Coagulative Disorders in Critically Ill COVID-19 Patients with Acute Distress Respiratory Syndrome: A Critical Review


 , , , ,
, , , , 
 ,
,  ,
, 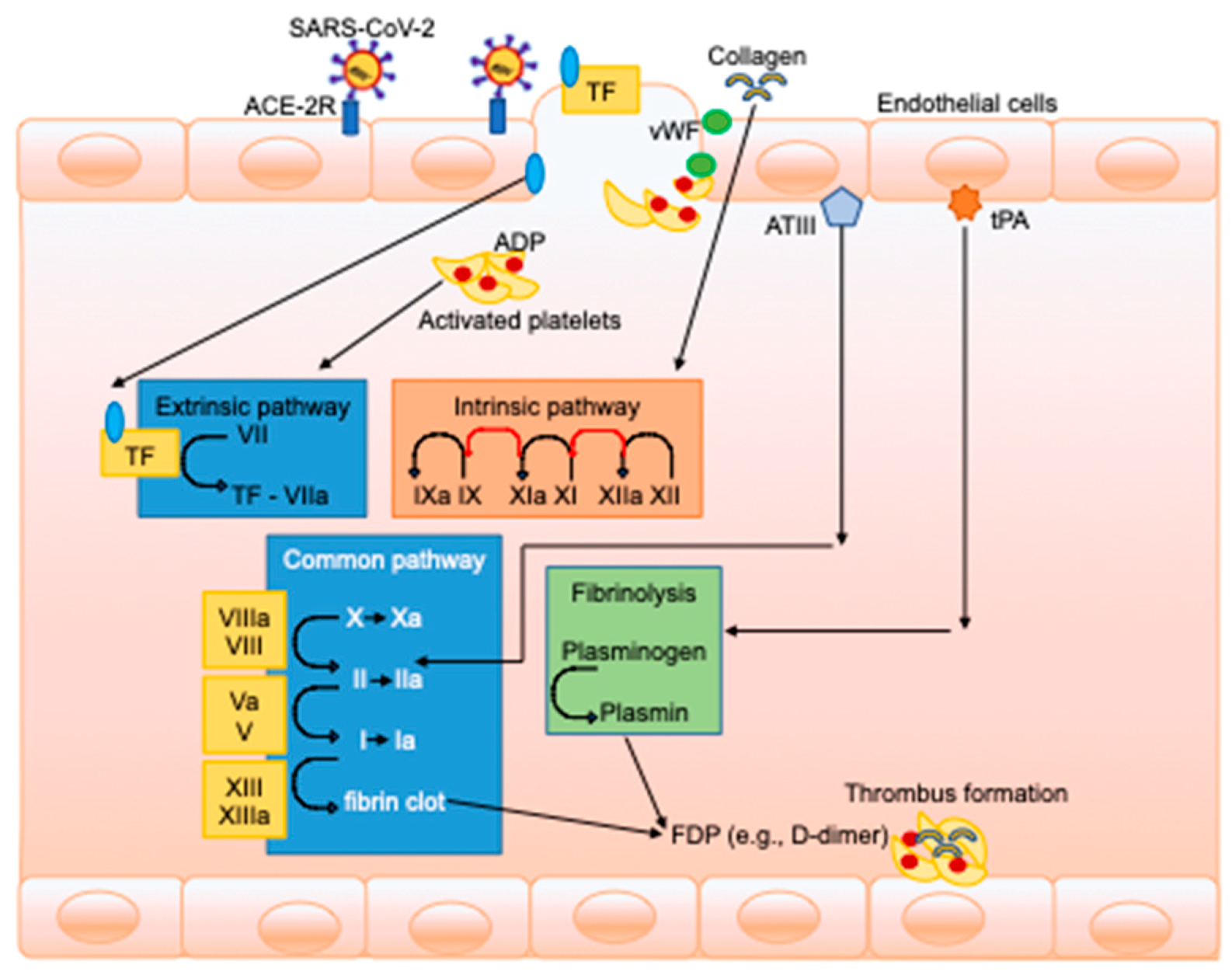{kind=link}
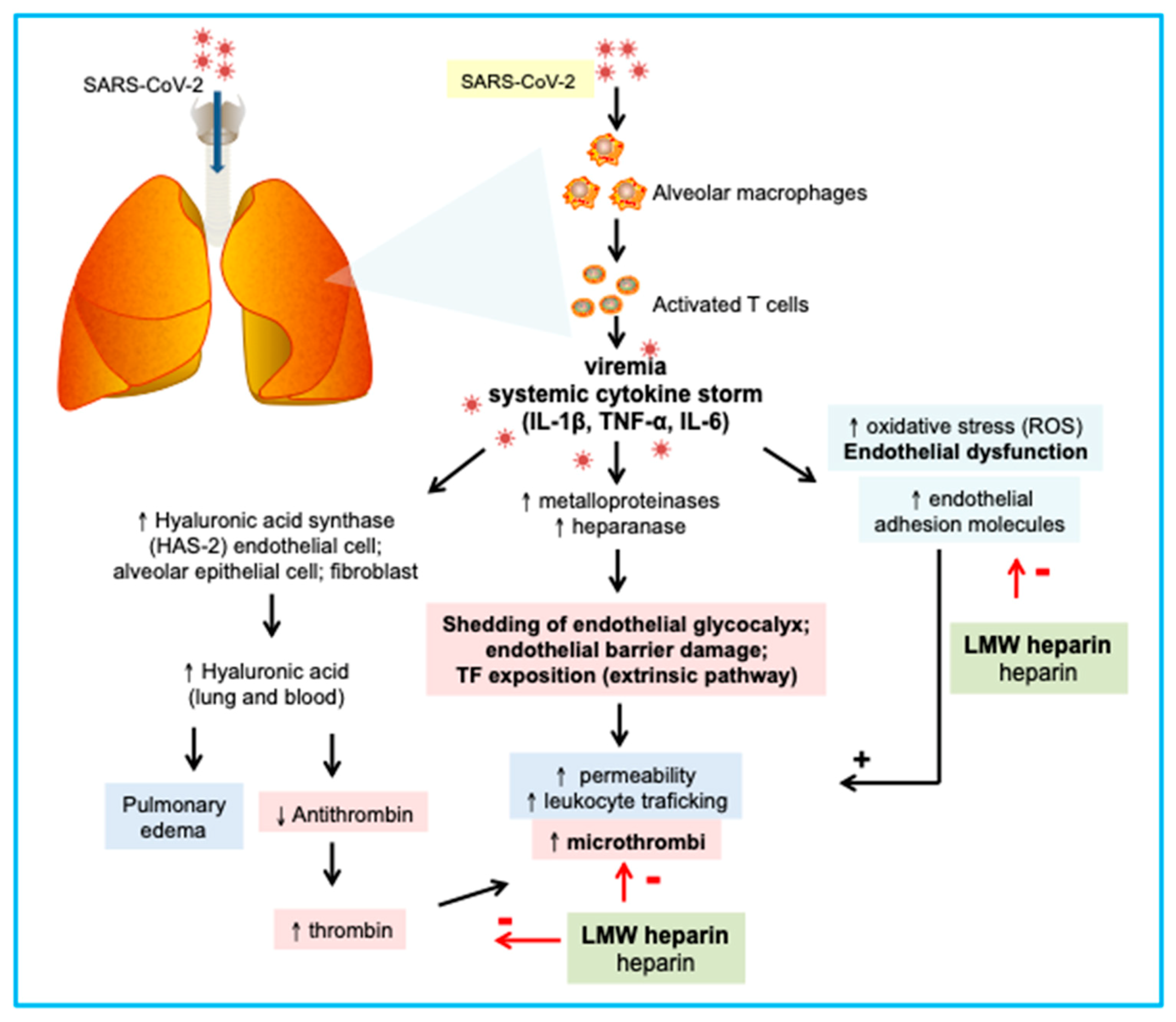{kind=link}
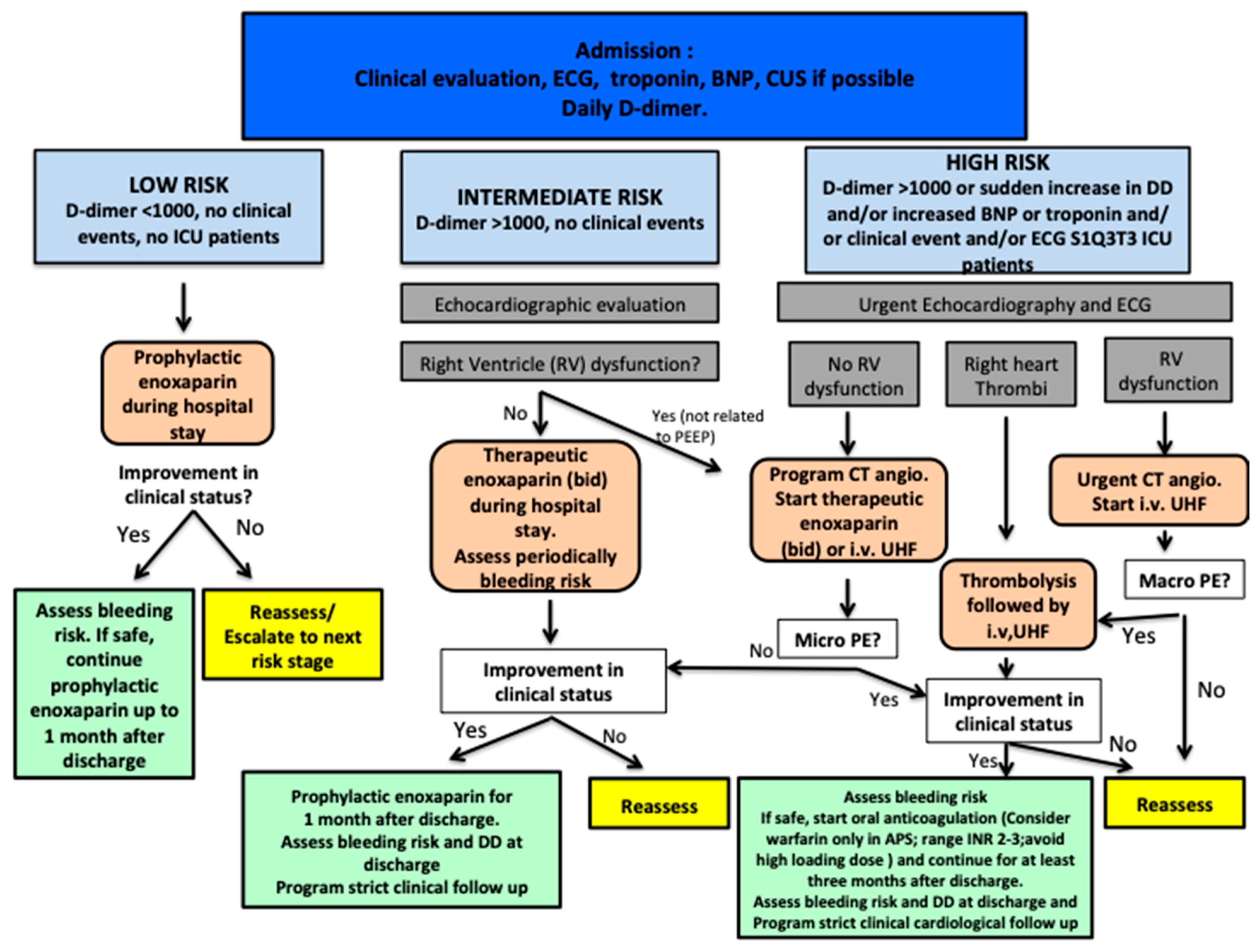{kind=link}
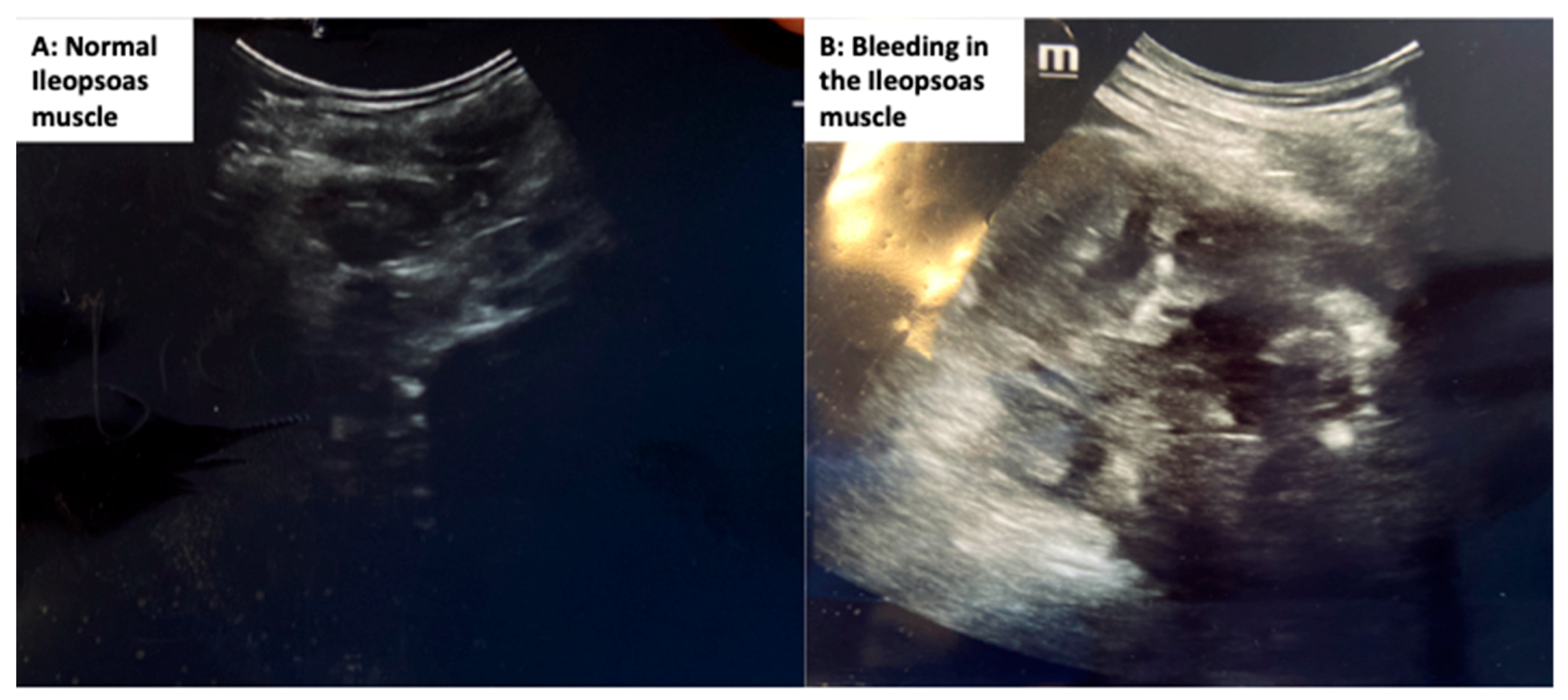{kind=link}
Abstract
1. Introduction
2. Pathophysiology of Coagulative Derangements in COVID-19 Patients
3. Understanding the Role of Platelets and Inflammation in Pneumonia Pathogenesis
4. COVID-19 and Hyper/Hypocoagulable States
5. Clinical Evidence, Manifestations, and Treatment of Coagulative Disorders in COVID-19 Patients
6. Coagulation Markers and Treatment Proposals
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE2 | angiotensin converting enzyme 2 |
| ARDS | Acute distress respiratory syndrome |
| aPTT | activated partial thromboplastin time |
| ARDS | acute respiratory distress syndrome |
| AST | aspartate transaminase |
| AT | antithrombin |
| CD | cluster differentiation |
| CI | confidence interval |
| CMV | cytomegalovirus |
| COVID-19 | coronavirus disease 2019 |
| CRP | C-reactive protein |
| CT | computed tomography |
| CVT | deep venous thrombosis |
| DIC | disseminated intravascular coagulation |
| FDPs | fibrin degradation products |
| HIV | human immunodeficiency virus |
| HR | hazard ratio |
| ICU | intensive care unit |
| IL | interleukin |
| INR | international normalized ratio |
| IVIG | intravenous immunoglobulin |
| LMWH | low molecular weight heparin |
| MOF | multiorgan failure |
| OR | odds ratio |
| PEEP | positive end-expiratory pressure |
| PT | prothrombin time |
| R | reaction time |
| ROS | reactive oxygen species |
| ROTEM | rotational thromboelastometry |
| rPA | tissue plasminogen activator |
| SARS-CoV-2 | severe coronavirus disease-2 |
| TEG | thromboelastography |
| TF | tissue factor |
| TFPI | tissue factor pathway inhibitor |
| TNF-α | tumor necrosis factor-α |
| UFH | unfractionated heparin |
