The Multilevel Structure of Sulfonated Syndiotactic-Polystyrene Model Polyelectrolyte Membranes Resolved by Extended Q-Range Contrast Variation SANS


Abstract
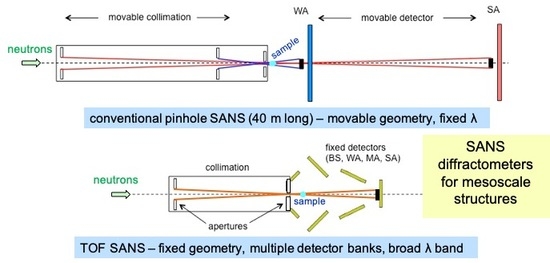
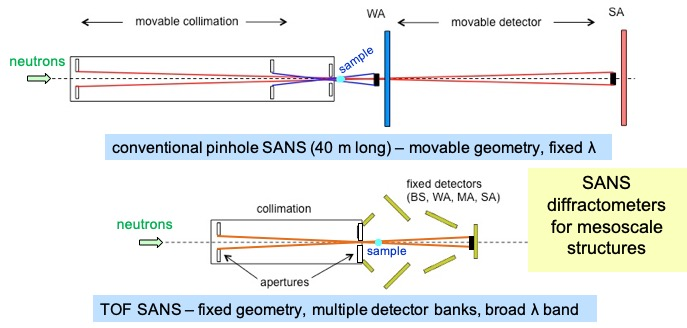
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.3. Data Analysis
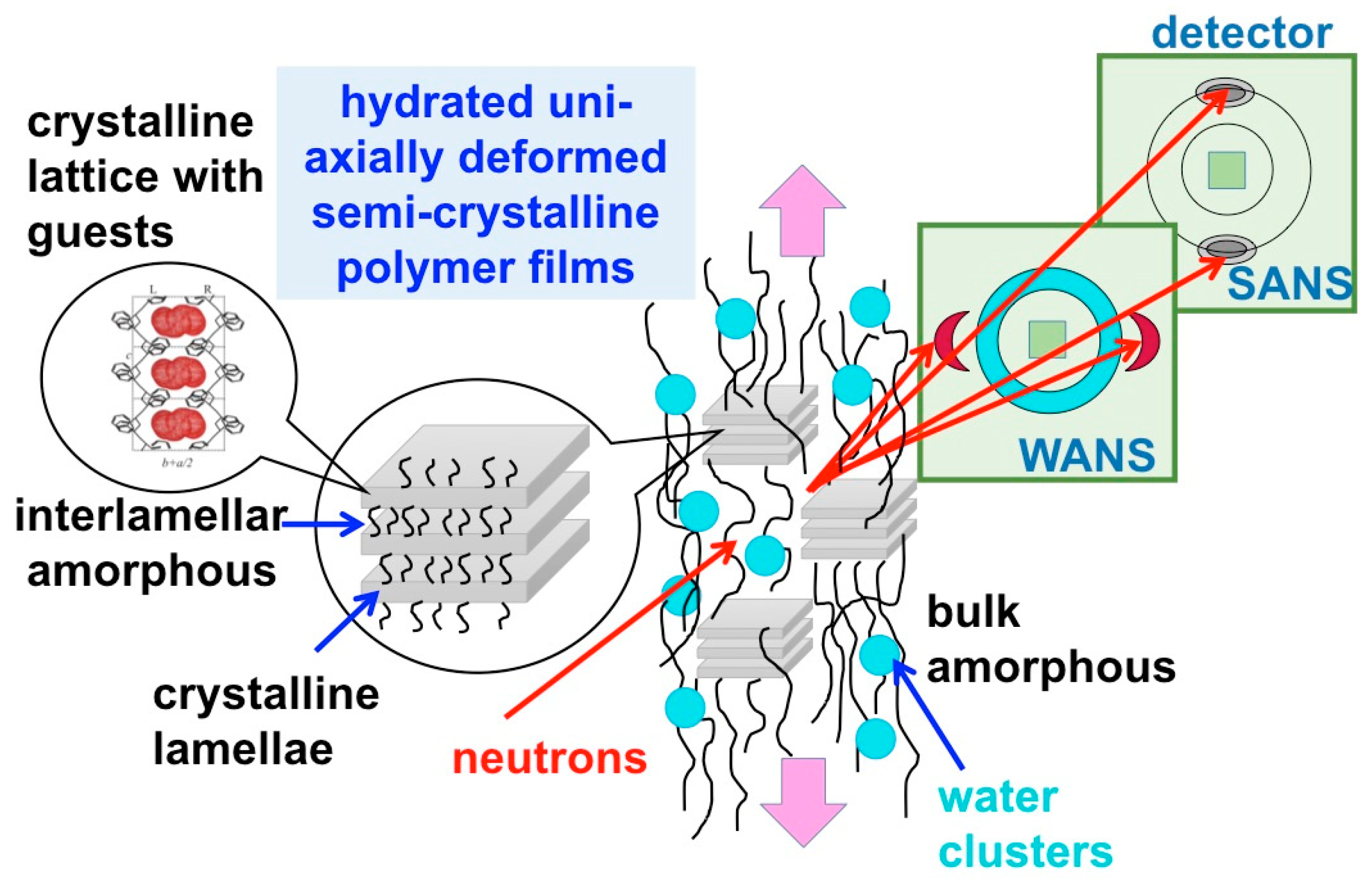
3. Results and Discussion
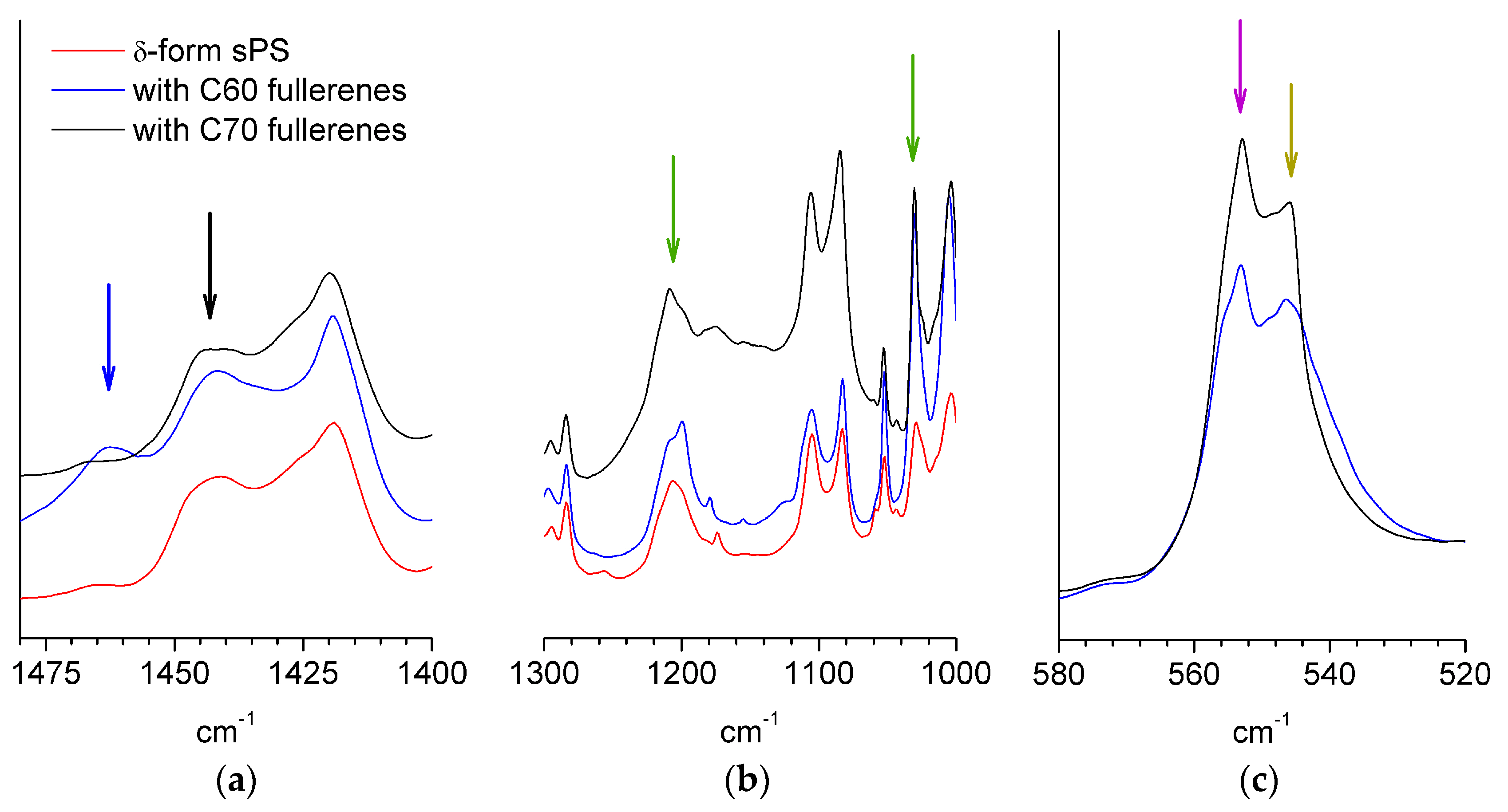
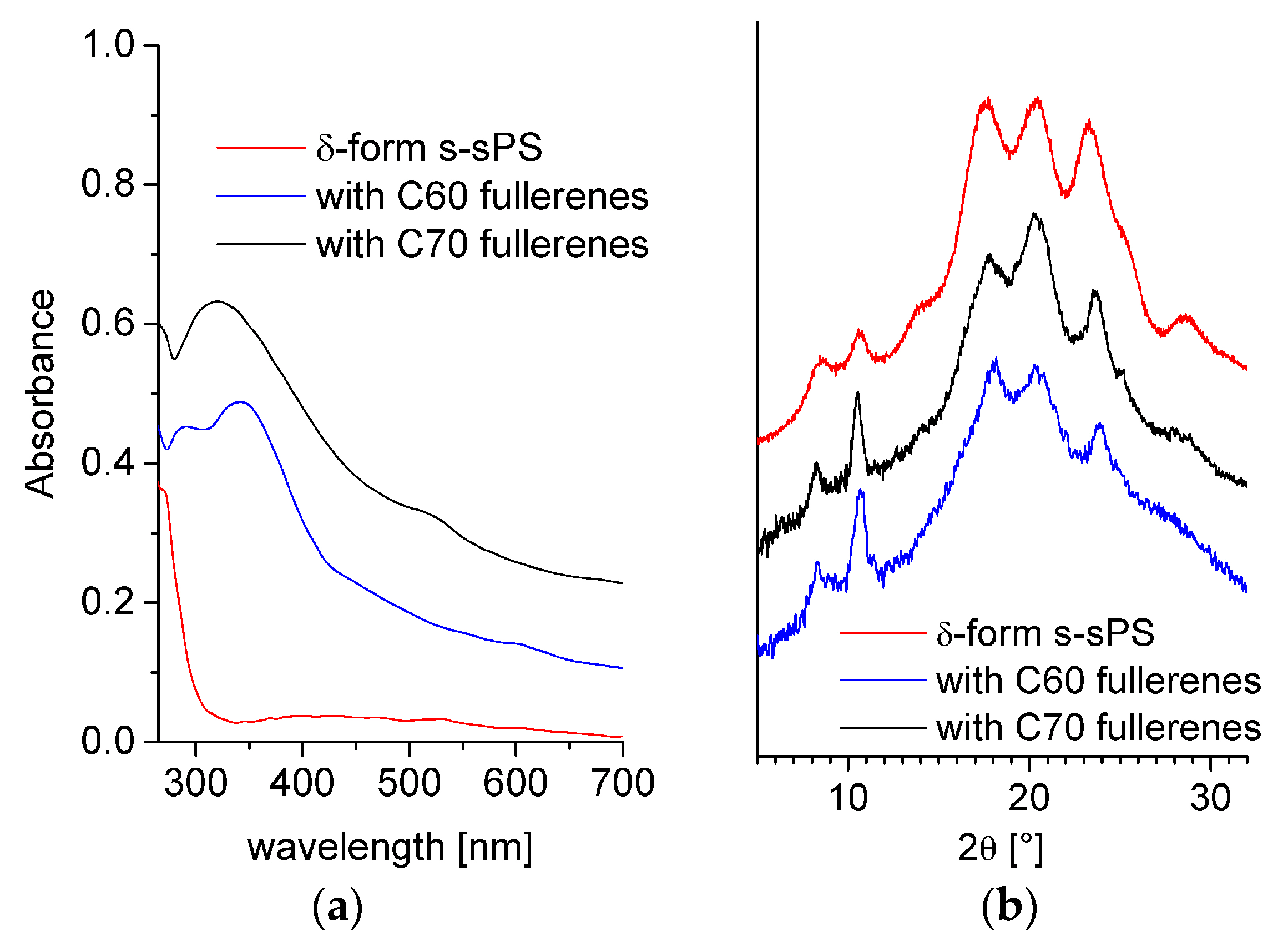
3.1. Composition and Crystallinity Characterization.
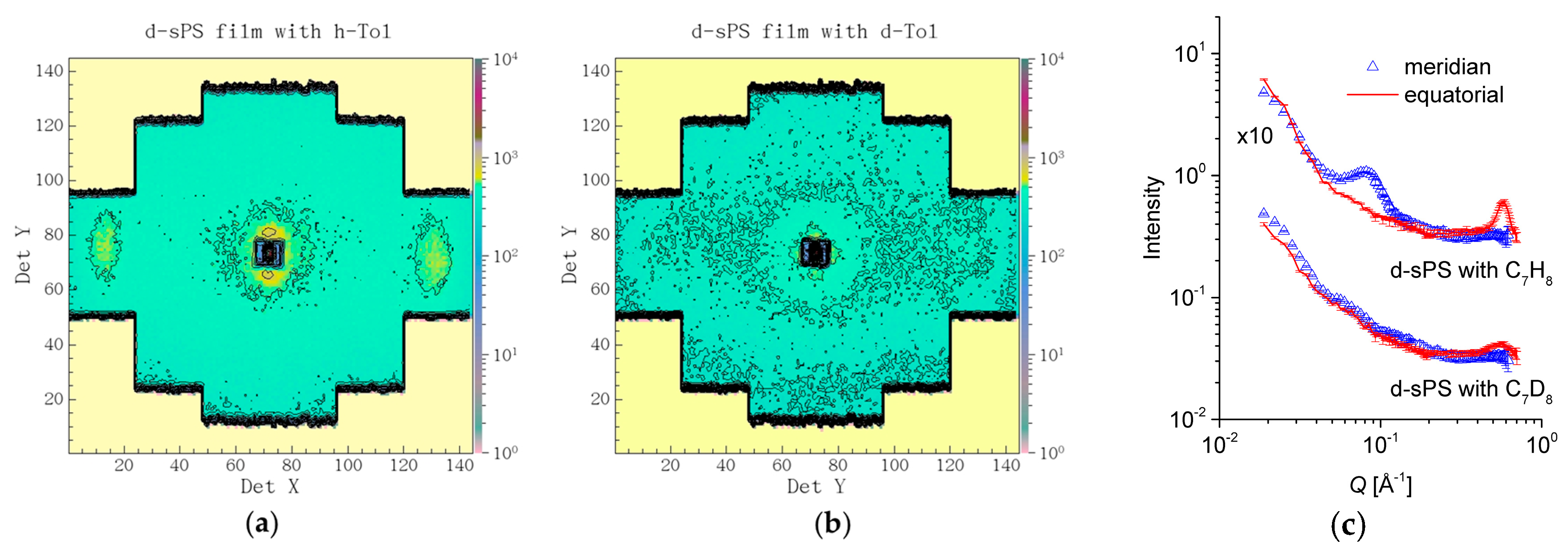
3.2. SANS on Dry Films
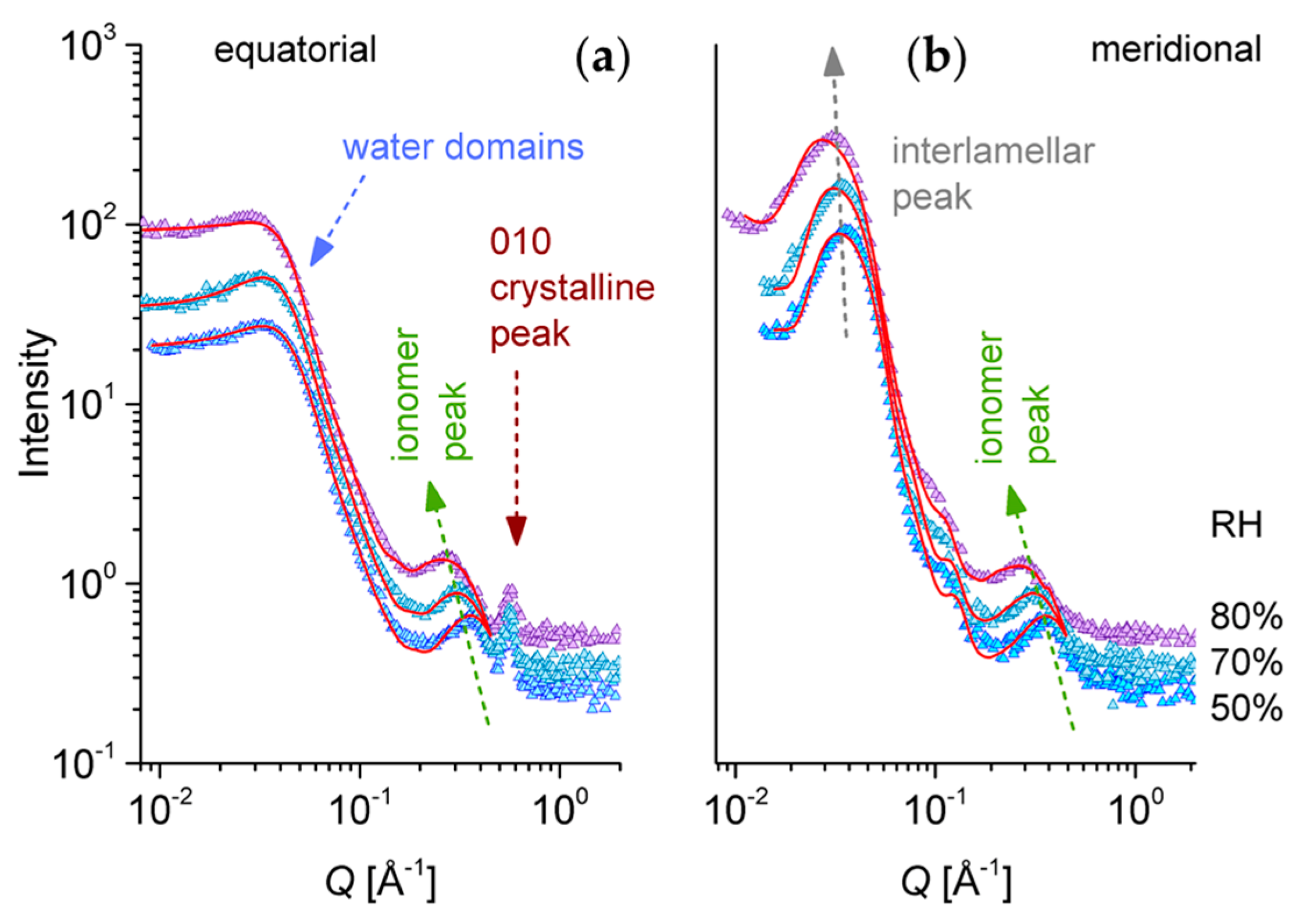
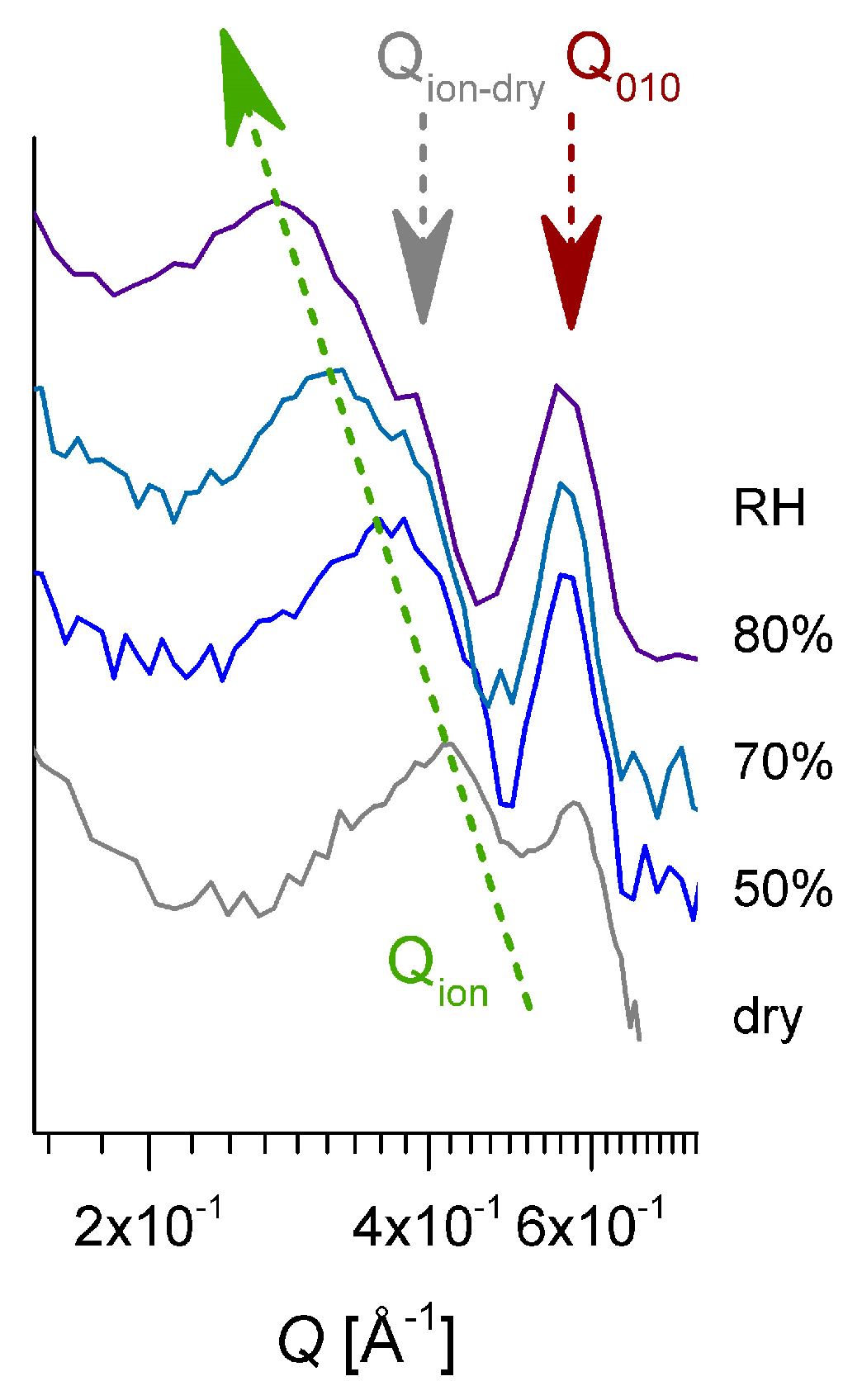
3.3. SANS on Hydrated Films—Variation of Hydration Level
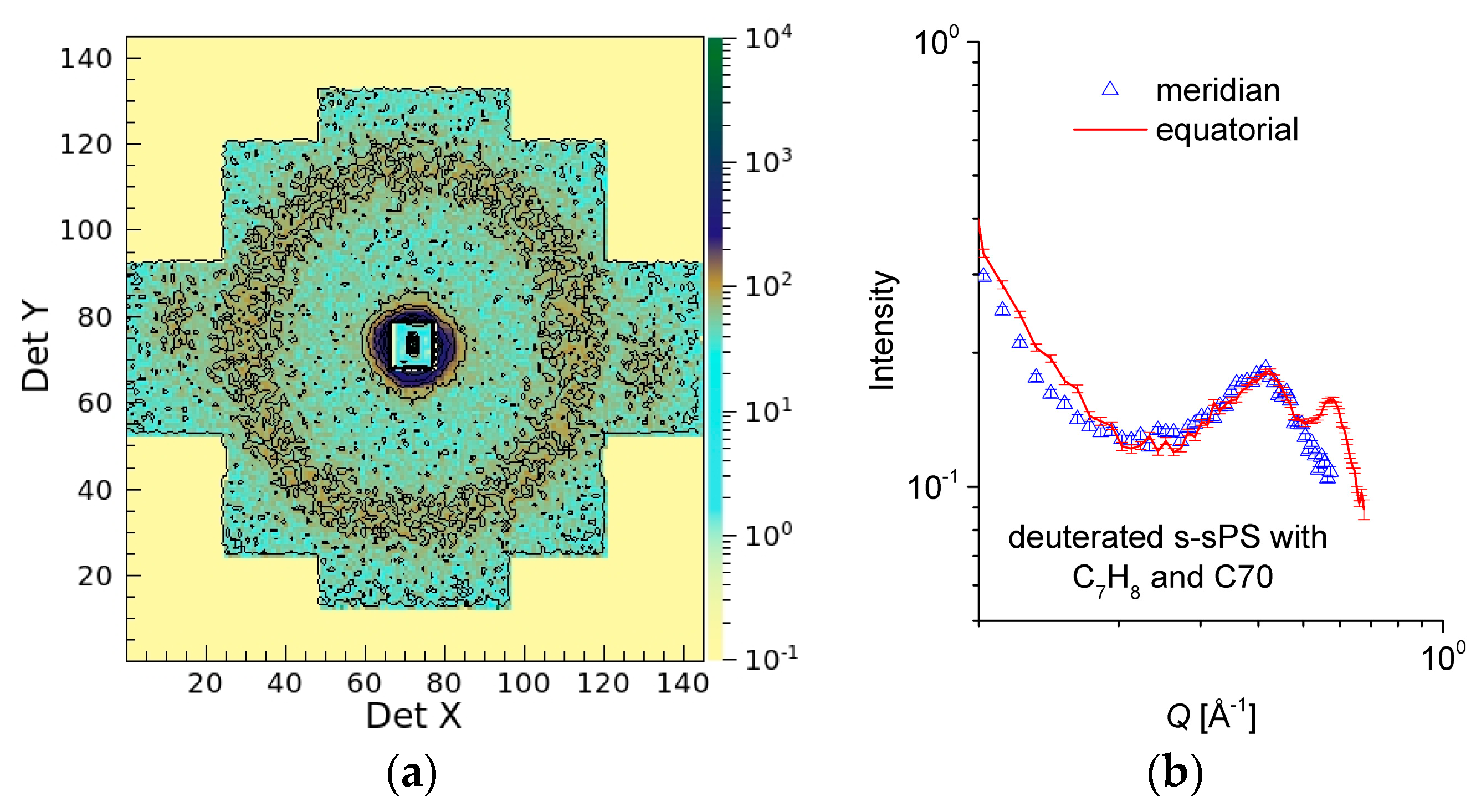
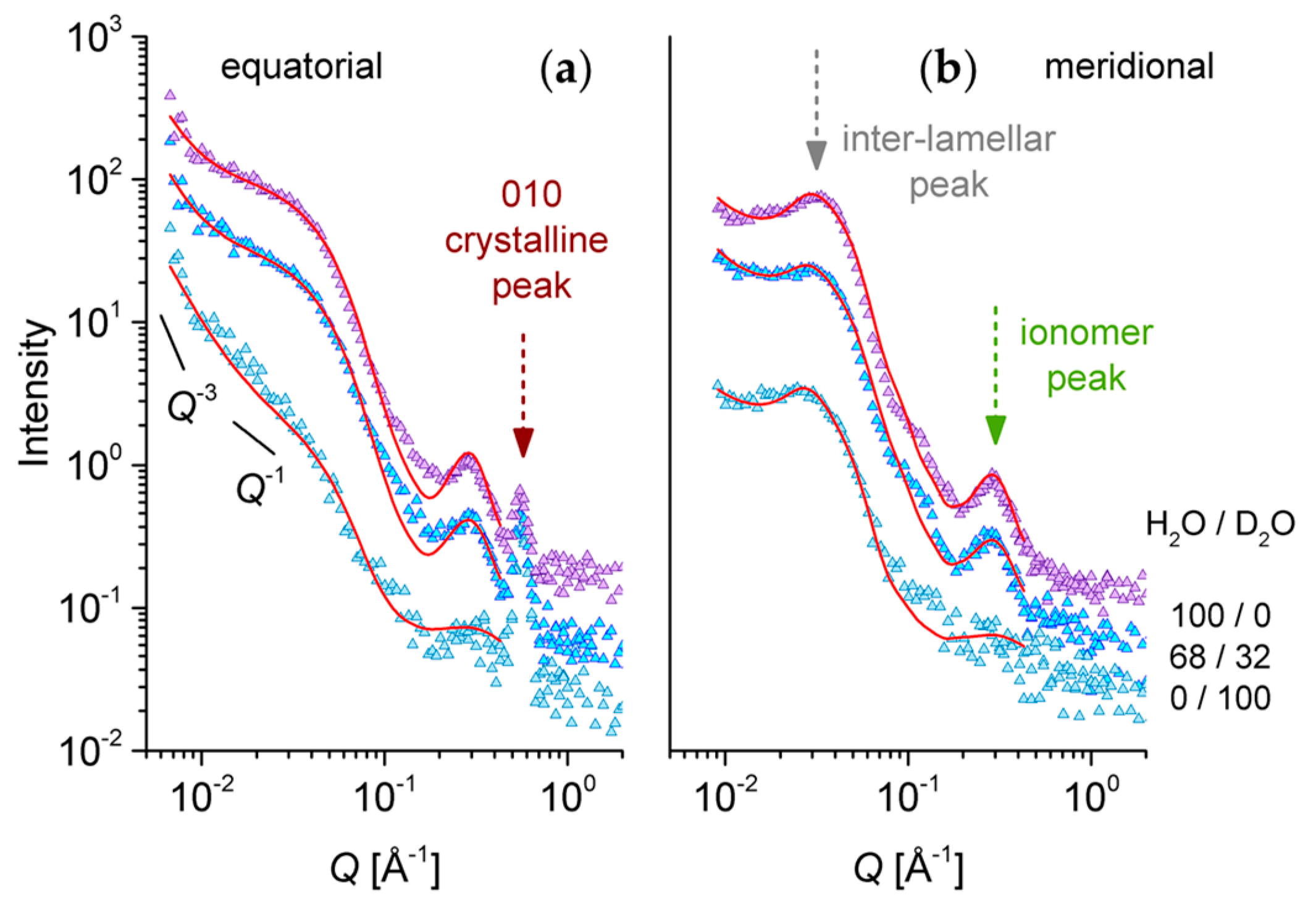
3.4. SANS on Hydrated Films—Neutron Contrast Variation
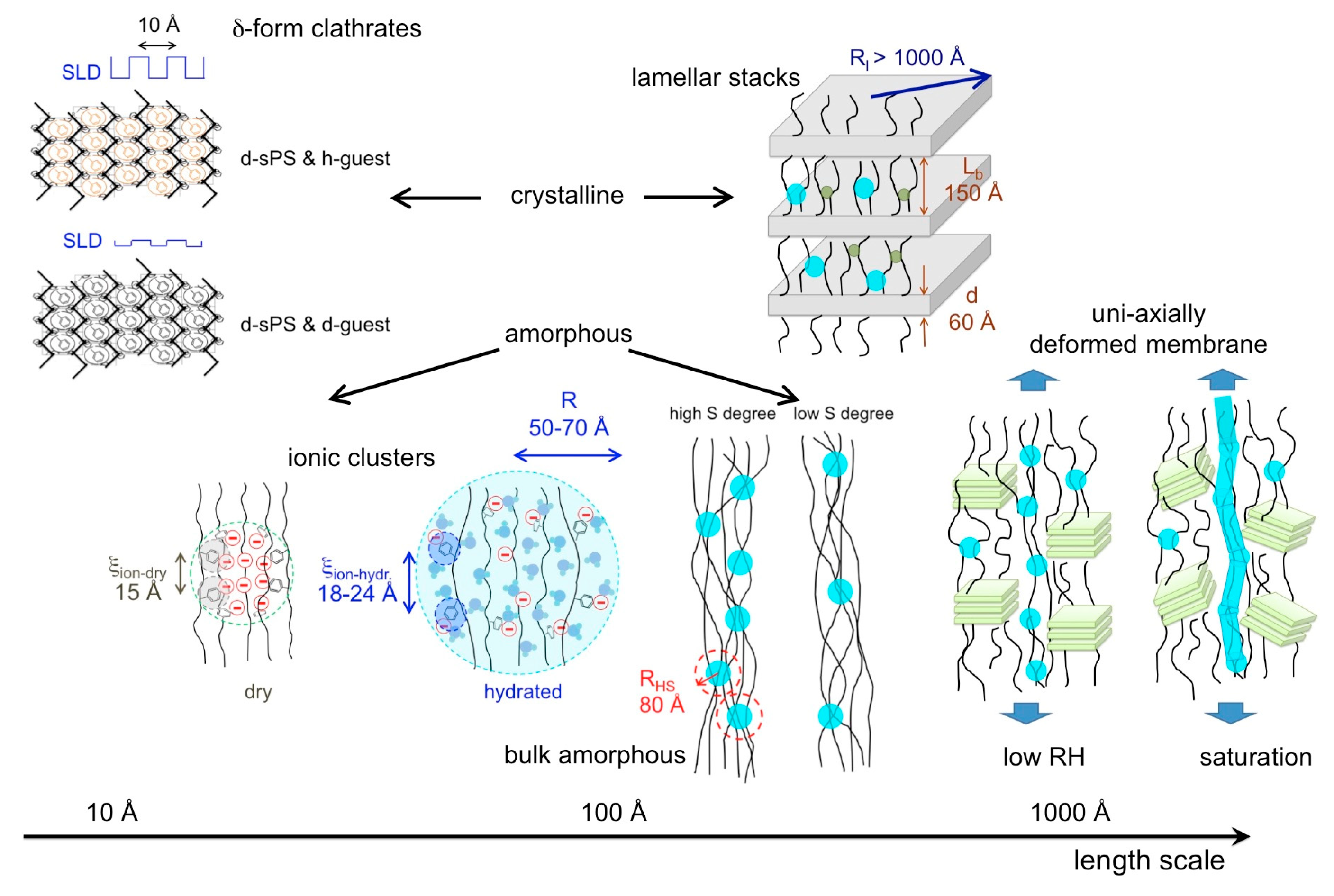
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Eberle, U.; Mueller, B.; Von Helmolt, R. Fuel Cell Electric Vehicles and Hydrogen Infrastructure: Status 2012. Energy Environ. Sci. 2012, 5, 8780–8797. [Google Scholar] [CrossRef]
- Barbir, F. Fuel Cells Applications. In PEM Fuel Cells, 2nd ed.; Barbir, F., Ed.; Elsevier Inc.: Philadelphia, PA, USA, 2013; pp. 373–434. [Google Scholar]
- Kraytsberg, A.; Ein-Eli, Y. Review of Advanced Materials for Proton Exchange Membrane Fuel Cells. Energy Fuels 2014, 28, 7303–7330. [Google Scholar] [CrossRef]
- Hayashi, A.; Nishihara, M.; Matsuda, J.; Sasaki, K. Polymer electrolyte Fuel Cells (PEFCs). In Hydrogen Energy Engineering. Green Energy and Technology; Sasaki, K., Li, H.-W., Hayashi, A., Yamabe, J., Ogura, T., Lyth, S.M., Eds.; Springer: Tokyo, Japan, 2016; pp. 301–311. [Google Scholar]
- Kreuer, K.-D.; Paddison, S.J.; Spohr, E.; Schuster, M. Transport in Proton Conductors for Fuel-Cell Applications: Simulations, Elementary Reactions, and Phenomenology. Chem. Rev. 2004, 104, 4637–4678. [Google Scholar] [CrossRef]
- Okada, T. Theory for Water Management in Membranes for Polymer Electrolyte Fuel Cells: Part 1 The Effect of Impurity Ions at the Anode Side on the Membrane Performance. J. Electroanal. Chem. 1999, 465, 1–17. [Google Scholar] [CrossRef]
