wallace, H.J.; Stacey, M.C. Levels of tumor necrosis factor-alpha (TNF-alpha) and soluble TNF receptors in chronic venous leg ulcers--correlations to healing status. J. Investig. Dermatol. 1998, 110, 292–296. [Google Scholar] [CrossRef]
- Peschen, M.; Grenz, H.; Grothe, C.; Schöpf, E.; Vanscheidt, W. Patterns of epidermal growth factor receptor, basic fibroblast growth factor and transforming growth factor-beta3 expression in skin with chronic venous insufficiency. Eur. J. Dermatol. 1998, 8, 334–338. [Google Scholar]
- Peschen, M.; Grenz, H.; Brand-Saberi, B.; Bunaes, M.; Simon, J.C.; Schöpf, E.; Vanscheidt, W. Increased expression of platelet-derived growth factor receptor alpha and beta and vascular endothelial growth factor in the skin of patients with chronic venous insufficiency. Arch. Dermatol. Res. 1998, 290, 291–297. [Google Scholar] [CrossRef]
- Li, Y.Q.; Doyle, J.W.; Roth, T.P.; Dunn, R.M.; Lawrence, W.T. IL-10 and GM-CSF expression and the presence of antigen-presenting cells in chronic venous ulcers. J. Surg. Res. 1998, 79, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.E.; Roth, T.P.; Dunn, R.M.; Doyle, J.W. Comparison of IL-10 levels in chronic venous insufficiency ulcers and autologous donor tissue. Arch. Dermatol. Res. 1998, 290, 669–673. [Google Scholar] [CrossRef]
- Pappas, P.J.; You, R.; Rameshwar, P.; Gorti, R.; DeFouw, D.O.; Phillips, C.K.; Padberg, F.T., Jr.; Silva, M.B., Jr.; Simonian, G.T.; Hobson, R.W., 2nd; et al. Dermal tissue fibrosis in patients with chronic venous insufficiency is associated with increased transforming growth factor-beta1 gene expression and protein production. J. Vasc. Surg. 1999, 30, 1129–1145. [Google Scholar] [CrossRef]
- Saalbach, A.; Wetzig, T.; Haustein, U.F.; Anderegg, U. Detection of human soluble Thy-1 in serum by ELISA. Fibroblasts and activated endothelial cells are a possible source of soluble Thy-1 in serum. Cell Tissue Res. 1999, 298, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Trengove, N.J.; Bielefeldt-Ohmann, H.; Stacey, M.C. Mitogenic activity and cytokine levels in non-healing and healing chronic leg ulcers. Wound Repair Regen. 2000, 8, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Lauer, G.; Sollberg, S.; Cole, M.; Flamme, I.; Stürzebecher, J.; Mann, K.; Krieg, T.; Eming, S.A. Expression and proteolysis of vascular endothelial growth factor is increased in chronic wounds. J. Investig. Dermatol. 2000, 115, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Cowin, A.J.; Hatzirodos, N.; Holding, C.A.; Dunaiski, V.; Harries, R.H.; Rayner, T.E.; Fitridge, R.; Cooter, R.D.; Schultz, G.S.; Belford, D.A. Effect of healing on the expression of transforming growth factor beta(s) and their receptors in chronic venous leg ulcers. J. Investig. Dermatol. 2001, 117, 1282–1289. [Google Scholar] [CrossRef]
- Murphy, M.A.; Joyce, W.P.; Condron, C.; Bouchier-Hayes, D. A reduction in serum cytokine levels parallels healing of venous ulcers in patients undergoing compression therapy. Eur. J. Vasc. Endovasc. Surg. 2002, 23, 349–352. [Google Scholar] [CrossRef]
- Jude, E.B.; Blakytny, R.; Bulmer, J.; Boulton, A.J.M.; Ferguson, M.W.J. Transforming growth factor-beta 1, 2, 3 and receptor type I and II in diabetic foot ulcers. Diabet. Med. 2002, 19, 440–447. [Google Scholar] [CrossRef]
- Kim, B.-C.; Kim, H.T.; Park, S.H.; Cha, J.-S.; Yufit, T.; Kim, S.-J.; Falanga, V. Fibroblasts from chronic wounds show altered TGF-beta-signaling and decreased TGF-beta Type II receptor expression. J. Cell Physiol. 2003, 195, 331–336. [Google Scholar] [CrossRef]
- Tian, Y.-W.; Stacey, M.C. Cytokines and growth factors in keratinocytes and sweat glands in chronic venous leg ulcers. An immunohistochemical study. Wound Repair Regen. 2003, 11, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Senet, P.; Bon, F.-X.; Benbunan, M.; Bussel, A.; Traineau, R.; Calvo, F.; Dubertret, L.; Dosquet, C. Randomized trial and local biological effect of autologous platelets used as adjuvant therapy for chronic venous leg ulcers. J. Vasc. Surg. 2003, 38, 1342–1348. [Google Scholar] [CrossRef]
- Nayeri, F.; Olsson, H.; Peterson, C.; Sundqvist, T. Hepatocyte growth factor; expression, concentration and biological activity in chronic leg ulcers. J. Dermatol. Sci. 2005, 37, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Galkowska, H.; Olszewski, W.L.; Wojewodzka, U. Keratinocyte and dermal vascular endothelial cell capacities remain unimpaired in the margin of chronic venous ulcer. Arch. Dermatol. Res. 2005, 296, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ye, C.; Yin, H.; Ye, J.; Wang, S. RANTES expression in venous ulceration of lower limbs. Nan Fang Yi Ke Da Xue Xue Bao 2008, 28, 861–862. [Google Scholar]
- Gohel, M.S.; Windhaber, R.A.; Tarlton, J.F.; Whyman, M.R.; Poskitt, K.R. The relationship between cytokine concentrations and wound healing in chronic venous ulceration. J. Vasc. Surg. 2008, 48, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Beidler, S.K.; Douillet, C.D.; Berndt, D.F.; Keagy, B.A.; Rich, P.B.; Marston, W.A. Inflammatory cytokine levels in chronic venous insufficiency ulcer tissue before and after compression therapy. J. Vasc. Surg. 2009, 49, 1013–1020. [Google Scholar] [CrossRef]
- Charles, C.A.; Romanelli, P.; Martinez, Z.B.; Ma, F.; Roberts, B.; Kirsner, R.S. Tumor necrosis factor-alfa in nonhealing venous leg ulcers. J. Am. Acad. Dermatol. 2009, 60, 951–955. [Google Scholar] [CrossRef]
- Chen, L.; Liu, D.; Xiong, J.; Ye, C.; Yin, H.; Wang, S. The value of regulated upon activation normal T cell expressed and secreted in the pathogenesis of varicose ulcer of lower limbs. Zhonghua Yi Xue Za Zhi 2009, 89, 1460–1463. [Google Scholar]
- Karatepe, O.; Unal, O.; Ugurlucan, M.; Kemik, A.; Karahan, S.; Aksoy, M.; Kurtoglu, M. The impact of valvular oxidative stress on the development of venous stasis ulcer valvular oxidative stress and venous ulcers. Angiology 2010, 61, 283–288. [Google Scholar] [CrossRef]
