Preventing Silica Scale Formation Using Hydroxide Ions Generated by Water Electrolysis



Abstract
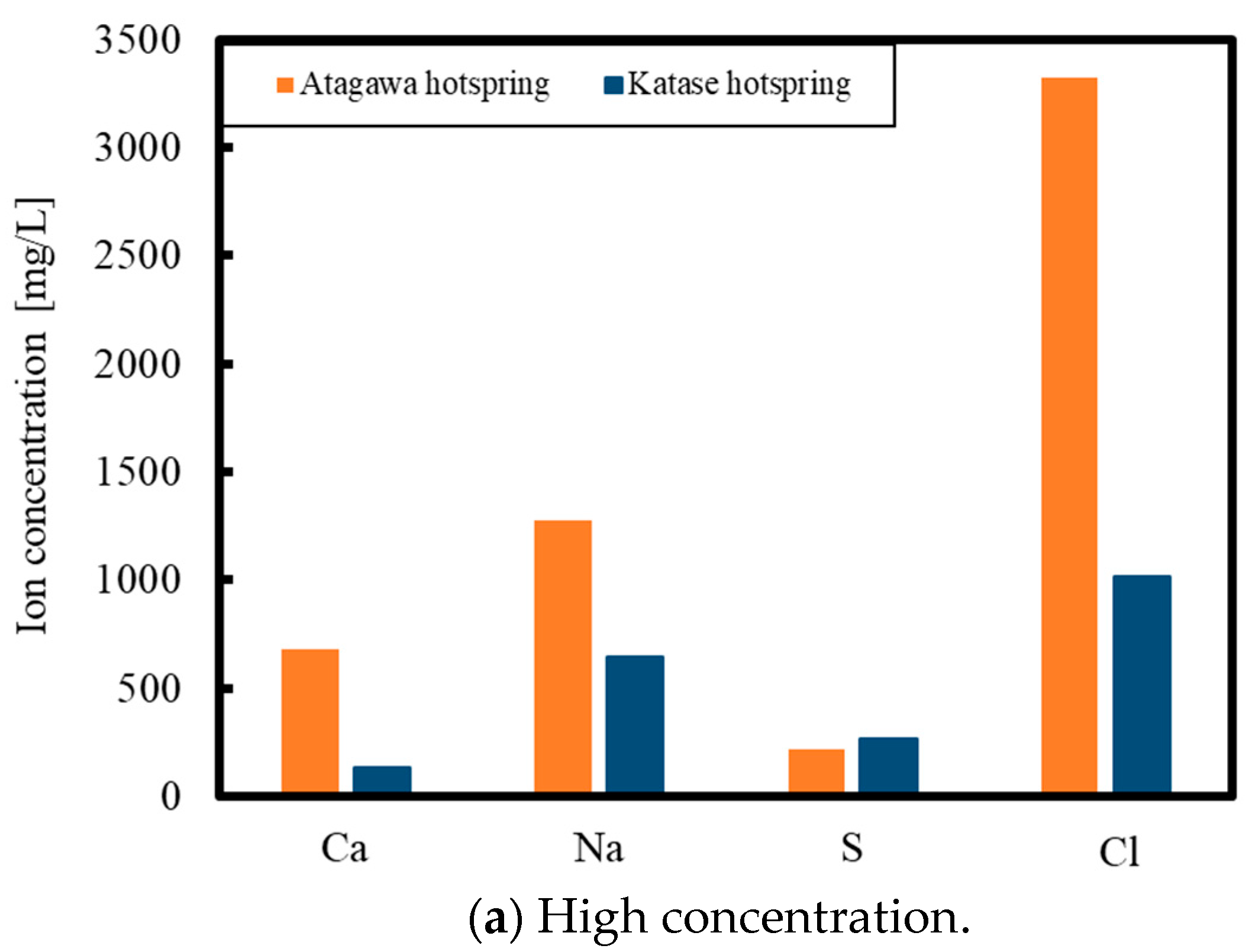
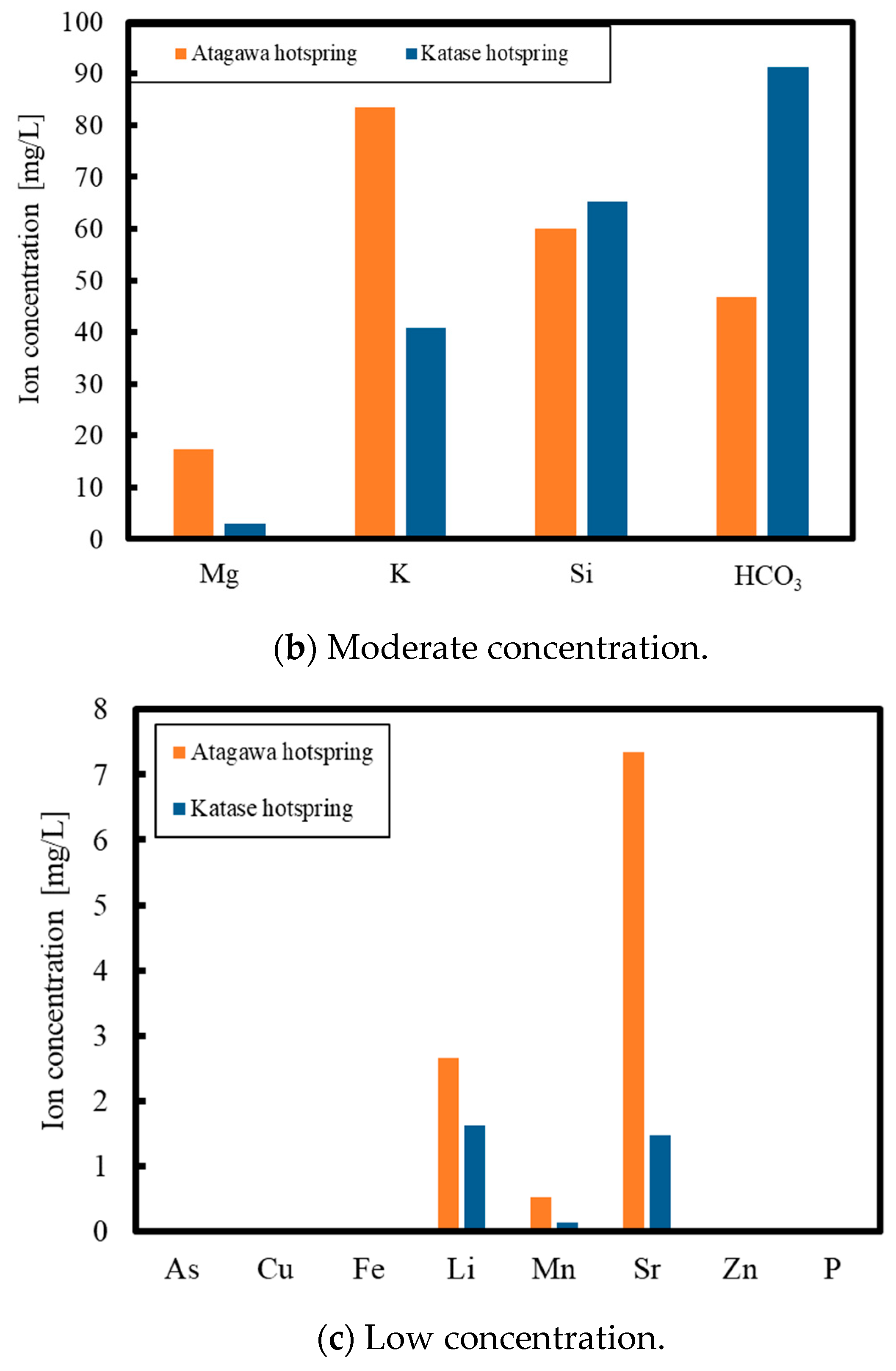
1. Introduction
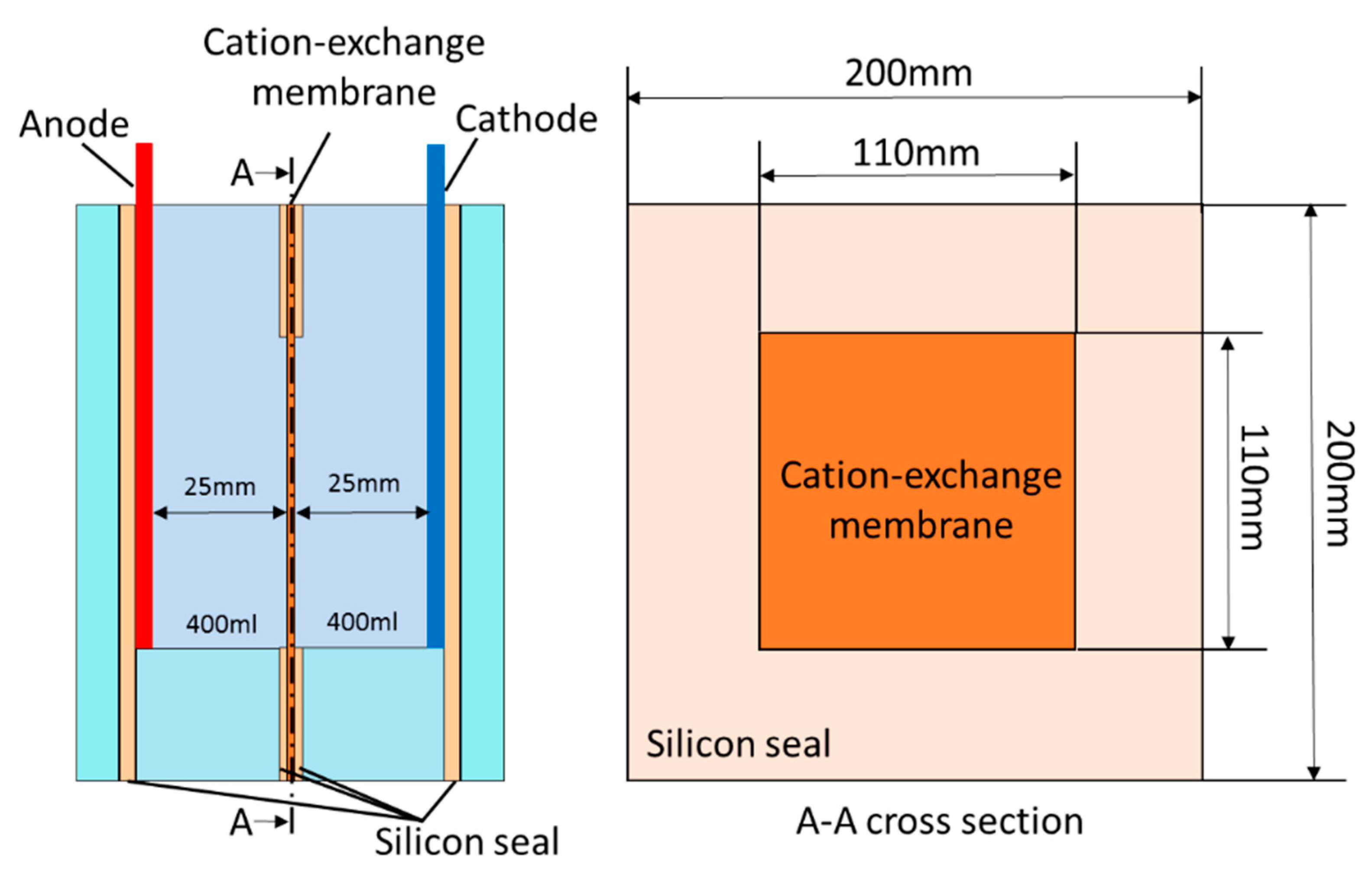
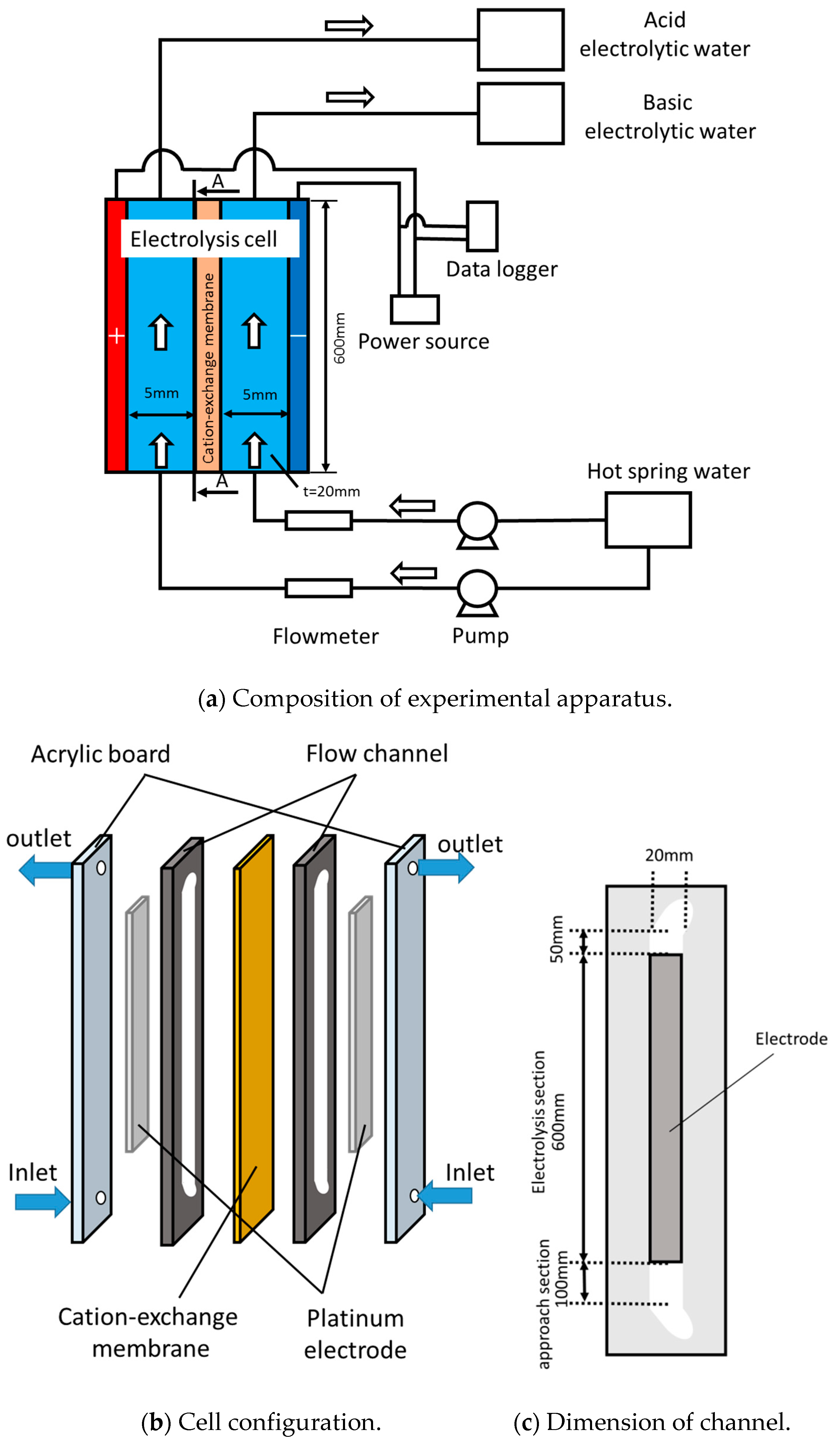
2. Experimental Methods
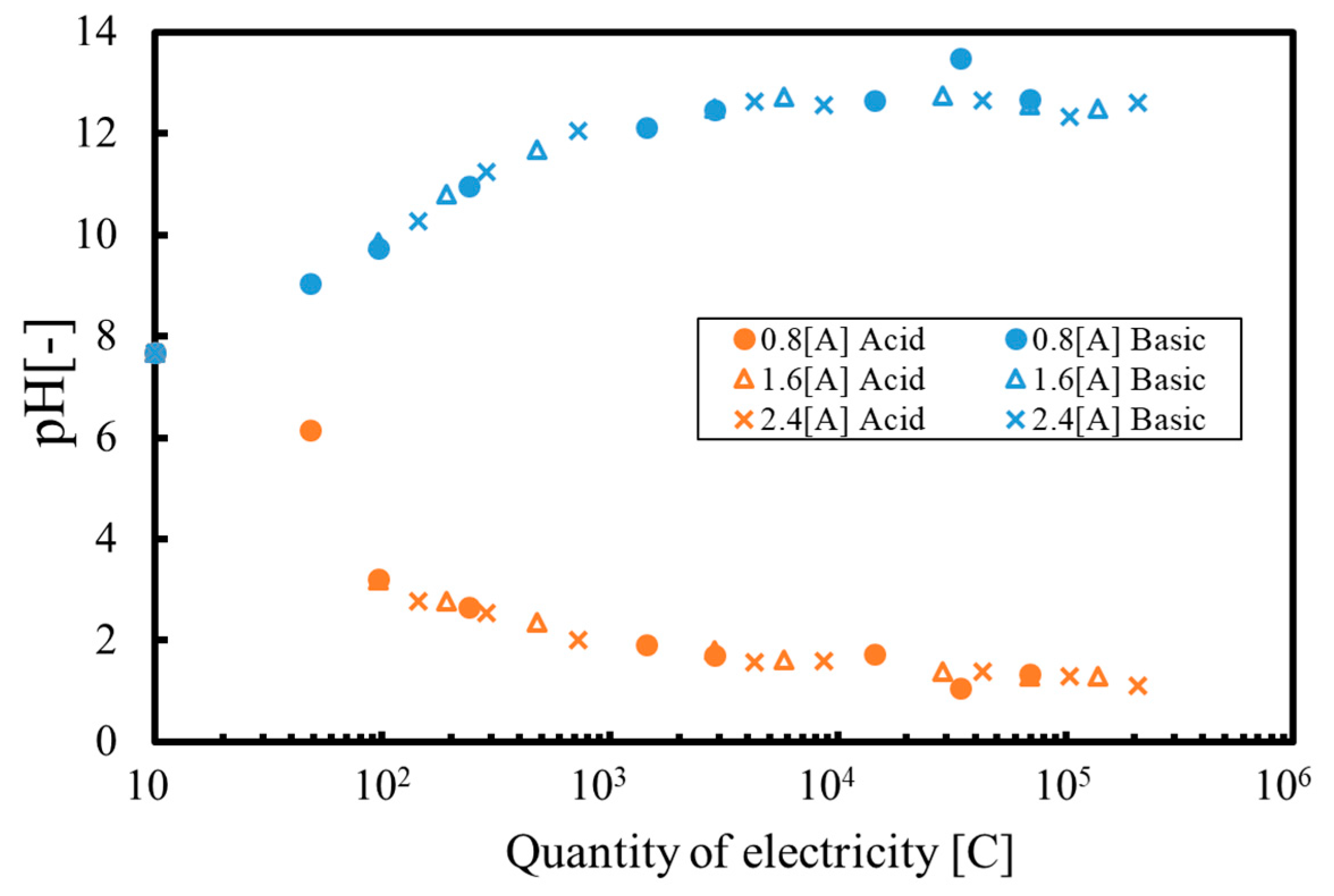
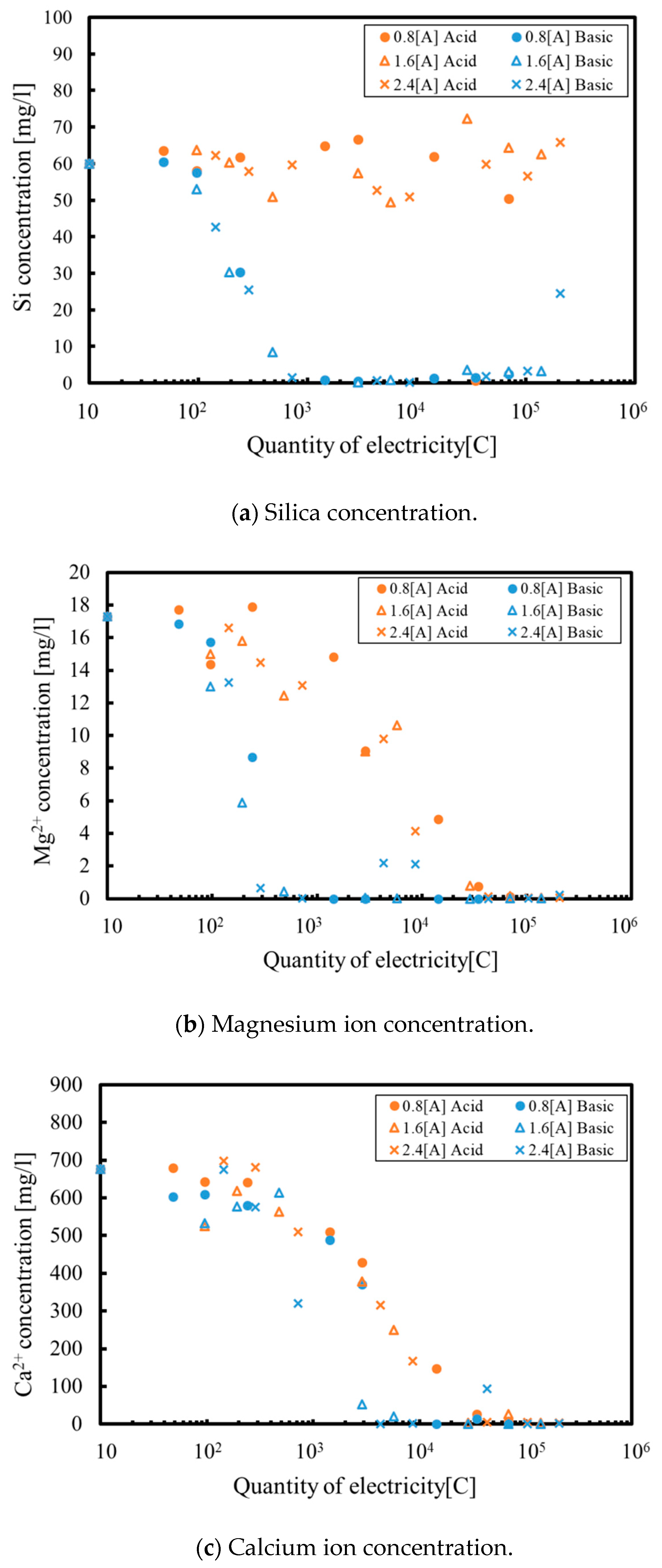
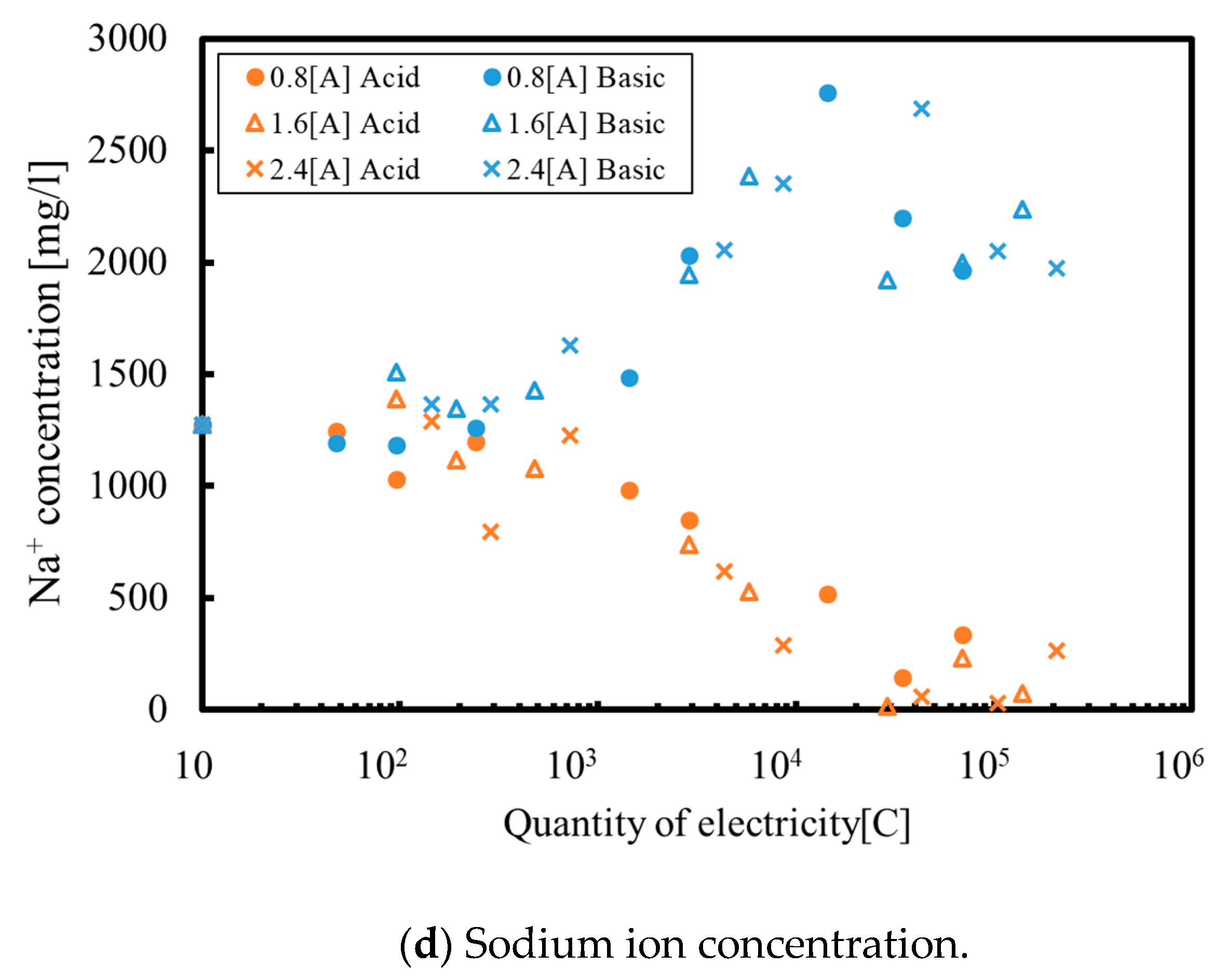
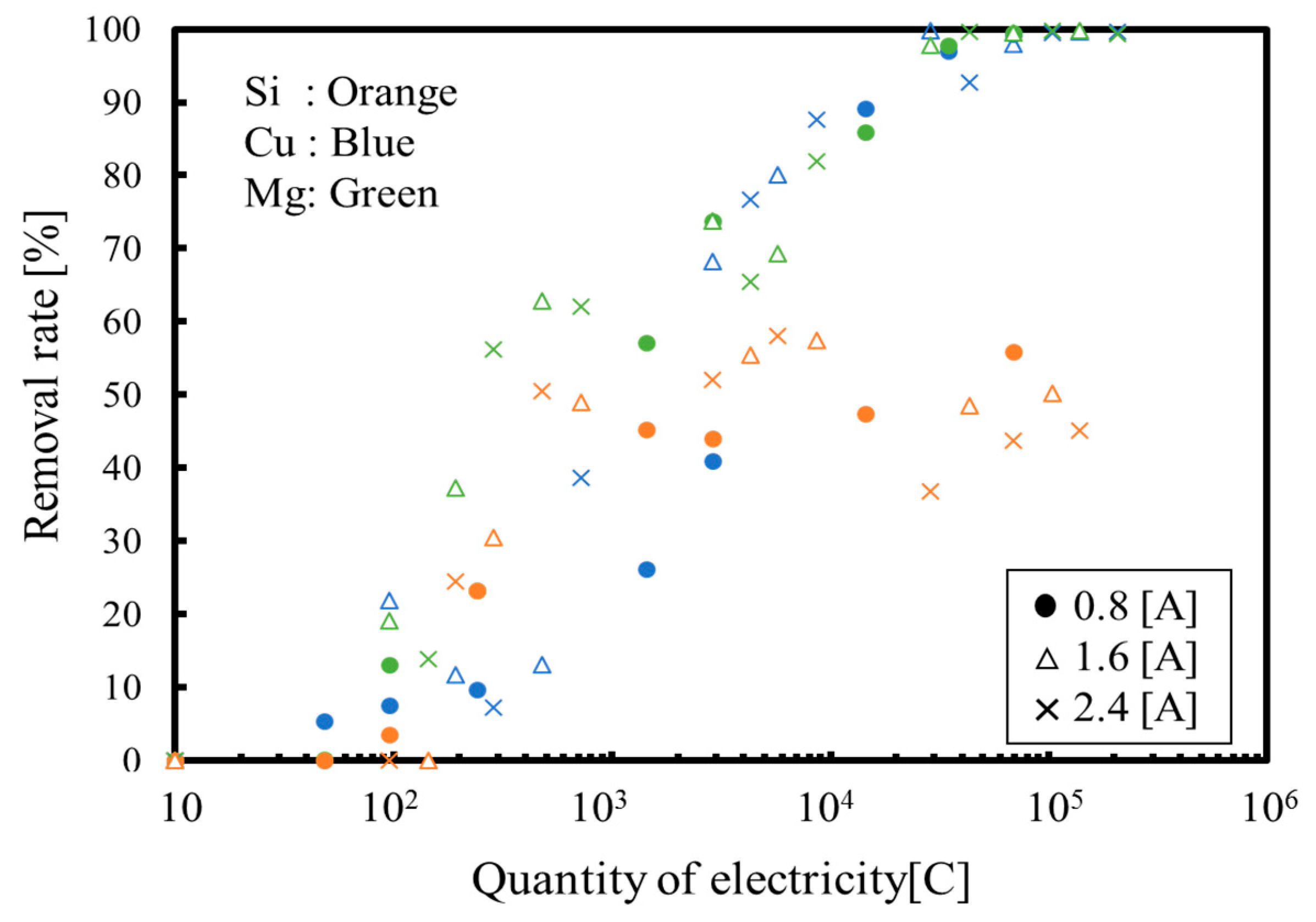
3. Results and Discussion
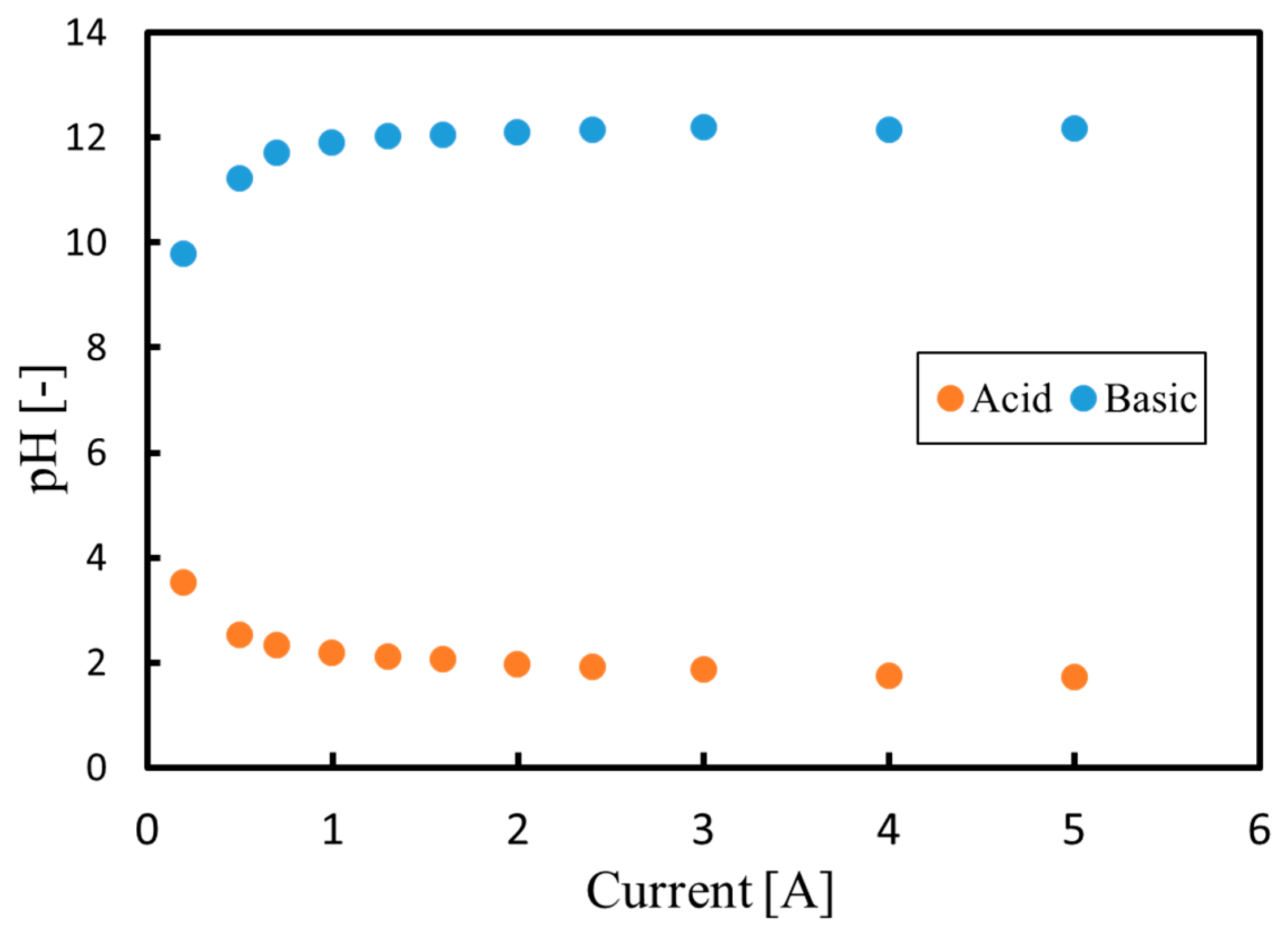
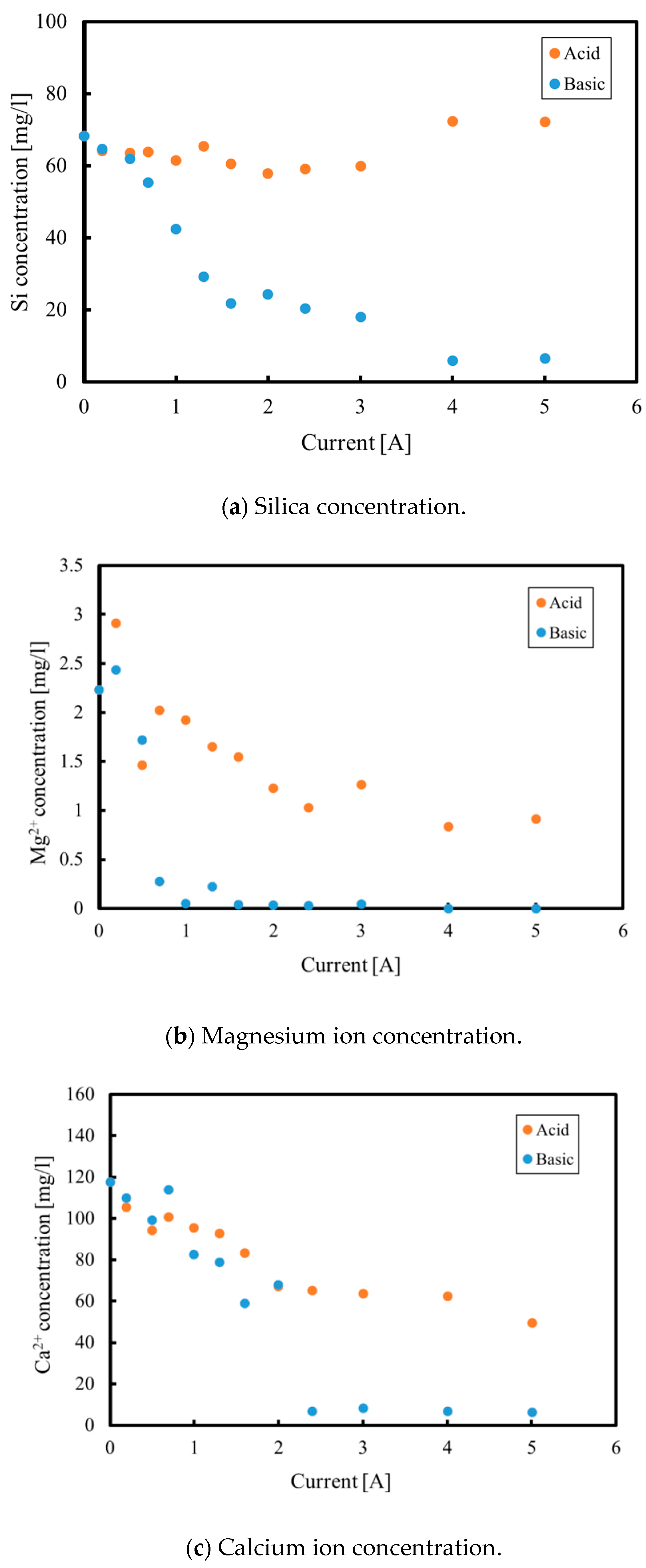
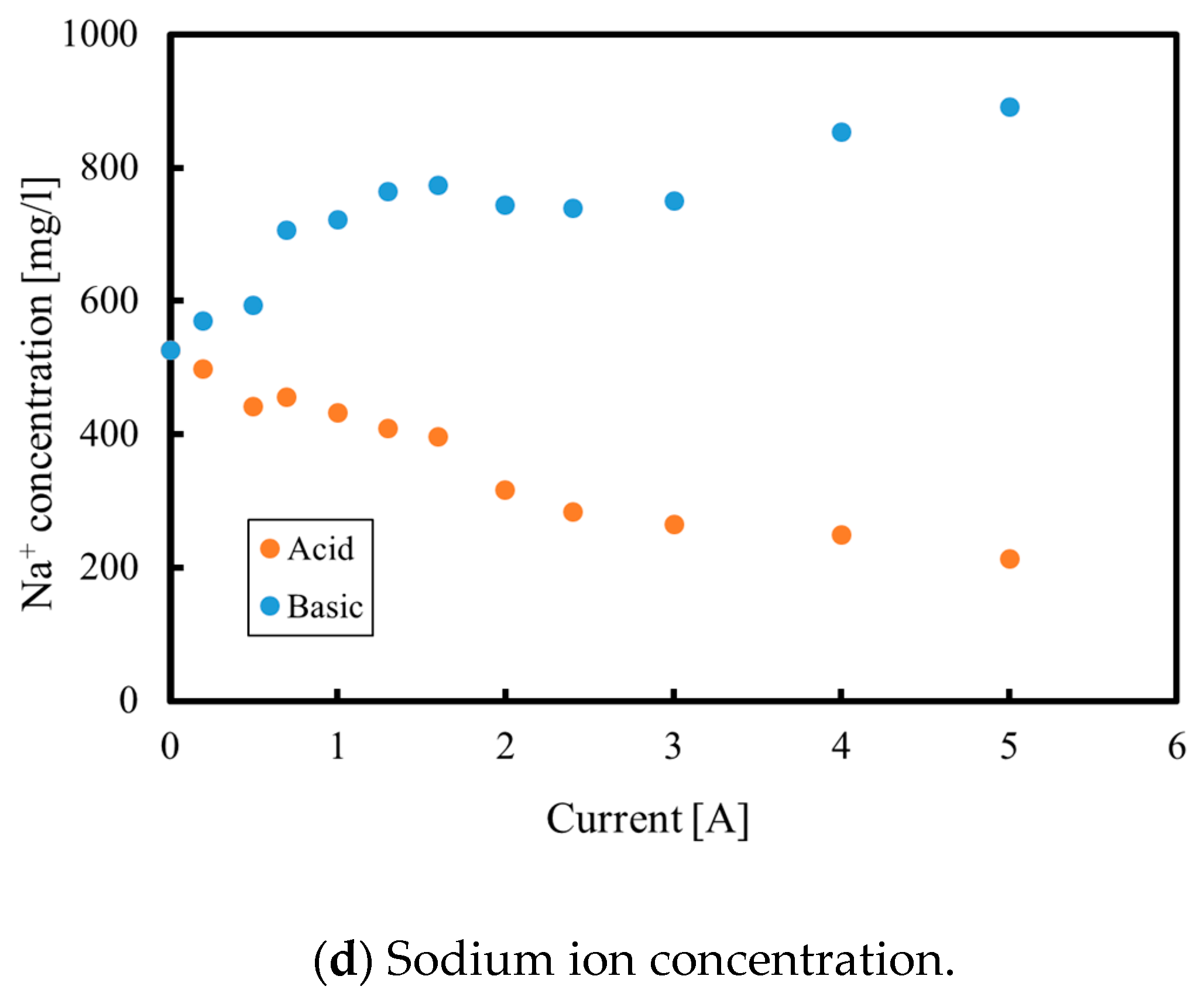
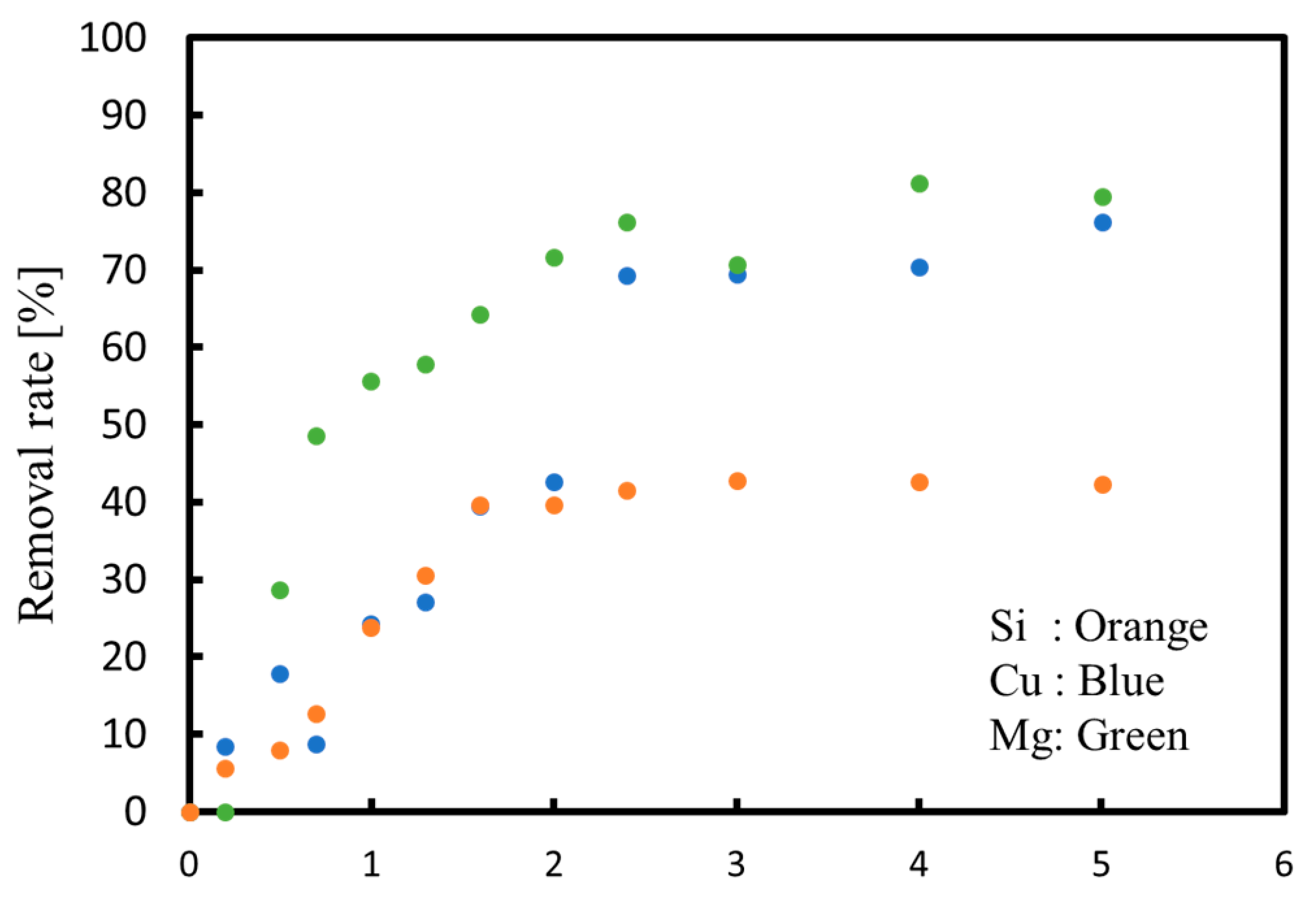
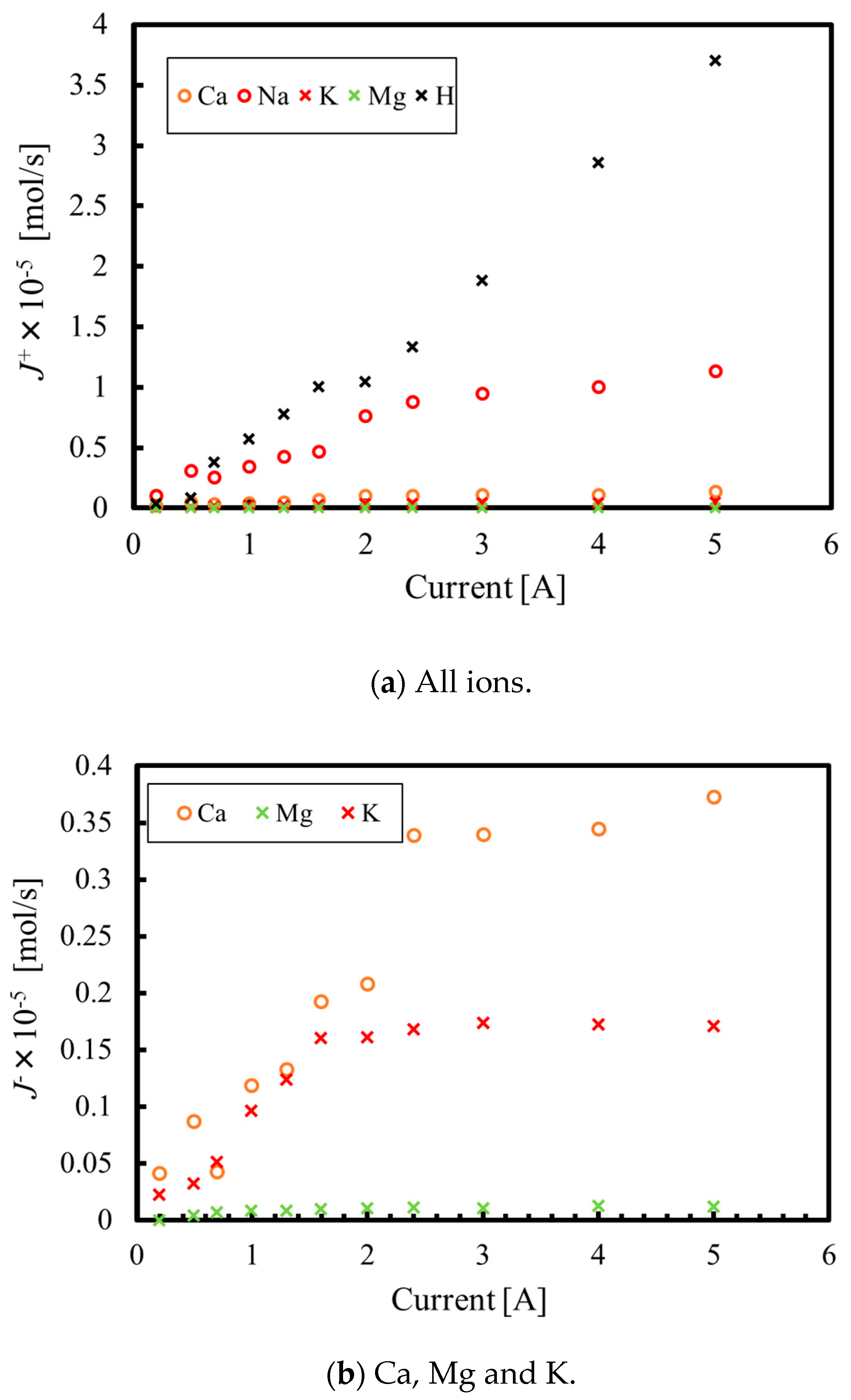
3.1. Batch Process
3.2. Continuous Process
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fridleifsson, I.B. Geothermal energy for the benefit of the people. Renew. Sustain. Energy Rev. 2001, 5, 299–312. [Google Scholar] [CrossRef]
- The Geothermal Research Society of Japan. Handbook of Geothermal Energy; Ohmsha, Ltd.: Tokyo, Japan, 2014. (In Japanese) [Google Scholar]