- St-Pierre, J.; Wilkinson, D.P.; Knights, S.; Bos, M.L. Relationships Between Water Management, Contamination and Lifetime Degradation in PEFC. J. New Mater. Electrochem. Syst. 2000, 3, 99–106. [Google Scholar]
- Huguet, P.; Morin, A.; Gebel, G.; Deabate, S.; Sutor, A.K.; Peng, Z. In Situ Analysis of Water Management in Operating Fuell Cells by Confocal Raman Spectroscopy. Electrochem. Commun. 2011, 13, 418–422. [Google Scholar] [CrossRef]
- Kusoglu, A.; Weber, A.Z. New Insights into Perfluorinated Sulfonic-Acid Ionomers. Chem. Rev. 2017, 117, 987–1104. [Google Scholar] [CrossRef]
- Smitha, B.; Sridhar, A.; Khan, A.A. Solid Polymer Electrolyte Membranes for Fuel Cell Applications—A Review. J. Membrane Sci. 2005, 259, 10–26. [Google Scholar] [CrossRef]
- Sahu, A.K.; Jalajakshi, A.; Pitchumani, S.; Shridhar, P.; Shukla, A.K. Endurance of Nafion-Composite Membranes in PEFCs Operating at Elevated Temperature under Low Relative-Humidity. J. Chem. Sci. 2012, 124, 529–536. [Google Scholar] [CrossRef]
- Vinothkannan, M.; Hariprasad, R.; Ramakrishnan, S.; Kim, A.R.; Yoo, D.J. Potential Bifunctional Filler (CeO2-ACNTs) for Nafion Matrix toward Extended Electrochemical Power Density and Durability in Proton-Exchange Membrane Fuel Cells Operating at reduced Relative Humidity. ACS Sustain. Chem. Eng. 2019, 7, 12847–12857. [Google Scholar] [CrossRef]
- Kim, A.R.; Gabunada, J.C.; Yoo, D.J. Amelioration in Physicochemical Properties and Single Cell Performance of Sulfonated Poly (ether ether ketone) Block Copolymer Composite Membrane using Sulfonated Carbon Nanotubes for Intermediate Humidity Fuel Cells. Int. J. Energy Res. 2019, 43, 2974–2989. [Google Scholar] [CrossRef]
- Kim, A.R.; Vinothkannan, M.; Yoo, D.J. Sulfonated Fluorinated Multi-block Copolymer Hybrid Containing Sulfonated (Poly Ether Ether Ketone) and Graphene Oxide: A Ternary Hybrid Membrane Architecture for Electrolyte Applications in Proton Exchange Membrane Fuel Cells. J. Energy. Chem. 2018, 27, 1247–1260. [Google Scholar] [CrossRef]
- Kim, A.R.; Vinothkannan, M.; Park, C.J.; Yoo, D.J. Alleviating the Mechanical and Thermal Degradations of Highly Sulfonated Poly (Ether Ether Ketone) Blocks via Copolymerization with Hydrophobic Unit for Intermediate Humidity Fuel Cells. Polymers 2018, 10, 1346. [Google Scholar] [CrossRef] [PubMed]