- Pukstad, B.S.; Ryan, L.; Flo, T.H.; Stenvik, J.; Moseley, R.; Harding, K.; Thomas, D.W.; Espevik, T. Non-healing is associated with persistent stimulation of the innate immune response in chronic venous leg ulcers. J. Dermatol. Sci. 2010, 59, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Trøstrup, H.; Lundquist, R.; Christensen, L.H.; Jorgensen, L.N.; Karlsmark, T.; Haab, B.B.; Agren, M.S. S100A8/A9 deficiency in nonhealing venous leg ulcers uncovered by multiplexed antibody microarray profiling. Br. J. Dermatol. 2011, 165, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Zillmer, R.; Trostrup, H.; Karlsmark, T.; Ifversen, P.; Agren, M.S. Duration of wound fluid secretion from chronic venous leg ulcers is critical for interleukin-1alpha, interleukin-1beta, interleukin-8 levels and fibroblast activation. Arch. Dermatol. Res. 2011, 303, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Kroeze, K.L.; Vink, L.; de Boer, E.M.; Scheper, R.J.; van Montfrans, C.; Gibbs, S. Simple wound exudate collection method identifies bioactive cytokines and chemokines in (arterio) venous ulcers. Wound Repair Regen. 2012, 20, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Ma, Y.; Brogan, M.S. Chronic and non-healing wounds: The story of vascular endothelial growth factor. Med. Hypotheses 2015, 85, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Filkor, K.; Nemeth, T.; Nagy, I.; Kondorosi, E.; Urban, E.; Kemeny, L.; Szolnoky, G. The expression of inflammatory cytokines, TAM tyrosine kinase receptors and their ligands is upregulated in venous leg ulcer patients: A novel insight into chronic wound immunity. Int. Wound J. 2016, 13, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Serra, R.; Grande, R.; Buffone, G.; Molinari, V.; Perri, P.; Perri, A.; Amato, B.; Colosimo, M.; de Franciscis, S. Extracellular matrix assessment of infected chronic venous leg ulcers: Role of metalloproteinases and inflammatory cytokines. Int. Wound J. 2016, 13, 53–58. [Google Scholar] [CrossRef]
- Serra, R.; Grande, R.; Butrico, L.; Buffone, G.; Calio, F.G.; Squillace, A.; Rizzo, B.A.; Massara, M.; Spinelli, F.; Ferrarese, A.G.; et al. Effects of a new nutraceutical substance on clinical and molecular parameters in patients with chronic venous ulceration. Int. Wound J. 2016, 13, 88–96. [Google Scholar] [CrossRef]
- Goto, T.; Tamai, N.; Nakagami, G.; Kitamura, A.; Naito, A.; Hirokawa, M.; Shimokawa, C.; Takahashi, K.; Umemoto, J.; Sanada, H. Can Wound Exudate from Venous Leg Ulcers Measure Wound Pain Status? A Pilot Study. PLoS ONE 2016, 11, e0167478. [Google Scholar] [CrossRef]
- Trostrup, H.; Holstein, P.; Christophersen, L.; Jorgensen, B.; Karlsmark, T.; Hoiby, N.; Moser, C.; Agren, M.S. S100A8/A9 is an important host defence mediator in neuropathic foot ulcers in patients with type 2 diabetes mellitus. Arch. Dermatol. Res. 2016, 308, 347–355. [Google Scholar] [CrossRef]
- De Francesco, F.; Graziano, A.; Trovato, L.; Ceccarelli, G.; Romano, M.; Marcarelli, M.; Cusella De Angelis, G.M.; Cillo, U.; Riccio, M.; Ferraro, G.A. A Regenerative Approach with Dermal Micrografts in the Treatment of Chronic Ulcers. Stem Cell Rev. Rep. 2017, 13, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Ligi, D.; Croce, L.; Mosti, G.; Raffetto, J.D.; Mannello, F. Chronic Venous Insufficiency: Transforming Growth Factor-β Isoforms and Soluble Endoglin Concentration in Different States of Wound Healing. Int. J. Mol. Sci. 2017, 18, 2206. [Google Scholar] [CrossRef] [PubMed]
- Chimento, S.; Billero, V.; Cavallin, L.; Romanelli, M.; Nadji, M.; Romanelli, P. Evaluation of osteopontin expression in chronic wounds: A potential prognostic and therapeutic biomarker. J. Wound Care 2017, 26, S4–S8. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, E.; Bakondi, E.; Kovacs, K.; Hegedus, C.; Lakatos, P.; Robaszkiewicz, A.; Regdon, Z.; Virag, L.; Szabo, E. Redox Profiling Reveals Clear Differences between Molecular Patterns of Wound Fluids from Acute and Chronic Wounds. Oxid. Med. Cell. Longev. 2018, 2018, 5286785. [Google Scholar] [CrossRef] [PubMed]
- Krzystek-Korpacka, M.; Kedzior, K.; Maslowski, L.; Mierzchala, M.; Bednarz-Misa, I.; Bronowicka-Szydelko, A.; Kubiak, J.; Gacka, M.; Placzkowska, S.; Gamian, A. Impact of chronic wounds of various etiology on systemic profiles of key inflammatory cytokines, chemokines and growth factors, and their interplay. Adv. Clin. Exp. Med. Off. Organ Wroc. Med. Univ. 2019, 28, 1301–1309. [Google Scholar] [CrossRef]
- Ligi, D.; Croce, L.; Mannello, F. Chronic Venous Disorders: The Dangerous, the Good, and the Diverse. Int. J. Mol. Sci. 2018, 19, 2544. [Google Scholar] [CrossRef]
- Vaalamo, M.; Mattila, L.; Johansson, N.; Kariniemi, A.L.; Karjalainen-Lindsberg, M.L.; Kahari, V.M.; Saarialho-Kere, U. Distinct populations of stromal cells express collagenase-3 (MMP-13) and collagenase-1 (MMP-1) in chronic ulcers but not in normally healing wounds. J. Investig. Dermatol. 1997, 109, 96–101. [Google Scholar] [CrossRef]
- Vaalamo, M.; Leivo, T.; Saarialho-Kere, U. Differential expression of tissue inhibitors of metalloproteinases (TIMP-1, -2, -3, and -4) in normal and aberrant wound healing. Hum. Pathol. 1999, 30, 795–802. [Google Scholar] [CrossRef]
- Nwomeh, B.C.; Liang, H.X.; Cohen, I.K.; Yager, D.R. MMP-8 is the predominant collagenase in healing wounds and nonhealing ulcers. J. Surg. Res. 1999, 81, 189–195. [Google Scholar] [CrossRef]
- Tarlton, J.F.; Bailey, A.J.; Crawford, E.; Jones, D.; Moore, K.; Harding, K.D. Prognostic value of markers of collagen remodeling in venous ulcers. Wound Repair Regen. 1999, 7, 347–355. [Google Scholar] [CrossRef]
- Saito, S.; Trovato, M.J.; You, R.; Lal, B.K.; Fasehun, F.; Padberg, F.T., Jr.; Hobson, R.W., 2nd; Duran, W.N.; Pappas, P.J. Role of matrix metalloproteinases 1, 2, and 9 and tissue inhibitor of matrix metalloproteinase-1 in chronic venous insufficiency. J. Vasc. Surg. 2001, 34, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Mirastschijski, U.; Impola, U.; Jahkola, T.; Karlsmark, T.; MS, A.G.; Saarialho-Kere, U. Ectopic localization of matrix metalloproteinase-9 in chronic cutaneous wounds. Hum. Pathol. 2002, 33, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Norgauer, J.; Hildenbrand, T.; Idzko, M.; Panther, E.; Bandemir, E.; Hartmann, M.; Vanscheidt, W.; Herouy, Y. Elevated expression of extracellular matrix metalloproteinase inducer (CD147) and membrane-type matrix metalloproteinases in venous leg ulcers. Br. J. Dermatol. 2002, 147, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Impola, U.; Jeskanen, L.; Ravanti, L.; Syrjanen, S.; Baldursson, B.; Kahari, V.M.; Saarialho-Kere, U. Expression of matrix metalloproteinase (MMP)-7 and MMP-13 and loss of MMP-19 and p16 are associated with malignant progression in chronic wounds. Br. J. Dermatol. 2005, 152, 720–726. [Google Scholar] [CrossRef]