- Yanagisawa, N. Case Study of Calcium Carbonate Scale at EGS and Hot Spring Binary System. In Proceedings of the World Geothermal Congress 2015, Melbourne, Australia, 19–25 April 2015; pp. 1–5. [Google Scholar]
- Grassiani, M. Siliceous scaling aspects of geothermal power generation using binary cycle heat recovery. Bull. d’Hydrogiologie 1999, 7, 379–391. [Google Scholar]
- Bremere, I.; Kennedy, M.; Mhyio, S.; Geert-Jan Witkamp, A.J.; Schippers, J. Prevention of silica scale in membrane systems: Removal of monomer and polymer silica. Desalination 2000, 132, 89–100. [Google Scholar] [CrossRef]
- Cho, Y.I.; Lee, S.H.; Kim, W.; Suh, S. Physical water treatment for the mitigation of mineral fouling in cooling-tower water applications. In Proceedings of the Heat Exchanger Fouling and Cleaning: Fundamentals and Applications, Santa Fe, NM, USA, 18–22 May 2003. [Google Scholar]
- Haklidir, M.; Haklidir, F. Fuzzy Control of Calcium Carbonate and Silica Scales in Geothermal Systems. In Proceedings of the World Geothermal Congress 2015, Melbourne, Australia, 19–25 April 2015. [Google Scholar]