| VTE | venous thromboembolism |
| vWF | von Willebrand factor |
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Robba, C.; Battaglini, D.; Pelosi, P.; Rocco, P.R.M. Multiple organ dysfunction in SARS-CoV-2: MODS-CoV-2. Expert Rev. Respir. Med. 2020, 14, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Shi, S.; Qin, M.; Shen, B.; Cai, Y.; Liu, T.; Yang, F.; Gong, W.; Liu, X.; Liang, J.; Zhao, Q.; et al. Association of Cardiac Injury With Mortality in Hospitalized Patients With COVID-19 in Wuhan, China. JAMA Cardiol. 2020, 5, 802. [Google Scholar] [CrossRef] [PubMed]
- Helms, J.; Tacquard, C.; Severac, F.; Leonard-Lorant, I.; Ohana, M.; Delabranche, X.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Fagot Gandet, F.; et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: A multicenter prospective cohort study. Intensive Care Med. 2020, 46, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, C. Thromboembolic Findings in COVID-19 Autopsies: Pulmonary Thrombosis or Embolism? Ann. Intern. Med. 2020, 173, 394–395. [Google Scholar] [CrossRef]
- Palta, S.; Saroa, R.; Palta, A. Overview of the coagulation system. Indian J. Anaesth. 2014, 58, 515–523. [Google Scholar] [CrossRef]
- Yang, Y.; Tang, H. Aberrant coagulation causes a hyper-inflammatory response in severe influenza pneumonia. Cell Mol. Immunol. 2016, 13, 432–442. [Google Scholar] [CrossRef]
- McGonagle, D.; O’Donnell, J.S.; Sharif, K.; Emery, P.; Bridgewood, C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2020, 2, e437–e445. [Google Scholar] [CrossRef]
- Tschöpe, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hubner, N.; et al. Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat. Rev. Cardiol. 2020, 1–25. [Google Scholar] [CrossRef]
- Biswas, I.; Khan, G.A. Coagulation Disorders in COVID-19: Role of Toll-like Receptors. J. Inflamm. Res. 2020, 13, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Tamagawa-Mineoka, R. Important roles of platelets as immune cells in the skin. J. Derm. Sci. 2015, 77, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Vieira-de-Abreu, A.; Campbell, R.A.; Weyrich, A.S.; Zimmerman, G.A. Platelets: Versatile effector cells in hemostasis, inflammation, and the immune continuum. Semin. Immunopathol. 2012, 34, 5–30. [Google Scholar] [CrossRef] [PubMed]