- Ulrich, D.; Lichtenegger, F.; Unglaub, F.; Smeets, R.; Pallua, N. Effect of chronic wound exudates and MMP-2/-9 inhibitor on angiogenesis in vitro. Plast. Reconstr. Surg. 2005, 116, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Zamboni, P.; Scapoli, G.; Lanzara, V.; Izzo, M.; Fortini, P.; Legnaro, R.; Palazzo, A.; Tognazzo, S.; Gemmati, D. Serum iron and matrix metalloproteinase-9 variations in limbs affected by chronic venous disease and venous leg ulcers. Dermatol. Surg. 2005, 31, 644–649, discussion 649. [Google Scholar] [CrossRef] [PubMed]
- Mwaura, B.; Mahendran, B.; Hynes, N.; Defreitas, D.; Avalos, G.; Adegbola, T.; Adham, M.; Connolly, C.E.; Sultan, S. The impact of differential expression of extracellular matrix metalloproteinase inducer, matrix metalloproteinase-2, tissue inhibitor of matrix metalloproteinase-2 and PDGF-AA on the chronicity of venous leg ulcers. Eur. J. Vasc. Endovasc. Surg. 2006, 31, 306–310. [Google Scholar] [CrossRef]
- Meyer, F.J.; Burnand, K.G.; Abisi, S.; Tekoppele, J.M.; van Els, B.; Smith, A. Effect of collagen turnover and matrix metalloproteinase activity on healing of venous leg ulcers. Br. J. Surg. 2008, 95, 319–325. [Google Scholar] [CrossRef]
- Subramaniam, K.; Pech, C.M.; Stacey, M.C.; Wallace, H.J. Induction of MMP-1, MMP-3 and TIMP-1 in normal dermal fibroblasts by chronic venous leg ulcer wound fluid. Int. Wound J. 2008, 5, 79–86. [Google Scholar] [CrossRef]
- Beidler, S.K.; Douillet, C.D.; Berndt, D.F.; Keagy, B.A.; Rich, P.B.; Marston, W.A. Multiplexed analysis of matrix metalloproteinases in leg ulcer tissue of patients with chronic venous insufficiency before and after compression therapy. Wound Repair Regen. 2008, 16, 642–648. [Google Scholar] [CrossRef]
- Moor, A.N.; Vachon, D.J.; Gould, L.J. Proteolytic activity in wound fluids and tissues derived from chronic venous leg ulcers. Wound Repair Regen. 2009, 17, 832–839. [Google Scholar] [CrossRef] [PubMed]
- Serra, R.; Buffone, G.; Falcone, D.; Molinari, V.; Scaramuzzino, M.; Gallelli, L.; de Franciscis, S. Chronic venous leg ulcers are associated with high levels of metalloproteinases-9 and neutrophil gelatinase-associated lipocalin. Wound Repair Regen. 2013, 21, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Grzela, T.; Niderla-Bielinska, J.; Litwiniuk, M.; White, R. The direct inhibition of MMP-2 and MMP-9 by an enzyme alginogel: A possible mechanism of healing support for venous leg ulcers. J. Wound Care 2014, 23, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Serra, R.; Gallelli, L.; Butrico, L.; Buffone, G.; Calio, F.G.; De Caridi, G.; Massara, M.; Barbetta, A.; Amato, B.; Labonia, M.; et al. From varices to venous ulceration: The story of chronic venous disease described by metalloproteinases. Int. Wound J. 2017, 14, 233–240. [Google Scholar] [CrossRef]
- Amato, B.; Coretti, G.; Compagna, R.; Amato, M.; Buffone, G.; Gigliotti, D.; Grande, R.; Serra, R.; de Franciscis, S. Role of matrix metalloproteinases in non-healing venous ulcers. Int. Wound J. 2015, 12, 641–645. [Google Scholar] [CrossRef]
- Caimi, G.; Ferrara, F.; Montana, M.; Muratori, I.; Amato, C.; Canino, B.; Lo Presti, R.; Hopps, E. Behaviour of the plasma concentration of gelatinases and their tissue inhibitors in subjects with venous leg ulcers. Clin. Hemorheol. Microcirc. 2015, 60, 309–316. [Google Scholar] [CrossRef]
- Ligi, D.; Mosti, G.; Croce, L.; Raffetto, J.D.; Mannello, F. Chronic venous disease—Part II: Proteolytic biomarkers in wound healing. Biochim. Biophys. Acta 2016, 1862, 1900–1908. [Google Scholar] [CrossRef]
- Serena, T.E.; Cullen, B.M.; Bayliff, S.W.; Gibson, M.C.; Carter, M.J.; Chen, L.; Yaakov, R.A.; Samies, J.; Sabo, M.; DeMarco, D.; et al. Defining a new diagnostic assessment parameter for wound care: Elevated protease activity, an indicator of nonhealing, for targeted protease-modulating treatment. Wound Repair Regen. 2016, 24, 589–595. [Google Scholar] [CrossRef]
- Ruf, M.T.; Andreoli, A.; Vujic, G.; Itin, P.; Pluschke, G.; Schmid, P. Exudate collection using wound sponges-An easy, non-invasive and reliable method to explore protease activities in ulcers. Wound Repair Regen. 2017, 25, 320–326. [Google Scholar] [CrossRef]
- Trostrup, H.; Holstein, P.; Karlsmark, T.; Moser, C.; Agren, M.S. Uncontrolled gelatin degradation in non-healing chronic wounds. J. Wound Care 2018, 27, 724–734. [Google Scholar] [CrossRef]
- Gillespie, D.L.; Writing Group III of the Pacific Vascular Symposium 6; Kistner, B.; Glass, C.; Bailey, B.; Chopra, A.; Ennis, B.; Marston, B.; Masuda, E.; Moneta, G.; et al. Venous ulcer diagnosis, treatment, and prevention of recurrences. J. Vasc. Surg. 2010, 52, 8S–14S. [Google Scholar] [CrossRef] [PubMed]
- Lal, B.K.; Saito, S.; Pappas, P.J.; Padberg, F.T., Jr.; Cerveira, J.J.; Hobson, R.W., 2nd; Durán, W.N. Altered proliferative responses of dermal fibroblasts to TGF-beta1 may contribute to chronic venous stasis ulcer. J. Vasc. Surg. 2003, 37, 1285–1293. [Google Scholar] [CrossRef]
- Mendez, M.V.; Raffetto, J.D.; Phillips, T.; Menzoian, J.O.; Park, H.Y. The proliferative capacity of neonatal skin fibroblasts is reduced after exposure to venous ulcer wound fluid: A potential mechanism for senescence in venous ulcers. J. Vasc. Surg. 1999, 30, 734–743. [Google Scholar] [CrossRef]
- Seidman, C.; Raffetto, J.D.; Marien, B.; Kroon, C.; Seah, C.C.; Menzoian, J.O. bFGF-induced alterations in cellular markers of senescence in growth-rescued fibroblasts from chronic venous ulcer and venous reflux patients. Ann. Vasc. Surg. 2003, 17, 239–244. [Google Scholar] [CrossRef]
- Parker, C.N.; Finlayson, K.J.; Shuter, P.; Edwards, H.E. Risk factors for delayed healing in venous leg ulcers: A review of the literature. Int. J. Clin. Pract. 2015, 69, 967–977. [Google Scholar] [CrossRef]
- Margolis, D.J.; Berlin, J.A.; Strom, B.L. Risk factors associated with the failure of a venous leg ulcer to heal. Arch. Dermatol. 1999, 135, 920–926. [Google Scholar] [CrossRef]