- Yildirim, N.; Yildirim, A. High Total Inorganic Carbon Concentration Dependent Carbonate Scaling and Mitigation System in Moderate to High Enthalpy Geothermal Fields in Turkey. In Proceedings of the World Geothermal Congress, Melbourne, Australia, 25–29 April 2015. [Google Scholar]
- Matsuki, K.; Okumura, K.; Sugimoto, F. Removal of geothermal scales with high speed water jets. J. Geothorm. Res. Soc. Jpn. 1987, 9, 255–270. [Google Scholar]
- Sano, Y.; Nakashima, D. Prevention of calcium carbonate scale using electrolyzed water. Int. J. Heat Mass Transf. 2018, 127, 1147–1156. [Google Scholar] [CrossRef]
- Sugita, H.; Yamamoto, M.; Nakamura, M. Equilibrium concentration of dissolved silica solution. J. Geotherm. Res. Soc. Jpn. 1998, 20, 225–232. [Google Scholar]
- Neofotistou, E.; Demadis, K.D. Silica scale inhibition by polyaminoamide STARBURST® dendrimers, Colloids and Surfaces A: Physicochem. Eng. Asp. 2004, 242, 213–216. [Google Scholar] [CrossRef]
- Demadis, K.D. A structure/function study of polyaminoamide dendrimers as silica scale growth inhibitors. J. Chem. Technol. Biotechnol. 1995, 80, 630–640. [Google Scholar] [CrossRef]