- Esmon, C.T.; Xu, J.; Lupu, F. Innate immunity and coagulation. J. Thromb. Haemost. 2011, 9, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Stokes, K.Y.; Granger, D.N. Platelets: A critical link between inflammation and microvascular dysfunction. J. Physiol. 2012, 590, 1023–1034. [Google Scholar] [CrossRef]
- Klinger, M.H.F.; Jelkmann, W. Role of blood platelets in infection and inflammation. J. Interf. Cytokine Res. 2002, 22, 913–922. [Google Scholar] [CrossRef]
- Gawaz, M.; Langer, H.; May, A.E. Platelets in inflammation and atherogenesis. J. Clin. Investig. 2005, 115, 3378–3384. [Google Scholar] [CrossRef]
- Morrell, C.N.; Aggrey, A.A.; Chapman, L.M.; Modjeski, K.L. Emerging roles for platelets as immune and inflammatory cells. Blood 2014, 123, 2759–2767. [Google Scholar] [CrossRef]
- Zarbock, A.; Polanowska-Grabowska, R.K.; Ley, K. Platelet-neutrophil-interactions: Linking hemostasis and inflammation. Blood Rev. 2007, 21, 99–111. [Google Scholar] [CrossRef]
- Li, J.; Kim, K.; Barazia, A.; Tseng, A.; Cho, J. Platelet-neutrophil interactions under thromboinflammatory conditions. Cell Mol. Life Sci. 2015, 72, 2627–2643. [Google Scholar] [CrossRef] [PubMed]
- Van Gorp, E.C.M.; Suharti, C.; ten Cate, H.; Dolmans, W.M.V.; van der Meer, J.W.M.; Ten Cate, J.W.; Brandjes, D.P. Review: Infectious Diseases and Coagulation Disorders. J. Infect. Dis. 1999, 180, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Du, R.H.; Li, B.; Zheng, X.S.; Yang, X.L.; Hu, B.; Wang, Y.Y.; Xia, G.F.; Yan, B.; Shi, Z.L.; et al. Molecular and serological investigation of 2019-nCoV infected patients: Implication of multiple shedding routes. Emerg. Microbes Infect. 