- Klode, J.; Stoffels, I.; Körber, A.; Weindorf, M.; Dissemond, J. Relationship between the seasonal onset of chronic venous leg ulcers and climatic factors. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 1415–1419. [Google Scholar] [CrossRef]
- Stojadinovic, O.; Pastar, I.; Vukelic, S.; Mahoney, M.G.; Brennan, D.; Krzyzanowska, A.; Golinko, M.; Brem, H.; Tomic-Canic, M. Deregulation of keratinocyte differentiation and activation: A hallmark of venous ulcers. J. Cell Mol. Med. 2008, 12, 2675–2690. [Google Scholar] [CrossRef]
- Eming, S.A.; Koch, M.; Krieger, A.; Brachvogel, B.; Kreft, S.; Bruckner-Tuderman, L.; Krieg, T.; Shannon, J.D.; Fox, J.W. Differential proteomic analysis distinguishes tissue repair biomarker signatures in wound exudates obtained from normal healing and chronic wounds. J. Proteome Res. 2010, 9, 4758–4766. [Google Scholar] [CrossRef]
- Rayment, E.A.; Upton, Z.; Shooter, G.K. Increased matrix metalloproteinase-9 (MMP-9) activity observed in chronic wound fluid is related to the clinical severity of the ulcer. Br. J. Dermatol. 2008, 158, 951–961. [Google Scholar] [CrossRef]
- James, T.J.; Hughes, M.A.; Cherry, G.W.; Taylor, R.P. Simple biochemical markers to assess chronic wounds. Wound Repair Regen. 2000, 8, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Melikian, R.; O’Donnell, T.F., Jr.; Suarez, L.; Iafrati, M.D. Risk factors associated with the venous leg ulcer that fails to heal after 1 year of treatment. J. Vasc. Surg. Venous Lymphat. Disord. 2019, 7, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Partsch, H.; Mortimer, P. Compression for leg wounds. Br. J. Dermatol. 2015, 173, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Partsch, H. Compression heals leg ulcers due to abolishment of venous reflux. J. Wound Care 2019, 28, 427. [Google Scholar] [CrossRef]
- Gohel, M.S.; Heatley, F.; Liu, X.; Bradbury, A.; Bulbulia, R.; Cullum, N.; Epstein, D.M.; Nyamekye, I.; Poskitt, K.R.; Renton, S.; et al. A Randomized Trial of Early Endovenous Ablation in Venous Ulceration. N. Engl. J. Med. 2018, 378, 2105–2114. [Google Scholar] [CrossRef]
- Epstein, D.M.; Gohel, M.S.; Heatley, F.; Liu, X.; Bradbury, A.; Bulbulia, R.; Cullum, N.; Nyamekye, I.; Poskitt, K.R.; Renton, S.; et al. Cost-effectiveness analysis of a randomized clinical trial of early versus deferred endovenous ablation of superficial venous reflux in patients with venous ulceration. Br. J. Surg. 2019, 106, 555–562. [Google Scholar] [CrossRef]
- Lin, Z.C.; Loveland, P.M.; Johnston, R.V.; Bruce, M.; Weller, C.D. Subfascial endoscopic perforator surgery (SEPS) for treating venous leg ulcers. Cochrane Database Syst. Rev. 2019, 3, CD012164. [Google Scholar] [CrossRef]
- Neglen, P. Chronic venous obstruction: Diagnostic considerations and therapeutic role of percutaneous iliac stenting. Vascular 2007, 15, 273–280. [Google Scholar] [CrossRef]
- Neglen, P.; Hollis, K.C.; Raju, S. Combined saphenous ablation and iliac stent placement for complex severe chronic venous disease. J. Vasc. Surg. 2006, 44, 828–833. [Google Scholar] [CrossRef]
- Raju, S.; Kirk, O.K.; Jones, T.L. Endovenous management of venous leg ulcers. J. Vasc. Surg. Venous Lymphat. Disord. 2013, 1, 165–172. [Google Scholar] [CrossRef]
- Wang, W.; Yu, Z.; Chen, Y.-X. Stenting for chronic obstructive venous disease: A current comprehensive meta-analysis and systematic review. Phlebology 2016, 31, 376–389. [Google Scholar] [CrossRef]
- Raffetto, J.D. Dermal pathology, cellular biology, and inflammation in chronic venous disease. Thromb. Res. 2009, 123 (Suppl. 4), S66–S71. [Google Scholar] [CrossRef]
- Raffetto, J.D.; Marston, W.A. Venous ulcer: What is new? Plast. Reconstr. Surg. 2011, 127 (Suppl. 1), 279S–288S. [Google Scholar] [CrossRef] [PubMed]
- Mannello, F.; Raffetto, J.D. Matrix metalloproteinase activity and glycosaminoglycans in chronic venous disease: The linkage among cell biology, pathology and translational research. Am. J. Transl. Res. 2011, 3, 149–158. [Google Scholar]
- Regulski, M.; Jacobstein, D.A.; Petranto, R.D.; Migliori, V.J.; Nair, G.; Pfeiffer, D. A retrospective analysis of a human cellular repair matrix for the treatment of chronic wounds. Ostomy/Wound Manag. 2013, 59, 38–43. [Google Scholar]
- Ananian, C.E.; Davis, R.D.; Johnson, E.L.; Regulski, M.J.; Reyzelman, A.M.; Saunders, M.C.; Danilkovitch, A. Wound Closure Outcomes Suggest Clinical Equivalency Between Lyopreserved and Cryopreserved Placental Membranes Containing Viable Cells. Adv. Wound Care 2019, 8, 546–554. [Google Scholar] [CrossRef]
- Serena, T.E.; Carter, M.J.; Le, L.T.; Sabo, M.J.; DiMarco, D.T.; EpiFix VLU Study Group. A multicenter, randomized, controlled clinical trial evaluating the use of dehydrated human amnion/chorion membrane allografts and multilayer compression therapy vs. multilayer compression therapy alone in the treatment of venous leg ulcers. Wound Repair Regen. 2014, 22, 688–693. [Google Scholar] [CrossRef]
- Ghatnekar, G.S.; Grek, C.L.; Armstrong, D.G.; Desai, S.C.; Gourdie, R.G. The effect of a connexin43-based Peptide on the healing of chronic venous leg ulcers: A multicenter, randomized trial. J. Investig. Dermatol. 2015, 135, 289–298. [Google Scholar] [CrossRef]
- Bianchi, C.; Cazzell, S.; Vayser, D.; Reyzelman, A.M.; Dosluoglu, H.; Tovmassian, G.; EpiFix VLU Study Group. A multicentre randomised controlled trial evaluating the efficacy of dehydrated human amnion/chorion membrane (EpiFix((R))) allograft for the treatment of venous leg ulcers. Int. Wound J. 2018, 15, 114–122. [Google Scholar] [CrossRef]
- Ghatnekar, G.S.; O’Quinn, M.P.; Jourdan, L.J.; Gurjarpadhye, A.A.; Draughn, R.L.; Gourdie, R.G. Connexin43 carboxyl-terminal peptides reduce scar progenitor and promote regenerative healing following skin wounding. Regen. Med. 2009, 4, 205–223. [Google Scholar] [CrossRef]
- Montgomery, J.; Ghatnekar, G.S.; Grek, C.L.; Moyer, K.E.; Gourdie, R.G. Connexin 43-Based Therapeutics for Dermal Wound Healing. Int. J. Mol. Sci. 2018, 19, 1778. [Google Scholar] [CrossRef] [PubMed]