- Salvador Cob, S.; Hofs, B.; Maffezzoni, C.; Adamus, J.; Siegers, W.G.; Cornelissen, E.R.; Genceli Güner, F.E.; Witkamp, G.J. Silica removal to prevent silica scaling in reverse osmosis membranes. Desalination 2014, 344, 137–143. [Google Scholar] [CrossRef]
- Latour, I.; Miranda, R.; Blanco, A. Silica removal with sparingly soluble magnesium compounds. Part I Sep. Purif. Technol. 2014, 138, 210–218. [Google Scholar] [CrossRef]
- Latour, I.; Miranda, R.; Blanco, A. Silica removal with sparingly soluble magnesium compounds. Part II Sep. Purif. Technol. 2015, 149, 331–338. [Google Scholar] [CrossRef]
- Sugita, H.; Bando, Y.; Nakamura, M. Removal of Silica from Geothermal Brine by Seeding Method Using Silica Gel. J. Chem. Eng. Jpn. 1998, 31, 150–152. [Google Scholar] [CrossRef]
- Latour, I.; Miranda, R.; Carceller, R.; Blanco, A. Efficiency of polyaluminum nitrate sulphate-polyamine hybrid coagulants for silica removal. Desalin. Water Treat. 2016, 57, 17973–17984. [Google Scholar] [CrossRef]