2020, 9, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lan, Y.; Yuan, X.; Deng, X.; Li, Y.; Cai, X.; Li, L.; He, R.; Tan, Y.; Deng, X.; et al. Detectable 2019-nCoV viral RNA in blood is a strong indicator for the further clinical severity. Emerg. Microbes Infect. 2020, 9, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Gralinski, L.E.; Bankhead, A.; Jeng, S.; Menachery, V.D.; Proll, S.; Belisle, S.E.; Matzke, M.; Webb-Robertson, B.J.M.; Luna, M.L.; Shukla, A.K.; et al. Mechanisms of Severe Acute Respiratory Syndrome Coronavirus-Induced Acute Lung Injury. MBio 2013, 4, e00271-13. [Google Scholar] [CrossRef]
- Berri, F.; Rimmelzwaan, G.F.; Hanss, M.; Albina, E.; Foucault-Grunenwald, M.L.; Lê, V.B.; Vogelzang-van Trierum, E.; Gil, P.; Camerer, E.; Martinez, D.; et al. Plasminogen Controls Inflammation and Pathogenesis of Influenza Virus Infections via Fibrinolysis. PLoS Pathog. 2013, 9, e1003229. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef]
- Ling, Y.; Xu, S.-B.; Lin, Y.-X.; Tian, D.; Zhu, Z.-Q.; Dai, F.-H.; Wu, F.; Song, Z.-G.; Huang, W.; Chen, J.; et al. Persistence and clearance of viral RNA in 2019 novel coronavirus disease rehabilitation patients. Chin. Med. J. 2020, 133, 1039–1043. [Google Scholar] [CrossRef]
- Loelius, S.G.; Lannan, K.L.; Blumberg, N.; Phipps, R.P.; Spinelli, S.L. The HIV protease inhibitor, ritonavir, dysregulates human platelet function in vitro. Throm Res. 2018, 169, 96–104. [Google Scholar] [CrossRef]
- Slutsky, A.S.; Ranieri, V.M. Ventilator-Induced Lung Injury. N. Engl. J. Med. 2013, 369, 2126–2136. [Google Scholar] [CrossRef]
- Li, T. Diagnosis and clinical management of severe acute respiratory syndrome Coronavirus 2 (SARS-CoV-2) infection: An operational recommendation of Peking Union Medical College Hospital (V2.0): Working Group of 2019 Novel Coronavirus, Peking Union Medical College Hospital. Emerg. Microbes Infect. 2020, 9, 582–585. [Google Scholar] [PubMed]