- Coccheri, S.; Mannello, F. Development and use of sulodexide in vascular diseases: Implications for treatment. Drug Des. Dev. Ther. 2013, 8, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Carroll, B.J.; Piazza, G.; Goldhaber, S.Z. Sulodexide in venous disease. J. Thromb. Haemost. JTH 2019, 17, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Scondotto, G.; Aloisi, D.; Ferrari, P.; Martini, L. Treatment of venous leg ulcers with sulodexide. Angiology 1999, 50, 883–889. [Google Scholar] [CrossRef]
- Coccheri, S.; Scondotto, G.; Agnelli, G.; Palazzini, E.; Zamboni, V.; Arterial Arm of the Suavis Group. Sulodexide in the treatment of intermittent claudication. Results of a randomized, double-blind, multicentre, placebo-controlled study. Eur. Heart J. 2002, 23, 1057–1065. [Google Scholar] [CrossRef]
- Coccheri, S.; Scondotto, G.; Agnelli, G.; Aloisi, D.; Palazzini, E.; Zamboni, V.; Venous arm of the SUAVIS Group. Randomised, double blind, multicentre, placebo controlled study of sulodexide in the treatment of venous leg ulcers. Thromb. Haemost. 2002, 87, 947–952. [Google Scholar]
- Urbanek, T.; Zbigniew, K.; Begier-Krasinska, B.; Baum, E.; Breborowicz, A. Sulodexide suppresses inflammation in patients with chronic venous insufficiency. Int. Angiol. 2015, 34, 589–596. [Google Scholar]
- Gabryel, B.; Jarzabek, K.; Machnik, G.; Adamczyk, J.; Belowski, D.; Obuchowicz, E.; Urbanek, T. Superoxide dismutase 1 and glutathione peroxidase 1 are involved in the protective effect of sulodexide on vascular endothelial cells exposed to oxygen-glucose deprivation. Microvasc. Res. 2016, 103, 26–35. [Google Scholar] [CrossRef]
- De Felice, F.; Megiorni, F.; Pietrantoni, I.; Tini, P.; Lessiani, G.; Mastroiacovo, D.; Mattana, P.; Antinozzi, C.; Di Luigi, L.; Delle Monache, S.; et al. Sulodexide counteracts endothelial dysfunction induced by metabolic or non-metabolic stresses through activation of the autophagic program. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 2669–2680. [Google Scholar] [CrossRef]
- Raffetto, J.D.; Yu, W.; Wang, X.; Calanni, F.; Mattana, P.; Khalil, R.A. Sulodexide Improves Contraction and Decreases Matrix Metalloproteinase-2 and -9 in Veins Under Prolonged Stretch. J. Cardiovasc. Pharmacol. 2020, 75, 211–221. [Google Scholar] [CrossRef]
- Ligi, D.; Maniscalco, R.; Mannello, F. New Frontiers for an Old Drug: What Is New on the Pleiotropic Effect of Sulodexide in Chronic Venous Disease. J. Cardiovasc. Pharmacol. 2020, 75, 208–210. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.R.; Silveira, I.A.; Oliveira, B. Treatment of venous ulcers with growth factors: Systematic review and meta-analysis. Rev. Bras. Enferm. 2019, 72, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhang, D.; Tan, L.; Huang, H. Silver dressings for the healing of venous leg ulcer: A meta-analysis and systematic review. Medicine 2020, 99, e22164. [Google Scholar] [CrossRef] [PubMed]
- Dissemond, J.; Augustin, M.; Dietlein, M.; Faust, U.; Keuthage, W.; Lobmann, R.; Munter, K.C.; Strohal, R.; Stucker, M.; Traber, J.; et al. Efficacy of MMP-inhibiting wound dressings in the treatment of chronic wounds: A systematic review. J. Wound Care 2020, 29, 102–118. [Google Scholar] [CrossRef]
- Norman, G.; Westby, M.J.; Rithalia, A.D.; Stubbs, N.; Soares, M.O.; Dumville, J.C. Dressings and topical agents for treating venous leg ulcers. Cochrane Database Syst. Rev. 2018, 6, CD012583. [Google Scholar] [CrossRef]
- Otero, G.; Agorio, C.; Sujanov, A.; Echarte, L.; Tchekmedyian, A.; Montelongo, M.; Menyou, A.; Rodriguez, A.; Diaz, L.; Rodriguez, I.; et al. Autologous bone marrow-derived cells for venous leg ulcers treatment: A pilot study. Cytotherapy 2019, 21, 189–199. [Google Scholar] [CrossRef]
- Zollino, I.; Campioni, D.; Sibilla, M.G.; Tessari, M.; Malagoni, A.M.; Zamboni, P. A phase II randomized clinical trial for the treatment of recalcitrant chronic leg ulcers using centrifuged adipose tissue containing progenitor cells. Cytotherapy 2019, 21, 200–211. [Google Scholar] [CrossRef]
- Lee, A.J.; Robertson, L.A.; Boghossian, S.M.; Allan, P.L.; Ruckley, C.V.; Fowkes, F.G.; Evans, C.J. Progression of varicose veins and chronic venous insufficiency in the general population in the Edinburgh Vein Study. J. Vasc. Surg. Venous Lymphat. Disord. 2015, 3, 18–26. [Google Scholar] [CrossRef]
- Pannier, F.; Rabe, E. Progression in venous pathology. Phlebology 2015, 30, 95–97. [Google Scholar] [CrossRef]
- Zamboni, P.; Tognazzo, S.; Izzo, M.; Pancaldi, F.; Scapoli, G.L.; Liboni, A.; Gemmati, D. Hemochromatosis C282Y gene mutation increases the risk of venous leg ulceration. J. Vasc. Surg. 2005, 42, 309–314. [Google Scholar] [CrossRef]
- Gemmati, D.; Tognazzo, S.; Serino, M.L.; Fogato, L.; Carandina, S.; De Palma, M.; Izzo, M.; De Mattei, M.; Ongaro, A.; Scapoli, G.L.; et al. Factor XIII V34L polymorphism modulates the risk of chronic venous leg ulcer progression and extension. Wound Repair Regen. 2004, 12, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Gemmati, D.; Tognazzo, S.; Catozzi, L.; Federici, F.; De Palma, M.; Gianesini, S.; Scapoli, G.L.; De Mattei, M.; Liboni, A.; Zamboni, P. Influence of gene polymorphisms in ulcer healing process after superficial venous surgery. J. Vasc. Surg. 2006, 44, 554–562. [Google Scholar] [CrossRef]
- Zamboni, P.; Lanzara, S.; Mascoli, F.; Caggiati, A.; Liboni, A. Inflammation in venous disease. Int. Angiol. 2008, 27, 361–369. [Google Scholar]
- Bosanquet, D.C.; Sanders, A.J.; Ruge, F.; Lane, J.; Morris, C.A.; Jiang, W.G.; Harding, K.G. Development and validation of a gene expression test to identify hard-to-heal chronic venous leg ulcers. Br. J. Surg. 2019, 106, 1035–1042. [Google Scholar] [CrossRef]
- Marston, W.; Fish, D.; Unger, J.; Keagy, B. Incidence of and risk factors for iliocaval venous obstruction in patients with active or healed venous leg ulcers. J. Vasc. Surg. 2011, 53, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, P.F.; Hager, E.S.; Harlander-Locke, M.P.; Pace, N.; Jayaraj, A.; Yohann, A.; Kalbaugh, C.; Marston, W.; Kabnick, L.; Saqib, N.; et al. Treatment of superficial and perforator reflux and deep venous stenosis improves healing of chronic venous leg ulcers. J. Vasc. Surg. Venous Lymphat. Disord. 2020, 8, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Gianesini, S.; Obi, A.; Onida, S.; Baccellieri, D.; Bissacco, D.; Borsuk, D.; Campisi, C.; Campisi, C.C.; Cavezzi, A.; Chi, Y.W.; et al. Global guidelines trends and controversies in lower limb venous and lymphatic disease: Narrative literature revision and experts’ opinions following the vWINter international meeting in Phlebology, Lymphology & Aesthetics, 23–25 January 2019. Phlebology 2019, 34, 4–66. [Google Scholar] [CrossRef]