- Miranda, R.; Latour, I.; Hörsken, A.; Jarabo, R.; Blanco, A. Enhanced silica removal by polyamine- and polyacrylamide-polyaluminum hybrid coagulants. Chem. Eng. Technol. 2015, 38, 2045–2053. [Google Scholar] [CrossRef]
- Gabelich, C.J.; Williams, M.D.; Rahardianto, A.; Franklin, J.C.; Cohen, Y. High recovery reverse osmosis desalination using intermediate chemical demineralization. J. Membr. Sci. 2007, 301, 131–141. [Google Scholar] [CrossRef]
- Sheikholeslami, R.; al-Mutaz, I.S.; Koo, T.; Young, A. Pretreatment and the effects of cations and anions on prevention of silica fouling. Desalination 2001, 139, 83–95. [Google Scholar] [CrossRef]
- Milne, N.A.; O’Reilly, T.; Sanciolo, P.; Ostarcevic, E.; Beighton, M.; Taylor, K.; Mullett, M.; Tarquin, A.J.; Gray, S.R. Chemistry of silica scale mitigation for RO desalination with particular reference to remote operations. Water Res. 2014, 65, 107–133. [Google Scholar] [CrossRef]
- Den, W.; Wang, C.-J. Removal of silica from brackish water by electrocoagulation pretreatment to prevent fouling of reverse osmosis membranes. Sep. Purif. Technol. 2008, 59, 318–325. [Google Scholar] [CrossRef]
