Effective Conversion of Amide to Carboxylic Acid on Polymers of Intrinsic Microporosity (PIM-1) with Nitrous Acid



Abstract
1. Introduction
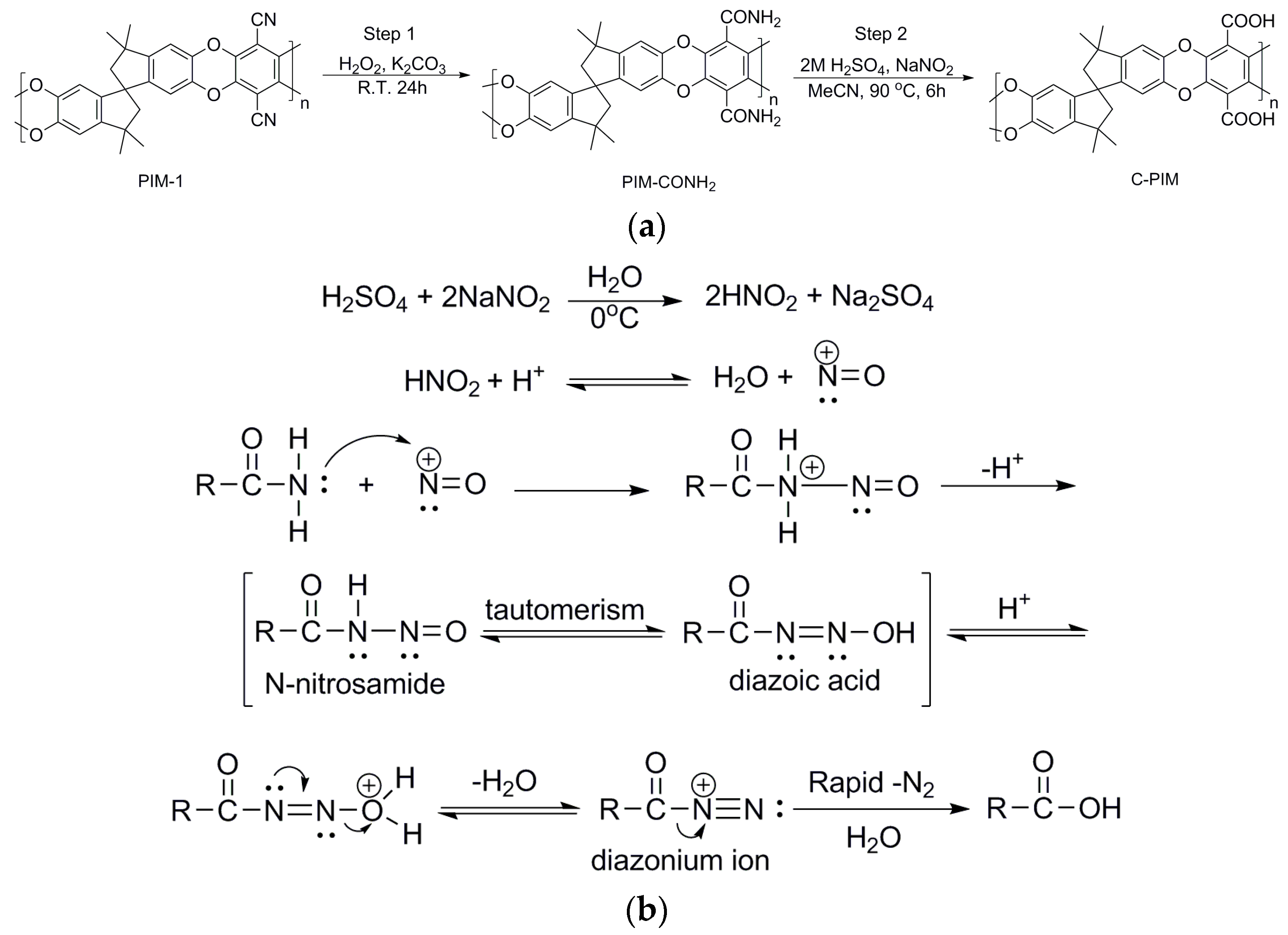