- Hickner, M.A.; Ghassemi, H.; Kim, Y.S.; Einsla, B.R.; McGrath, J.E. Alternative Polymer System for Proton Exchange Membranes (PEMs). Chem. Rev. 2004, 104, 4587–4612. [Google Scholar] [CrossRef] [PubMed]
- Hihashihara, T.; Matsumoto, K.; Ueda, M. Sulfonated Aromatic Hydrocarbon Polymers as Proton Exchange Membranes for Fuel Cells. Polymer 2009, 50, 5341–5357. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Ma, L.; Cai, W.; Cheng, H. Recent Developments on Alternative Proton Exchange Membranes: Strategies for Systematic Performance Improvement. Energy Technol. 2015, 3, 675–691. [Google Scholar] [CrossRef]
- Yoshida, M.; Zhao, Y.; Yoshizawa-Fujita, M.; Ohira, A.; Takeoka, Y.; Koizumi, S.; Rikukawa, M. PGF-NMR and SANS Studies in Cation Exchange Membranes Based on Sulfonated Polyphenylene Multiblock Copolymers. ECS Trans. 2013, 50, 1045–1053. [Google Scholar] [CrossRef]
- Feng, S.; Kondo, S.; Kaseyama, T.; Nakazawa, T.; Kikuchi, T.; Selyanchyn, R.; Fujikawa, S.; Christiani, L.; Sasaki, K.; Nishihara, M. Development of Polymer-Polymer Type Charge-Transfer Blend Membranes for Fuel Cell Application. J. Membr. Sci. 2018, 548, 223–231. [Google Scholar] [CrossRef]
- Boriello, A.; Agoretti, P.; Ambrosio, L.; Fasano, G.; Pellegrino, M.; Venditto, V.; Guerra, G. Syndiotactic Polystyrene Films with Sulfonated Amorphous Phase and Nanoporous Crystalline Phase. Chem. Mater. 2009, 21, 3191–3196. [Google Scholar] [CrossRef]
- Saga, S.; Matsumoto, H.; Saito, K.; Minagawa, M.; Tanioka, A. Polyelectrolyte Membranes Based on Hydrocarbon Polymer Containing Fullerenes. J. Power Sources 2008, 176, 16–22. [Google Scholar] [CrossRef]
- Fasano, G.; Califano, R.; Pellegrino, M.; Venditto, V.; Guerra, G.; Boriello, A.; Ambrosio, L.; Sansone, L. Semicrystalline proton-conductive membranes with sulfonated amorphous phases. Int. J. Hydrog. Energy 2011, 36, 8038–8044. [Google Scholar]
- Jaymand, M. Recent Progress in the Chemical Modification of Syndiotactic Polystyrene. Polym. Chem. 2014, 5, 2663–2690. [Google Scholar] [CrossRef]
- Tashiro, K.; Ueno, I.; Yoshioka, A.; Kobayashi, M. Molecular Mechanism of Solvent-Induced Crystallization of Syndiotactic Polystyrene Glass. 1. Time-Resolved Measurements of Infrared/Raman Spectra and X-Ray Diffraction. Macromolecules 2001, 34, 310–315. [Google Scholar] [CrossRef]