- Holt, P.K.; Barton, G.W.; Wark, M.; Mitchell, C.A. A quantitative comparison between chemical dosing and electrocoagulation. Colloids Surf. A Physicochem. Eng. Asp. 2002, 211, 233–248. [Google Scholar] [CrossRef]
- Merzouk, B.; Madani, K.; Sekki, A. Using electrocoagulation-electroflotation technology to treat synthetic solution and textile wastewater, two case studies. Desalination 2010, 250, 573–577. [Google Scholar] [CrossRef]
- Helen Annal, G.T.; Vishnu, P.K. Electrochemical Synthesis of Metal Oxides and Hydroxides. Chem. Mater. 2000, 12, 1195–1204. [Google Scholar]
- Mousty, C.; Walcarius, A. Electrochemically assisted deposition by local pH tuning: A versatile tool to generate ordered mesoporous silica thin films and layered double hydroxide materials. J. Solid State Electrochem. 2015, 19, 1905–1931. [Google Scholar] [CrossRef]
- Sibottier, E.; Sayen, S.; Gabori, F.; Walcarius, A. Factors Affecting the Preparation and Properties of Electrodeposited Silica Thin Films Functionalized with Amine or Thiol Groups. Langmuir 2006, 22, 8366–8373. [Google Scholar] [CrossRef] [PubMed]