- Lin, L.; Lu, L.; Cao, W.; Li, T. Hypothesis for potential pathogenesis of SARS-CoV-2 infection–a review of immune changes in patients with viral pneumonia. Emerg. Microbes Infect. 2020, 9, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Tomazini, B.M.; Maia, I.S.; Cavalcanti, A.B.; Berwanger, O.; Rosa, R.G.; Veiga, V.C.; Avezum, A.; Lopes, R.D.; Bueno, F.R.; Silva, M.V.A.; et al. Effect of Dexamethasone on Days Alive and Ventilator-Free in Patients With Moderate or Severe Acute Respiratory Distress Syndrome and COVID-19. JAMA 2020, 324, 1307. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Ten Cate, H.; van der Poll, T.; van Deventer, S.J. Pathogenesis of Disseminated Intravascular Coagulation in Sepsis. JAMA 1993, 270, 975–979. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S.M.; Wu, A.; To, K.F.; Lee, N.; Lam, C.W.K.; Wong, C.K.; Chan, P.K.S.; Ng, H.L.M.; Hui, D.S.; Tam, J.S.; et al. Haematological manifestations in patients with severe acute respiratory syndrome: Retrospective analysis. BMJ 2003, 326, 1358–1362. [Google Scholar] [CrossRef]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Zu, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 1–11. [Google Scholar] [CrossRef]
- Deng, Y.; Liu, W.; Liu, K.; Fang, Y.-Y.; Shang, J.; Zhou, L.; Wang, K.; Leng, F.; Wei, S.; Chen, L.; et al. Clinical characteristics of fatal and recovered cases of coronavirus disease 2019 (COVID-19) in Wuhan, China: A retrospective study. Chin. Med. J. 2020, 133, 1261–1267. [Google Scholar] [CrossRef]
- Poissy, J.; Goutay, J.; Caplan, M.; Parmentier, E.; Duburcq, T.; Lassalle, F.; Jeanpierre, E.; Rauch, A.; Labreuche, J.; Susen, S.; et al. Pulmonary Embolism in Patients with COVID-19. Circulation 2020, 142, 184–186. [Google Scholar] [CrossRef]
- Xu, J.F.; Wang, L.; Zhao, L.; Li, F.; Liu, J.; Zhang, L.; Li, Q.; Gu, J.; Liang, S.; Zhao, Q.; et al. Risk assessment of venous thromboembolism and bleeding in COVID-19 patients. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Llitjos, J.; Leclerc, M.; Chochois, C.; Monsallier, J.; Ramakers, M.; Auvray, M.; Merouani, K. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J. Thromb. Haemost. 2020, 18, 1743–1746. [Google Scholar] [CrossRef] [PubMed]