- Raut, P.; Liang, W.; Chen, Y.-M.; Zhu, Y.; Jana, S.C. Syndiotactic Polystyrene-Based Ionogel Membranes for High Temperature Electrochemical Applications. ACS Appl. Mater. Interfaces 2017, 9, 30933–30942. [Google Scholar] [CrossRef]
- Kreuer, K.-D.; Portale, G. A Critical Revision of the Nano-Morphology of Proton Conducting Ionomers and Polyelectrolytes for Fuel Cell Applications. Adv. Funct. Mater. 2013, 23, 5390–5397. [Google Scholar] [CrossRef]
- Schiavone, M.-M.; Tarallo, O.; Di Girolamo, R.; Caporaso, L.; Appavou, M.-S.; Revay, Z.; Radulescu, A. Structure and Morphology of Model Polymer Electrolyte Membranes based on Sulfonated Syndiotactic-Polystyrene in the δ Co-crystalline Phase Resolved by Small-Angle Neutron Scattering. Solid State Ionics 2018, 320, 392–406. [Google Scholar] [CrossRef]
- De Rosa, C.; Guerra, G.; Petraccone, V.; Pirozzi, B. Crystal Structure of the Emptied Clathrate Form (δe Form) of Syndiotactic Polystyrene. Macromolecules 1997, 30, 4147–4152. [Google Scholar] [CrossRef]
- Albunia, A.R.; d’Aniello, C.; Guerra, G. Three Different Co-crystalline Phases of Syndiotactic Polystyrene with a Nitroxide Radical. Cryst. Eng. Comm. 2010, 12, 3942–3949. [Google Scholar] [CrossRef]
- Kaneko, F.; Radulescu, A.; Ute, K. Time-resolved SANS Studies on Guest Exchange Processes in Co-Crystals of Syndiotactic Polystyrene. Polymer 2013, 54, 3145–3149. [Google Scholar] [CrossRef]
- Rambabu, G.; Bhat, S.D. Sulfonated Fullerene in SPEEK Matrix and its Impact on the Membrane Electrolyte Properties in Direct Methanol Fuel Cells. Electrochim. Acta 2015, 176, 657–669. [Google Scholar] [CrossRef]
- Hermans, P.H.; Weidinger, A. Quantitative Investigation of the X-Ray Diffraction Picture of Some Typical Rayon Specimens Part I. Textil. Res. 1961, 31, 558–571. [Google Scholar] [CrossRef]
- Radulescu, A.; Szekely, N.K.; Appavou, M.-S.; Pipich, V.; Kohnke, T.; Ossovyi, V.; Staringer, S.; Schneider, G.J.; Amann, M.; Zhang-Haagen, B.; et al. Studying Soft-Matter and Biological Systems over a Wide Length-Scale from Nanometer and Micrometer Sizes at the Small-Angle Neutron Diffractometer KWS-2. J. Vis. Exp. 2016, e54639. [Google Scholar] [CrossRef] [PubMed]