- Dahm, K.T.; Myrhaug, H.T.; Stromme, H.; Fure, B.; Brurberg, K.G. Effects of preventive use of compression stockings for elderly with chronic venous insufficiency and swollen legs: A systematic review and meta-analysis. BMC Geriatr. 2019, 19, 76. [Google Scholar] [CrossRef]
- Lurie, F.; Lal, B.K.; Antignani, P.L.; Blebea, J.; Bush, R.; Caprini, J.; Davies, A.; Forrestal, M.; Jacobowitz, G.; Kalodiki, E.; et al. Compression therapy after invasive treatment of superficial veins of the lower extremities: Clinical practice guidelines of the American Venous Forum, Society for Vascular Surgery, American College of Phlebology, Society for Vascular Medicine, and International Union of Phlebology. J. Vasc. Surg. Venous Lymphat. Disord. 2019, 7, 17–28. [Google Scholar] [CrossRef]
- Djalalov, S.; Sehatzadeh, S.; Keast, D.H.; Wong, W.W. Economic evaluation of compression stockings for the prevention of venous leg ulcer recurrence in Ontario. J. Wound Care 2020, 29, 141–151. [Google Scholar] [CrossRef]
- Rabe, E.; Partsch, H.; Hafner, J.; Lattimer, C.; Mosti, G.; Neumann, M.; Urbanek, T.; Huebner, M.; Gaillard, S.; Carpentier, P. Indications for medical compression stockings in venous and lymphatic disorders: An evidence-based consensus statement. Phlebology 2018, 33, 163–184. [Google Scholar] [CrossRef] [PubMed]
- Raffetto, J.D.; Eberhardt, R.T.; Dean, S.M.; Ligi, D.; Mannello, F. Pharmacologic treatment to improve venous leg ulcer healing. J. Vasc. Surg. Venous Lymphat. Disord. 2016, 4, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Marston, W.A.; Crowner, J.; Kouri, A.; Kalbaugh, C.A. Incidence of venous leg ulcer healing and recurrence after treatment with endovenous laser ablation. J. Vasc. Surg. Venous Lymphat. Disord. 2017, 5, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Gohel, M.S.; Barwell, J.R.; Taylor, M.; Chant, T.; Foy, C.; Earnshaw, J.J.; Heather, B.P.; Mitchell, D.C.; Whyman, M.R.; Poskitt, K.R. Long term results of compression therapy alone versus compression plus surgery in chronic venous ulceration (ESCHAR): Randomised controlled trial. BMJ 2007, 335, 83. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ulcer Type | Location | ClinicalPresentation |
| Venous ulcer | Gaiter region of the lower leg (anterior to medial malleolus, pretibial lower third of leg, occasionally lateral malleolus) | Single or multiple lesions; shallow depth; irregular shaped edges with well-defined margins; exudates yellow-white in color; commonly with granulation and fibrinous tissue and rarely with necrotic tissue; associated pain may be absent, mild, or extreme; lower extremity edema; eczema and pruritus; hemosiderin deposition or lipodermatosclerosis; dilated and tortuous superficial veins; inverted champagne bottle appearance of the lower leg |
| Arterial ulcer | Distal extremities and sites of trauma (e.g., over the toes, heels, and bony prominences) | Sharply demarcated borders; base yellow, brown, grey, or black in color and usually does not bleed; pale, dry, non-granulating and often necrotic wound bed; the surrounding skin may exhibit erythema, may be cool to touch, and may be hairless and thin; substantial pain, often severe, worsens in decubitus position or when walking; intermittent claudication (leg pain with exercise or at rest); toe nails become opaque and may be lost or hypertrophic; gangrene of the extremities may occur; reduction of capillary refill time; low exudate unless ulcers are infected |
| Lymphatic ulcer | Frequently in the ankle area but may develop in the trauma sites | Shallow depth; regular shaped; flat edge; rosy base; may be oozing, moist, or blistered; lymphorrhea; edema with buffalo hump on the dorsum of the foot and a positive Stemmer’s sign; the skin is translucent, cold, pale, unpigmented, and rarely fibrosclerotic. |
| Vasculitic ulcer | Multifocal or atypical areas. | Sharply marginated; ulcers can be single or multiple with necrosis and fibrin congestion; morphology depends on the size of the vessels and extent of the vascular bed affected; usually fever, weight loss, fatigue joint pain, and rash; reticulated erythema; widespread purpura; the skin surrounding ulcer is normal both before and after ulcer development; painful ulcer |
| Atypical ulcers | Cutaneous and characterized by an atypical wound bed, edges, and perilesional skin: the clinical aspects are correlated with different etiologies. | The wound bed is often exuberant or vegetative, with hyper-granulation tissue or necrotic tissue. Wound edges are undermined or exuberant. Perilesional skin may present with inflammation or satellite lesions. They are caused by inflammatory, neoplastic, vasculopathic, hematological, infectious, and drug-induced etiologies. Approximately 20% of these ulcers are caused by rare etiologies. |
| Ulcer Type | Location | Clinical Presentation |
| Venous ulcer | Gaiter region of the lower leg (anterior to medial malleolus, pretibial lower third of leg, occasionally lateral malleolus) | Single or multiple lesions; shallow depth; irregular shaped edges with well-defined margins; exudates yellow-white in color; commonly with granulation and fibrinous tissue and rarely with necrotic tissue; associated pain may be absent, mild, or extreme; lower extremity edema; eczema and pruritus; hemosiderin deposition or lipodermatosclerosis; dilated and tortuous superficial veins; inverted champagne bottle appearance of the lower leg |
| Arterial ulcer | Distal extremities and sites of trauma (e.g., over the toes, heels, and bony prominences) | Sharply demarcated borders; base yellow, brown, grey, or black in color and usually does not bleed; pale, dry, non-granulating and often necrotic wound bed; the surrounding skin may exhibit erythema, may be cool to touch, and may be hairless and thin; substantial pain, often severe, worsens in decubitus position or when walking; intermittent claudication (leg pain with exercise or at rest); toe nails become opaque and may be lost or hypertrophic; gangrene of the extremities may occur; reduction of capillary refill time; low exudate unless ulcers are infected |