- Bompard, F.; Monnier, H.; Saab, I.; Tordjman, M.; Abdoul, H.; Fournier, L.; Sanchez, O.; Lorut, C.; Chassagnon, G.; Revel, M.-P. Pulmonary embolism in patients with COVID-19 pneumonia. Eur. Respir. J. 2020, 56, 2001365. [Google Scholar] [CrossRef] [PubMed]
- Marsden, P.A. Inflammation and Coagulation in the Cardiovascular System. Circ. Res. 2006, 99, 1152–1153. [Google Scholar] [CrossRef] [PubMed]
- Thachil, J.; Tang, N.; Gando, S.; Falanga, A.; Cattaneo, M.; Levi, M.; Clark, C.; Iba, T. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J. Thromb. Haemost. 2020, 18, 1023–1026. [Google Scholar] [CrossRef]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020, 18, 1094–1099. [Google Scholar] [CrossRef]
- Favaloro, E.J.; Lippi, G. Recommendations for Minimal Laboratory Testing Panels in Patients with COVID-19: Potential for Prognostic Monitoring. Semin Thromb. Hemost. 2020, 46, 379–382. [Google Scholar] [CrossRef]
- Levi, M.; Meijers, J.C. DIC: Which laboratory tests are most useful. Blood Rev. 2011, 25, 33–37. [Google Scholar] [CrossRef]
- Pavoni, V.; Gianesello, L.; Pazzi, M.; Stera, C.; Meconi, T.; Frigieri, F.C. Evaluation of coagulation function by rotation thromboelastometry in critically ill patients with severe COVID-19 pneumonia. J. Thromb. Thrombolysis. 2020, 50, 281–286. [Google Scholar] [CrossRef]
- Spiezia, L.; Boscolo, A.; Poletto, F.; Cerruti, L.; Tiberio, I.; Campello, E.; Navalesi, P.; Simioni, P. COVID-19-Related Severe Hypercoagulability in Patients Admitted to Intensive Care Unit for Acute Respiratory Failure. Thromb Haemost. 2020, 120, 998–1000. [Google Scholar] [CrossRef]
- Panigada, M.; Bottino, N.; Tagliabue, P.; Grasselli, G.; Novembrino, C.; Chantarangkul, V.; Pesenti, A.; Peyvandi, F.; Tripodi, A. Hypercoagulability of COVID-19 patients in intensive care unit: A report of thromboelastography findings and other parameters of hemostasis. J. Thromb. Haemost. 2020, 18, 1738–1742. [Google Scholar] [CrossRef]