- Takata, S.; Suzuki, J.; Shinohara, T.; Oku, T.; Tominaga, T.; Ohishi, K.; Iwase, H.; Nakatani, T.; Inamura, Y.; Ito, T.; et al. The design and Q Resolution of the Small and Wide Angle Neutron Scattering Instrument (TAIKAN) in J-Parc. JPS Conf. Proc. 2015, 8, 036020. [Google Scholar]
- DeCaluwe, S.C.; Kienzle, P.A.; Bhargava, P.; Baker, A.M.; Dura, J.A. Phase Segregation of Sulfonate Groups in Nafion interface Lamellae, Quantified via Neutron Reflectometry Fitting Techniques for Multi-layered Structures. Soft Matter 2014, 10, 5763–5776. [Google Scholar] [CrossRef]
- Pedersen, J.S. Determination of Size Distribution from Small-Angle Scattering Data for Systems with Effective Hard-Sphere Interactions. J. Appl. Cryst. 1994, 27, 595–608. [Google Scholar] [CrossRef]
- Radulescu, A.; Fetters, L.J.; Richter, D. Polymer-Driven Wax Crystal Control Using Partially Crystalline Polymeric Materials. Adv. Pol. Sci. 2008, 210, 1–100. [Google Scholar]
- Asano, A. Free Volume Study of Amorphous Polymers Detected by Solid-State 13C-NMR Linewidth Experiments. J. Chem. Phys. 2001, 115, 8665–8669. [Google Scholar] [CrossRef]
- Revay, Z. Determining Elemental Composition Using Prompt γ Activation Analysis. Anal. Chem. 2009, 81, 6851–6859. [Google Scholar] [CrossRef]
- Huang, T.; Jin, B.; Peng, R.F.; Chen, C.D.; Zheng, R.Z.; He, Y.; Chu, S.J. Synthesis and Characterization of [60]Fullerenes-Glycidyl Azide Polymer and Its Thermal Decomposition. Polymers 2015, 7, 896–908. [Google Scholar] [CrossRef]
- Wang, C.; Ruan, L.; Chang, X.-L.; Zhang, X.; Yang, S.-T.; Guo, X.; Yuan, H.; Guo, C.; Shi, W.; Sun, B.; et al. The Isotopic Effect of 13C-Labeled Large Carbon Cage (C70) Fullerens and Their Formation Process. RSC Adv. 2015, 5, 76949–76956. [Google Scholar] [CrossRef]
- Wu, Z.C.; Jelski, D.A.; George, T.F. Vibrational motions of Buckminsterfullerenes. Chem. Phys. Lett. 1987, 137, 291–294. [Google Scholar] [CrossRef]
- Weiss, R.A.; Sen, A.; Willis, C.L.; Pottick, L.A. Block Copolymer Ionomers: 1. Synthesis and Physical Properties of Sulphonated Poly (Styrene-Ethylene/Butylene-Styrene). Polymer 1991, 32, 1867–1874. [Google Scholar] [CrossRef]