| Lymphatic ulcer | Frequently in the ankle area but may develop in the trauma sites | Shallow depth; regular shaped; flat edge; rosy base; may be oozing, moist, or blistered; lymphorrhea; edema with buffalo hump on the dorsum of the foot and a positive Stemmer’s sign; the skin is translucent, cold, pale, unpigmented, and rarely fibrosclerotic. |
| Vasculitic ulcer | Multifocal or atypical areas. | Sharply marginated; ulcers can be single or multiple with necrosis and fibrin congestion; morphology depends on the size of the vessels and extent of the vascular bed affected; usually fever, weight loss, fatigue joint pain, and rash; reticulated erythema; widespread purpura; the skin surrounding ulcer is normal both before and after ulcer development; painful ulcer |
| Atypical ulcers | Cutaneous and characterized by an atypical wound bed, edges, and perilesional skin: the clinical aspects are correlated with different etiologies. | The wound bed is often exuberant or vegetative, with hyper-granulation tissue or necrotic tissue. Wound edges are undermined or exuberant. Perilesional skin may present with inflammation or satellite lesions. They are caused by inflammatory, neoplastic, vasculopathic, hematological, infectious, and drug-induced etiologies. Approximately 20% of these ulcers are caused by rare etiologies. |
| Ulcer Etiology | Ulcer Type |
|---|---|
| Vascular | Venous, arterial, lymphatic, vasculitis |
| Metabolic | Diabetes mellitus, gout, necrobiosis lipoidica, porphiria cutanea tarda, homocysteinuria, prolidase deficiency, hyperoxaluria, ulcerative colitis, avitaminosis, cutaneous calcinosis |
| Connective tissue disease | Inflammatory bowel disease, pyoderma gangrenosum, rheumatoid arthritis, generalized and localized scleroderma, systemic lupus erythematous, bullous pemphigoid, dermatomyositis, Sjogren’ syndrome, polyarteritis nodosa, leukocytoclastic vasculitis |
| Cutaneous microthrombocitic ulcers | Cryofibrinogenemia, cryoglobulinemia, antiphospholipid syndrome, coagulopathies, calciphylaxis, cholesterol embolization |
| Hematological disease | Sickle cell disease, leukemia, thrombocytosis, thalassemia, hereditary spherocytosis, glucose-6-phosphate dehydrogenase deficiency, essential thrombocythemia, granulocytopenia, polycythemia, monoclonal and polyclonal, dysproteinemia |
| Neoplastic disease | Basal cell carcinoma, squamous cell carcinoma, malignant melanoma, primary cutaneous B cell lymphoma, Marjolin’s ulcer, pseudoepitheliomatous hyperplasia, Kaposi’s sarcoma, angiosarcoma, Bowen’s disease, intra-epidermal carcinoma, papillomatosis cutis carcinoid, neoplasms of lymphoproliferative tissue, Hodgkin disease |
| Panniculitis | Necrobiosis lipoidica, erythema nodosum, erythema induratum |
| Traumatic | Pressure ulcers, radiation damage, thermal burns, decubitus |
| Iatrogenic | Drugs. |
| Atypical | Cutaneous ulcer, caused by inflammatory, neoplastic, vasculopathic, hematological, infectious, and drug-induced etiologies, with approximately 20% of these ulcers caused by rare etiologies |
| Martorell HYTILU | Hypertensive ischemic leg ulcer, stenotic subcutaneous arteriolosclerosis |
| Infection | Pyogenic, osteomyelitis, tuberculosis, syphilis, tropical disease, fungal disease, leishmaniasis, histoplasmosis, herpes, lupus vulgaris, amoebiasis, chromoblastomycosis, coccidiomycosis, viral |
| Main Findings | Specimens | Ref |
|---|---|---|
| ↑ TNF-α in ulcer vs. normal tissue | Ulcer tissue | [61] |
| ↑ TGF-β1 in ulcer fibrin cuff vs. normal tissue | Ulcer tissue | [62] |
| No changes of PDGF-AB, GM-CSF, IL-1α, IL-1β, IL-6, and bFGF in non-healing vs. healing ulcers | WF | [63] |
| ↑ IL-6 level; no changes IL-1β, IL-2, and TNF-α in ulcer vs. normal serum | Serum | [64] |
| ↓ TGF-β RII | Fibroblasts from venous ulcer | [65] |
| ↑IL-1ra, IL-6, and PAF in resting ulcer effluent vs. systemic blood; no changes in TNF-α and IL-1β | Blood | [66] |
| ↑ IL-1β, IP-10, and PF4; ↓ IL-1β, MIP-1β, and RANTES; and ↑ IL-1ra, IL-10, MCP-1, and MIP-1α in healing ulcers | Ulcer tissue and WF | [67] |
| ↑ TNF-α and p75 receptor in nonhealing vs. healing ulcers | WF | [68] |
| ↑ EGFR, bFGF, and TGF-β3 in ulcers vs. normal tissue | Ulcer tissue | [69] |
| ↑ PDGFR-α and PDGFR-β, VEGF | Ulcer tissue | [70] |
| ↑ IL-10; no change GM-CSF in ulcers vs. normal tissue | Ulcer tissue | [71] |
| ↑ IL-10 in ulcer vs. normal tissue | Ulcer tissue | [72] |
| ↑ TGF-β1 in ulcer vs. normal tissue | Ulcer tissue | [73] |
| ↑ sThy-1 in UWF vs. serum | WF and serum | [74] |
| ↑ IL-1, IL-6, and TNF-α in non-healing vs. healing; no change in PDGF, EGF, bFGF, and TGF-β | WF | [75] |
| ↑ VEGF in ulcer vs. normal tissue | Ulcer tissue | [76] |
| ↑ TGF-β1, -2, and -3; TGF-β RI; and RII in healing vs. non-healing ulcers | Ulcer tissue | [77] |
| ↑ VEGF and TNF in non-healing vs. healing | Serum | [78] |
| No changes in TGF-β1 in ulcer vs. normal tissue | Ulcer tissue | [79] |
| ↓ TGF-β RII in ulcer fibroblasts vs. normal tissue | Ulcer tissue | [80] |
| ↑ TNF-α, TNF-rI, IL-1α, IL-6, TGF-β1, PDGF-A, EGF, bFGF, and VEGF in fibroblast from ulcer edge vs. control ↑ PDGF-A and VEGF in non-healing vs. healing ulcers | Ulcer tissue | [81] |
| ↓ IL-8 in healing vs. non-healing ulcer | WF | [82] |
| ↑ c-met in ulcer vs. normal skin ↑ HGF in chronic vs. acute UWF | Ulcer tissueWF | [83] |
| ↑ IL-1α, IL-1β, IL-1ra, EGF, and PDGF-A in endothelial cells near vs. distant ulcer; no changes in IL-6, GM-CSF, and TNF-α | Ulcer tissue | [84] |
| ↑ RANTES mRNA ulcer vs. normal | Blood | [85] |
| ↑ TGF-β1 in healing vs. non-healing | WF and blood | [86] |
| ↑ TGF-β1 and IL-1ra, and ↓IFN-γ in healing ↑ IL-1α, IL-1β, IFN-β, IL-12p40, and GM-CSF in non-healing | Ulcer tissue | [87] |
| ↑ TNF-α in ulcer vs. normal tissue | Ulcer tissue | [88] |
| ↑ RANTES mRNA ulcer vs. normal | Blood | [89] |
| ↑ IL-6 and TNF-α in healed ulcer vs. normal tissue | Valve tissue | [90] |
| ↓ level of IL-8 and MIP-1α in non-healing ulcers ↓ level of IL-1α, IL-1β, and MIP-1δ in healing ulcers | WF | [91] |
| ↓ S100A8/A9 in nonhealing vs. healing | WF | [92] |