- Reikvam, H.; Steien, E.; Hauge, B.; Liseth, K.; Hagen, K.G.; Størkson, R.; Hervig, T. Thrombelastography. Transfus Apher. Sci. 2009, 40, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.C.; Meijers, J.C.M.; Vroom, M.B.; Juffermans, N.P. Utility of thromboelastography and/or thromboelastometry in adults with sepsis: A systematic review. Crit Care. 2014, 18, R30. [Google Scholar] [CrossRef] [PubMed]
- Ranucci, M.; Ballotta, A.; Di Dedda, U.; Bayshnikova, E.; Dei Poli, M.; Resta, M.; Falco, M.; Albano, G.; Menicanti, L. The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J. Thromb. Haemost. 2020, 18, 1747–1751. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Liu, R.; Shin, S.; Sinha, R.; Pogliano, J.; Pogliano, K.; Griffin, J.H.; Nizet, V.; Corriden, R. SCH79797 improves outcomes in experimental bacterial pneumonia by boosting neutrophil killing and direct antibiotic activity. J. Antimicrob. Chemother. 2018, 73, 1586–1594. [Google Scholar] [CrossRef] [PubMed]
- Artzner, T.; Clere-Jehl, R.; Schenck, M.; Greget, M.; Merdji, H.; De Marini, P.; Tuzin, N.; Helms, J.; Meziani, F. Spontaneous ilio-psoas hematomas complicating intensive care unit hospitalizations. PLoS ONE 2019, 14, e0211680. [Google Scholar] [CrossRef] [PubMed]
- Spyropoulos, A.C.; Levy, J.H.; Ageno, W.; Connors, J.M.; Hunt, B.J.; Iba, T.; Levi, M.; Samama, C.M.; Thachil, J.; Giannis, D.; et al. Scientific and Standardization Committee communication: Clinical guidance on the diagnosis, prevention, and treatment of venous thromboembolism in hospitalized patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Z.; Li, C.; Chen, Y.; Gerotziafas, G.; Zhang, Z.; Wan, J.; Liu, P.; Elalamy, I.; Wang, C. Prevention and Treatment of Venous Thromboembolism Associated with Coronavirus Disease 2019 Infection: A Consensus Statement before Guidelines. Thromb. Haemost. 2020, 120, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Marietta, M.; Ageno, W.; Artoni, A.; De Candia, E.; Gresele, P.; Marchetti, M.; Marcucci, R.; Tripodi, A. COVID-19 and haemostasis: A position paper from Italian Society on Thrombosis and Haemostasis (SISET). Blood Transfus. 2020, 18, 167–169. [Google Scholar]