- Sheng, L.; Higashihara, T.; Nakazawa, S.; Ueda, M. Polystyrenes Containing Flexible Alkysulfonated Side Chains as a Proton Exchange Membrane for Fuel Cell Applications. Polym. Chem. 2012, 3, 3289–3295. [Google Scholar] [CrossRef]
- Kobayashi, M.; Yoshioka, T.; Imai, M.; Itoh, Y. Structural Ordering on Physical Gelation of Syndiotactic Polystyrene Dispersed in Chloroform Studied by Time-Resolved Measurements of Small Angle Neutron Scattering (SANS) and Infrared Spectroscopy. Macromolecules 1995, 28, 7376–7385. [Google Scholar] [CrossRef]
- Bogdanov, A.A.; Voznyakovskii, A.P.; Pozdnya, A.O. UV-Vis Diagnostics of the PMMA-C60 Composite System and the Kinetics of its Thermal Decomposition. Nanosyst. Phys. Chem. Math. 2014, 5, 62–66. [Google Scholar]
- Scharff, P.; Risch, K.; Carta-Abelmann, L.; Dmytruk, I.M.; Bilyi, M.M.; Golub, O.A.; Khavryuchenko, A.V.; Buzaneva, E.V.; Aksenov, V.L.; Avdeev, M.V.; et al. Structure of C60 Fullerene in Water: Spectroscopic Data. Carbon 2004, 42, 1203–1206. [Google Scholar] [CrossRef]
- Chakraborty, C.; Malik, S.; Guenet, J.-M. Syndiotactic Polystyrene/Fullerene Composites: Elucidation of Structural Aspect. Macromol. Symp. 2011, 303, 56–62. [Google Scholar] [CrossRef]
- Kaneko, F.; Radulescu, A.; Ute, K. Time-Resolved Small-Angle Neutron Scattering Study on Guest-Exchange Processes in Co-Crystals of Syndiotactic Polystyrene. J. Appl. Cryst. 2014, 47, 6–13. [Google Scholar] [CrossRef]
- Zhao, Y.; Yoshida, M.; Oshima, T.; Koizumi, S.; Rikukawa, M.; Szekely, N.; Radulescu, A.; Richter, D. Elucidation of the Morphology of the Hydrocarbon Multi-Block Copolymer Electrolyte Membranes for Proton Exchange Fuel Cells. Polymer 2016, 86, 157–167. [Google Scholar] [CrossRef]
- Gebel, G. Structural evolution of water swollen perfluorosulfonated ionomers from dry membrane to solution. Polymer 2000, 41, 5829–5838. [Google Scholar] [CrossRef]
- Dattani, R.; Michels, R.; Nedoma, A.J.; Schweins, R.; Westacott, P.; Huber, K.; Cabral, J.T. Conformation and interactions of polystyrene and fullerenes in dilute to semidilute solutions. Macromolecules 2014, 47, 6113–6120. [Google Scholar] [CrossRef]
- Fumagalli, M.; Lyonnard, S.; Prajapati, G.; Berrod, Q.; Porcar, L.; Guillermo, A.; Gebel, G. Fast Water Diffusion and Long-Term Polymer Reorganization during Nafion Membrane Hydration Evidenced by Time-Resolved Small-Angle Neutron Scattering. J. Phys. Chem. B 2015, 119, 7068–7076. [Google Scholar] [CrossRef] [PubMed]