- Shiohara, Y.; Okado, A.; Abe, M.; Sagiyama, M. The Deposition Behavior of Electrodeposited Zinc-Silica Composite Coating from a Sodium Nitrate-added Bath and the Coating Characteristics. Tetsu-to-Hagane 1991, 77, 878–885. [Google Scholar] [CrossRef]
- Ji, J.; Cooper, W.C.; Dreisinger, D.B.; Peters, E. Surface pH measurements during nickel electrodeposition. J. Appl. Electrochem. 1995, 25, 642–650. [Google Scholar] [CrossRef]
- Natter, H.; Hempelmann, R. Nanocrystalline Copper by Pulsed Electrodeposition: The Effects of Organic Additives, Bath Temperature, and pH. J. Phys. Chem. 1996, 100, 19525–19532. [Google Scholar] [CrossRef]
- Sano, Y.; Hao, Y.; Kuwahara, F. Development of an electrolysis based system to continuously recover magnesium from seawater. Heliyon 2018, 4. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electric resistance | Ω·cm2 | 4.5 |
| Burst strength | Mpa | ≥0.40 |
| Thickness | Mm | 0.21 |
| Recommend Temperature | °C | ≤60 |
| Recommend pH | - | 0–14 |
| Element | Molar Fraction (%) | |
|---|---|---|
| 5 min Later | 24 h Later | |
| Oxygen | 68.58 | 72.05 |
| Sodium | 0.41 | 0.24 |
| Magnesium | 9.51 | 1.31 |
| Silica | 11.93 | 1.27 |
| Chlorine | 0.24 | - |
| Calcium | 9.06 | 25.13 |
| Manganese | 0.27 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sano, Y.; Yamaguchi, M. Preventing Silica Scale Formation Using Hydroxide Ions Generated by Water Electrolysis. Membranes 2019, 9, 154. https://doi.org/10.3390/membranes9110154
Sano Y, Yamaguchi M. Preventing Silica Scale Formation Using Hydroxide Ions Generated by Water Electrolysis. Membranes. 2019; 9(11):154. https://doi.org/10.3390/membranes9110154
Chicago/Turabian StyleSano, Yoshihiko, and Masataka Yamaguchi. 2019. "Preventing Silica Scale Formation Using Hydroxide Ions Generated by Water Electrolysis" Membranes 9, no. 11: 154. https://doi.org/10.3390/membranes9110154
APA StyleSano, Y., & Yamaguchi, M. (2019). Preventing Silica Scale Formation Using Hydroxide Ions Generated by Water Electrolysis. Membranes, 9(11), 154. https://doi.org/10.3390/membranes9110154