| ↑ IL-1α, IL-1β, and IL-8 in WF secreted for 24h vs. WF secreted for 1h | WF | [93] |
| ↑ IL-8, GRO-α, MIP-3α, PARC, HGF, IL-6, MIP-1α, MCP-1, bFGF, TGF-β, CTAK, RANTES, SDF-1, IL-10, and TNF-α | WF | [94] |
| ↑ sVEGFR-1 in non-healing venous wound ↓ VEGFR-2 | WFTissue and plasma | [95] |
| ↑ mRNA of TAM receptors and their ligands (Gas6 and ProS) in VLU patients vs. control probands ↑ IL-1α and CXCL-8 gene expression in non-responder vs. responder VLU patients | PBMCs from patients with VLU | [96] |
| ↓ IL-6, IL-8, VEGF, and TNF- α in relation to ulcer healing speed | Plasma | [97] |
| ↑ IL-1, IL-6, IL-8, VEGF, and TNF-α in infected ulcers vs. uninfected ulcers | Plasma and WF | [98] |
| ↑ NGF and S100A8/A9 in painful ulcers | WF | [99] |
| ↑ IL-1β, IL-1ra, IL-6, IL-8/CXCL8, IL-10, IL-12, IL-17, bFGF, G-CSF, GM-CSF, INF-γ, MCP-1/CCL2, MIP-1α,/CCL3, MIP-1β/CCL4, TNF-α, and VEGF ↑ Eotaxin/CCL11, IP-10/CXCL10, and RANTES/CCL5 | WFPlasma | [20] |
| ↓ S100A8/A9 in VLUs vs. DFUs ↑VEGF in VLUs vs. DFUs | WF | [100] |
| ↑ PDGF-AA, PDGF-AA receptor, PDGF-BB, and PDGF-BB receptor ↑ TGF-β in injured skin vs. healthy skin | Ulcer tissue | [101] |
| ↑TGF-β3 and soluble endoglin | WF | [102] |
| ↑OPN | Ulcer tissue | [103] |
| ↑LDH activity, IL-8, TNF-α, and VEGF in chronic wound vs. acute wound ↑ Nitrotyrosine and Poly(ADP-Ribose) | WFUlcer tissue | [104] |
| ↑GM-CSF, IRF5, TNF-α, IL-1β, and IL-6 in chronic non-healing ulcers | WF | [51] |
| ↑ MCP-1, IL-1β, IL-4, IL-6, IL-8, MIP-1α, FGF-2, and VEGF-A ↓ G-CSF and GM-CSF | Serum | [105] |
| Main Findings | Specimens | Refs |
|---|---|---|
| ↑ MMP-1 in migrating keratinocytes and superficial dermal cells in chronic compared to acute ulcers | Ulcer tissue and in vitro cell culture | [107] |
| ↑TIMP-1 and TIMP3 in proliferating keratinocytes and ↑TIMP-2 in migrating epithelium in acute compared to chronic wounds | Ulcer tissue | [108] |
| ↑ MMP-1 and MMP-8; ↓ TIMP-1 in nonhealing compared to healing ulcers | Ulcer tissue and WF | [109] |
| ↑ MMP-9 in non-healing compared to healing ulcers | WF | [110] |
| ↑MMP-1 mRNA (no changes in protein) in C4 and C6 stages compared to healthy skin; ↑TIMP-1 mRNA (no changes in protein) in C6; ↑active MMP-2 in C4 and C5 stages | Ulcer tissue | [111] |
| ↑MMP-2 and ↑MMP9 in epithelium/edge of acute wounds compared to healthy skin; MMP2 and MMP-9 localized in ulcer bed | Ulcer tissue | [112] |
| ↑EMMPRIN, ↑MMP2, ↑MT-1MMP, and ↑MT2-MMP in ulcer tissue compared to healthy skin | Ulcer tissue | [113] |
| ↑MMP-7, ↑MMP-12 (epithelium), and ↑MMP-13 in malignant ulcers | Ulcer tissue | [114] |
| ↑MMP-2, ↑MMP-9, and angiogenesis induction by wound fluid from chronic compared to acute wounds; ↓ angiogenesis when MMP-2 and MMP-9 were inhibited | WF | [115] |
| ↑ MMP-9 activation in C4–C6 patients compared to healthy subjects | Serum | [116] |
| No changes in MMP-2, TIMP-2, and EMMPRIN; ↑ PDGF-AA in healing compared to non-healing ulcers | Ulcer tissue and WF | [117] |
| ↑ total MMP in ulcer tissue compared to healthy skin; ↑ collagen turnover; ↑MMP-1 and no changes in total MMPs and MMP-3 in healing compared to resistant ulcers | Ulcer tissue | [118] |
| ↑ MMP-1, ↑ MMP-3, and ↓ TIMP-1 in fibroblast exposed to wound fluid from chronic compared to acute UWF | WF | [119] |
| No changes in MMP-9 in relation to ulcer healing | WF and venous blood | [86] |
| ↑ MMP1, 2, 3, 8, 9, 12, and 13 in ulcer tissue compared to normal skin; ↓ MMP-1, -2, -8, and -9 in healing ulcers | Ulcer tissue | [120] |
| ↑ MMP-2 and MMP-9 in ulcer compared to normal tissue | Valve tissue | [90] |
| ↑ MMP-2 and MMP-9 in UWF compared to tissue | WF and ulcer tissue | [121] |
| ↑MMP-9 in ulcers compared to healthy subjects | WF, plasma and ulcer tissue | [122] |
| ↑ MMP-1 and MMP-8 in patients with infected compared to uninfected ulcers; ↑ MMP-2 and MMP-9 in uninfected ulcers | WF and plasma | [97] |
| ↓ MMP-2 and MMP-9 in correlation to ulcer healing | WF | [123] |
| ↓ MMP-9 and NGAL in high-healing ulcer vs. low-healing ulcers | WF and plasma | [124] |
| ↑ MMP-1 and MMP-8 in non-healing wound vs. healing wound | WF and ulcer tissue | [125] |
| ↑ MMP-2, MMP-9, TIMP-1, and TIMP-2 venous leg ulcers vs. healthy controls ↓ MMP-9, TIMP-2, and MMP-9/TIMP-1 ratio in healing ulcers ↑ MMP-2, MMP-9, TIMP-1, TIMP-2, and MMP-2/TIMP-2 ratio in healing ulcers vs. healthy controls | Plasma | [126] |
| ↓ MMP-1, MMP-2, MMP-9, NGAL, and MMP-8 in relation to ulcer healing speed | Plasma | [98] |
| ↑ MMP-1 and MMP-8 in infected ulcers vs. uninfected ulcers ↑ MMP-2 and MMP-9 in uninfected ulcers vs. infected ulcers | Plasma | [97] |
| ↑ MMP-2, MMP-9, MMP-12, TIMP-1, and TIMP-2 in VLU during inflammation ↑ MMP-1, MMP-7, MMP-13, and TIMP-4 in VLU during granulating phases | WF | [127] |
| ↑ MMP-2, MMP-8, MMP-9, and HNE in chronic wound vs. healing wound | WF | [128] |
| ↑ MMP-1, MMP-8, ADAM-17, and ADAMTS-4 ↓ ADAMTS-5, TIMP-1, and TIMP-2 | Serum | [124] |
| ↑ MMP-9 in wound fluid vs. corresponding tissue | WF | [129] |
| ↓ TIMP-1 in chronic VLU vs. acute VLU | WF | [130] |
| ↑ MMP-13 in chronic non-healing wounds | WF | [51] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raffetto, J.D.; Ligi, D.; Maniscalco, R.; Khalil, R.A.; Mannello, F. Why Venous Leg Ulcers Have Difficulty Healing: Overview on Pathophysiology, Clinical Consequences, and Treatment. J. Clin. Med. 2021, 10, 29. https://doi.org/10.3390/jcm10010029
Raffetto JD, Ligi D, Maniscalco R, Khalil RA, Mannello F. Why Venous Leg Ulcers Have Difficulty Healing: Overview on Pathophysiology, Clinical Consequences, and Treatment. Journal of Clinical Medicine. 2021; 10(1):29. https://doi.org/10.3390/jcm10010029
Chicago/Turabian StyleRaffetto, Joseph D., Daniela Ligi, Rosanna Maniscalco, Raouf A. Khalil, and Ferdinando Mannello. 2021. "Why Venous Leg Ulcers Have Difficulty Healing: Overview on Pathophysiology, Clinical Consequences, and Treatment" Journal of Clinical Medicine 10, no. 1: 29. https://doi.org/10.3390/jcm10010029
APA StyleRaffetto, J. D., Ligi, D., Maniscalco, R., Khalil, R. A., & Mannello, F. (2021). Why Venous Leg Ulcers Have Difficulty Healing: Overview on Pathophysiology, Clinical Consequences, and Treatment. Journal of Clinical Medicine, 10(1), 29. https://doi.org/10.3390/jcm10010029