- Lyonnard, S. Structure and Transport Properties of Polymer Electrolyte Membranes Probed at Microscopic Scales. In Alternative energies, Advanced Structured Materials; Ferreira, G., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; Volume 34, pp. 163–193. [Google Scholar]
- Melnichenko, Y.B.; Wignall, G.D.; Compton, R.N.; Bakale, G. Characterization of Fullerenes and Fullerene Derivatives by Small-Angle Neutron Scattering and Transmission Measurements. J. Chem. Phys. 1999, 111, 4724–4728. [Google Scholar] [CrossRef][Green Version]
- He, L.; Garamus, V.M.; Funari, S.S.; Malfois, M.; Willumeit, R.; Niemeyer, B. Comparison of Small-Angle Scattering Methods for the Structural Analysis of Octyl-β-Maltopyranoside Micelles. Macromolecules 2002, 106, 7596–7604. [Google Scholar] [CrossRef]
- Stellbrink, J.; Rother, G.; Laurati, M.; Lund, R.; Willner, L.; Richter, D. Poly(ethylene-alt-propylene)-Poly (ethylene oxide) Diblock Copolymer Micelles: A Colloidal Model System with Tunable Softness. J. Phys. Condens. Matter 2004, 16, S3821–S3834. [Google Scholar] [CrossRef]
- Gebel, G.; Moore, B. Small-Angle Scattering Study of Short Pendant Chain Perfluorosulfonated Ionomer Membranes. Macromolecules 2000, 33, 4850–4855. [Google Scholar] [CrossRef]
- Gebel, G. Structure of Membranes for Fuel Cells: SANS and SAXS Analyses of Sulfonated PEEK Membranes and Solutions. Macromolecules 2013, 46, 6057–6066. [Google Scholar] [CrossRef]
- Kusoglu, A. Ionomer Thin Films in PEM Fuel Cells. In Encyclopedia of Sustainability Science and Technology; Meyers, R.A., Ed.; Springer: New York, NY, USA, 2018; pp. 1–23. [Google Scholar]
- Radulescu, A.; Schwahn, D.; Monkenbusch, M.; Fetters, L.J.; Richter, D. Structural Study of the Influence of Partially Crystalline Poly (ethylene butene) Random Copolymers on Paraffin Crystallization in Dilute Solutions. J. Pol. Sci. B Pol. Phys. 2004, 42, 3113–3132. [Google Scholar] [CrossRef]
- Wang, H.; Wu, C.; Wui, D.; Men, Y. Lamellar Thickness Dependence of Crystal Modification Selection in the Syndiotactic Polystyrene γ-to-α/β Phase Transition Process. Macromolecules 2018, 51, 497–503. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | SLD, 1010 cm−2 |