- Flaczyk, A.; Rosovsky, R.P.; Reed, C.T.; Bankhead-Kendall, B.K.; Bittner, E.A.; Chang, M.G. Comparison of published guidelines for management of coagulopathy and thrombosis in critically ill patients with COVID 19: Implications for clinical practice and future investigations. Crit. Care 2020, 24, 559. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robba, C.; Battaglini, D.; Ball, L.; Valbusa, A.; Porto, I.; Della Bona, R.; La Malfa, G.; Patroniti, N.; Brunetti, I.; Loconte, M.; et al. Coagulative Disorders in Critically Ill COVID-19 Patients with Acute Distress Respiratory Syndrome: A Critical Review. J. Clin. Med. 2021, 10, 140. https://doi.org/10.3390/jcm10010140
Robba C, Battaglini D, Ball L, Valbusa A, Porto I, Della Bona R, La Malfa G, Patroniti N, Brunetti I, Loconte M, et al. Coagulative Disorders in Critically Ill COVID-19 Patients with Acute Distress Respiratory Syndrome: A Critical Review. Journal of Clinical Medicine. 2021; 10(1):140. https://doi.org/10.3390/jcm10010140
Chicago/Turabian StyleRobba, Chiara, Denise Battaglini, Lorenzo Ball, Alberto Valbusa, Italo Porto, Roberta Della Bona, Giovanni La Malfa, Nicolò Patroniti, Iole Brunetti, Maurizio Loconte, and et al. 2021. "Coagulative Disorders in Critically Ill COVID-19 Patients with Acute Distress Respiratory Syndrome: A Critical Review" Journal of Clinical Medicine 10, no. 1: 140. https://doi.org/10.3390/jcm10010140
APA StyleRobba, C., Battaglini, D., Ball, L., Valbusa, A., Porto, I., Della Bona, R., La Malfa, G., Patroniti, N., Brunetti, I., Loconte, M., Bassetti, M., Giacobbe, D. R., Vena, A., Silva, C. L. M., Rocco, P. R. M., & Pelosi, P. (2021). Coagulative Disorders in Critically Ill COVID-19 Patients with Acute Distress Respiratory Syndrome: A Critical Review. Journal of Clinical Medicine, 10(1), 140. https://doi.org/10.3390/jcm10010140