|---|---|
| sPS (crystalline) | 6.47 |
| sPS (amorphous) | 6.00 |
| s-sPS (amorphous) | 6.34 |
| –SO3H | 1.32 (1.1 [36]) |
| D2O | 6.38 |
| H2O | −0.56 |
| d-Tol | 5.66 |
| h-Tol | 0.94 |
| C60 | 5.50 |
| C70 | 5.67 |
| Sample | Condition | R, Å/RHS, Å | (ϕwater)amorphous, % | ρlam, × 1010 cm−2 | Lb, Å/σD, Å | ρinter-lam, × 1010 cm−2 | (ϕwater)inter-lam, % |
|---|---|---|---|---|---|---|---|
| S = 46.3%, with C70 | RH = 50% 100 H2O/0 D2O | 55.0/73.0 | 4.53 | 6.012 | 130.6/107.9 | 5.989 | 5.09 |
| - | RH = 70% 100 H2O/0 D2O | 58.8/77.0 | 7.45 | 6.012 | 151.6/114.7 | 5.983 | 5.17 |
| - | RH = 80% 100 H2O/0 D2O | 68.6/85.0 | 12.63 | 6.012 | 172.0/120.5 | 5.981 | 5.20 |
| - | - | - | - | - | - | - | |
| S = 19.5%, with C60 | RH = 80% 100 H2O/0 D2O | 52/- | 18.20 | 6.003 | 159.2/99.3 | 5.989 | 5.10 |
| - | RH = 80 %68 H2O/32 D2O | 52/- | 17.85 | 6.003 | 155.6/106.7 | 5.993 | 7.20 |
| - | - | - | - | - | - | - | - |
| - | RH = 80% 0 H2O/100 D2O | 52/- | 18.50 | 6.003 | 135.4/110.9 | 5.999 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schiavone, M.-M.; Iwase, H.; Takata, S.-i.; Radulescu, A. The Multilevel Structure of Sulfonated Syndiotactic-Polystyrene Model Polyelectrolyte Membranes Resolved by Extended Q-Range Contrast Variation SANS. Membranes 2019, 9, 136. https://doi.org/10.3390/membranes9110136
Schiavone M-M, Iwase H, Takata S-i, Radulescu A. The Multilevel Structure of Sulfonated Syndiotactic-Polystyrene Model Polyelectrolyte Membranes Resolved by Extended Q-Range Contrast Variation SANS. Membranes. 2019; 9(11):136. https://doi.org/10.3390/membranes9110136
Chicago/Turabian StyleSchiavone, Maria-Maddalena, Hiroki Iwase, Shin-ichi Takata, and Aurel Radulescu. 2019. "The Multilevel Structure of Sulfonated Syndiotactic-Polystyrene Model Polyelectrolyte Membranes Resolved by Extended Q-Range Contrast Variation SANS" Membranes 9, no. 11: 136. https://doi.org/10.3390/membranes9110136
APA StyleSchiavone, M.-M., Iwase, H., Takata, S.-i., & Radulescu, A. (2019). The Multilevel Structure of Sulfonated Syndiotactic-Polystyrene Model Polyelectrolyte Membranes Resolved by Extended Q-Range Contrast Variation SANS. Membranes, 9(11), 136. https://doi.org/10.3390/membranes9110136