2. Materials and Methods
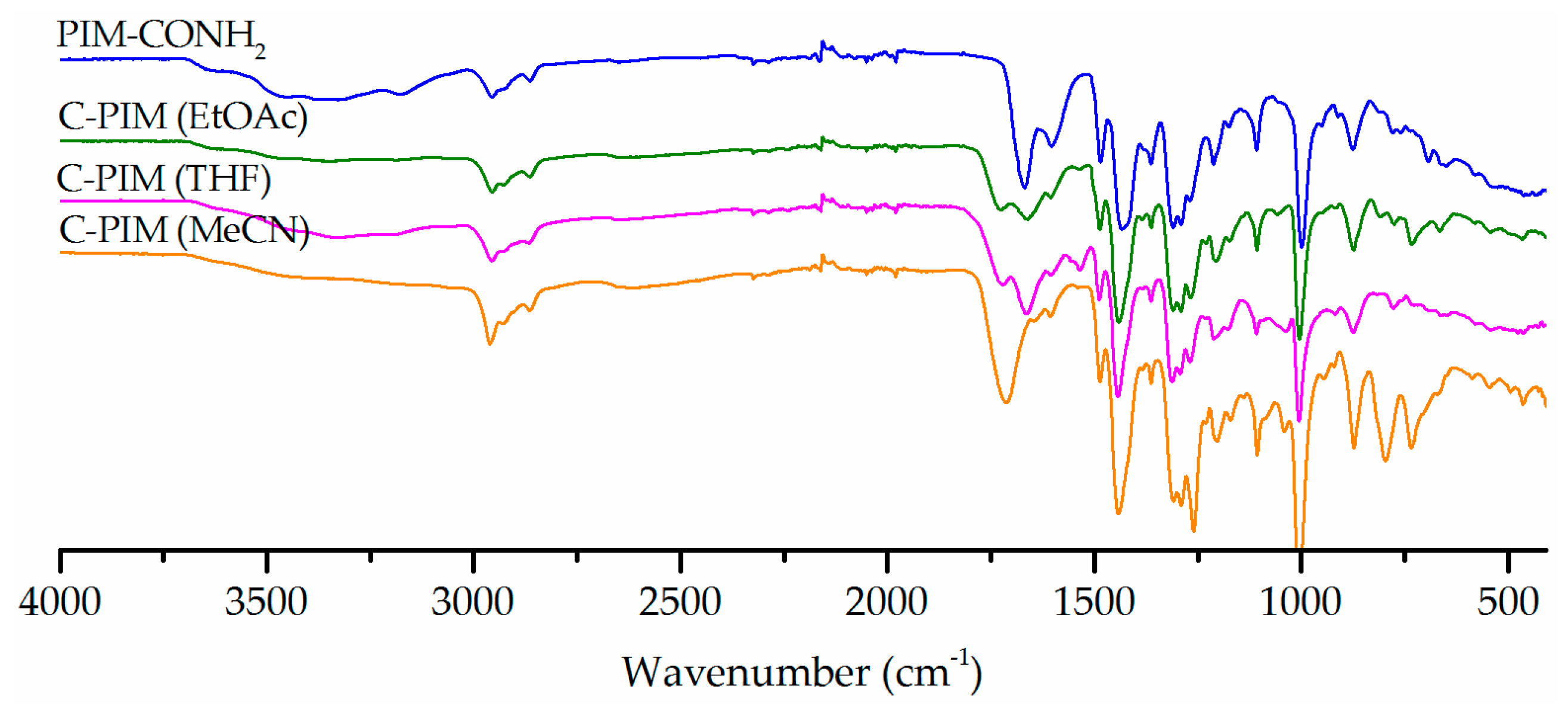
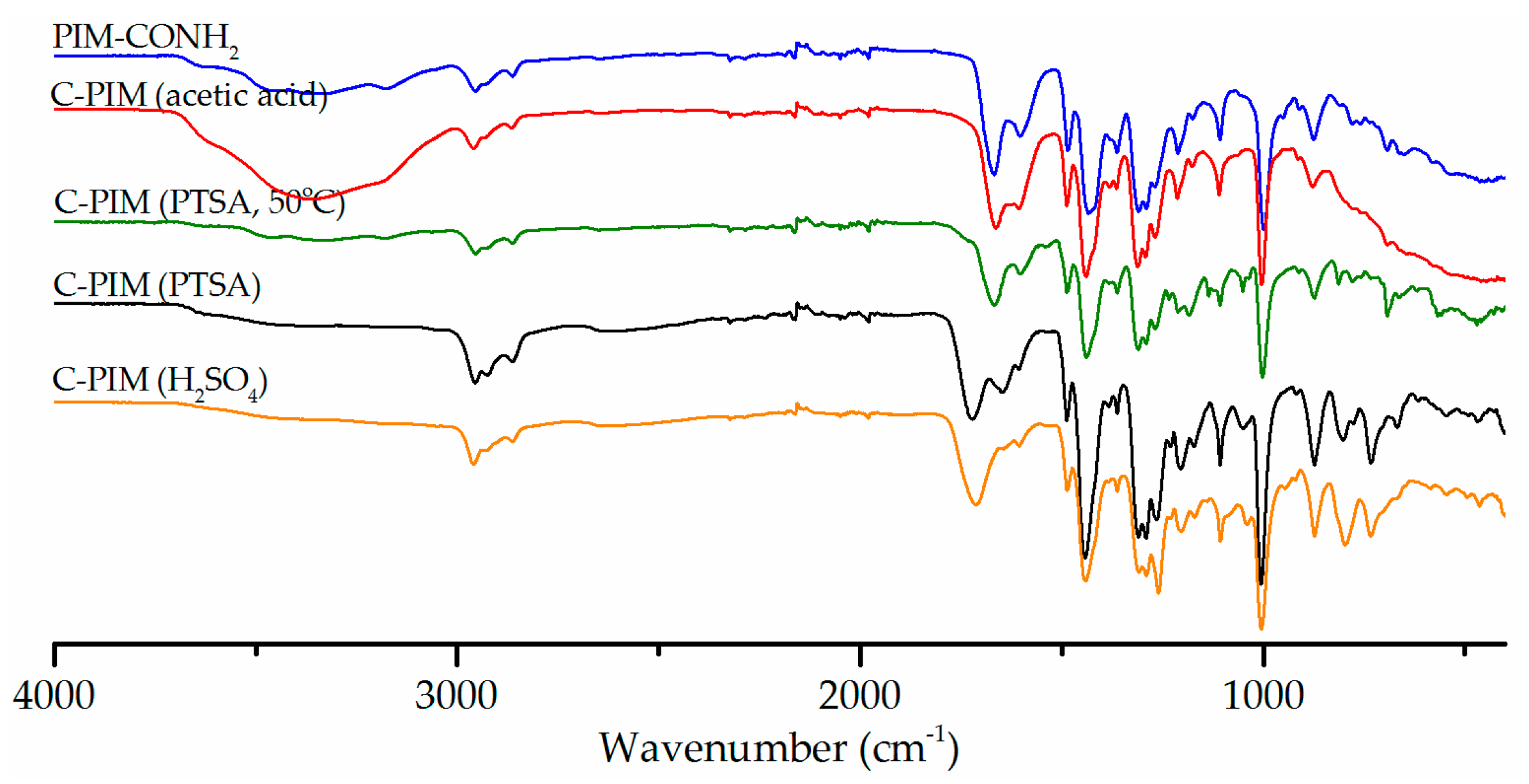
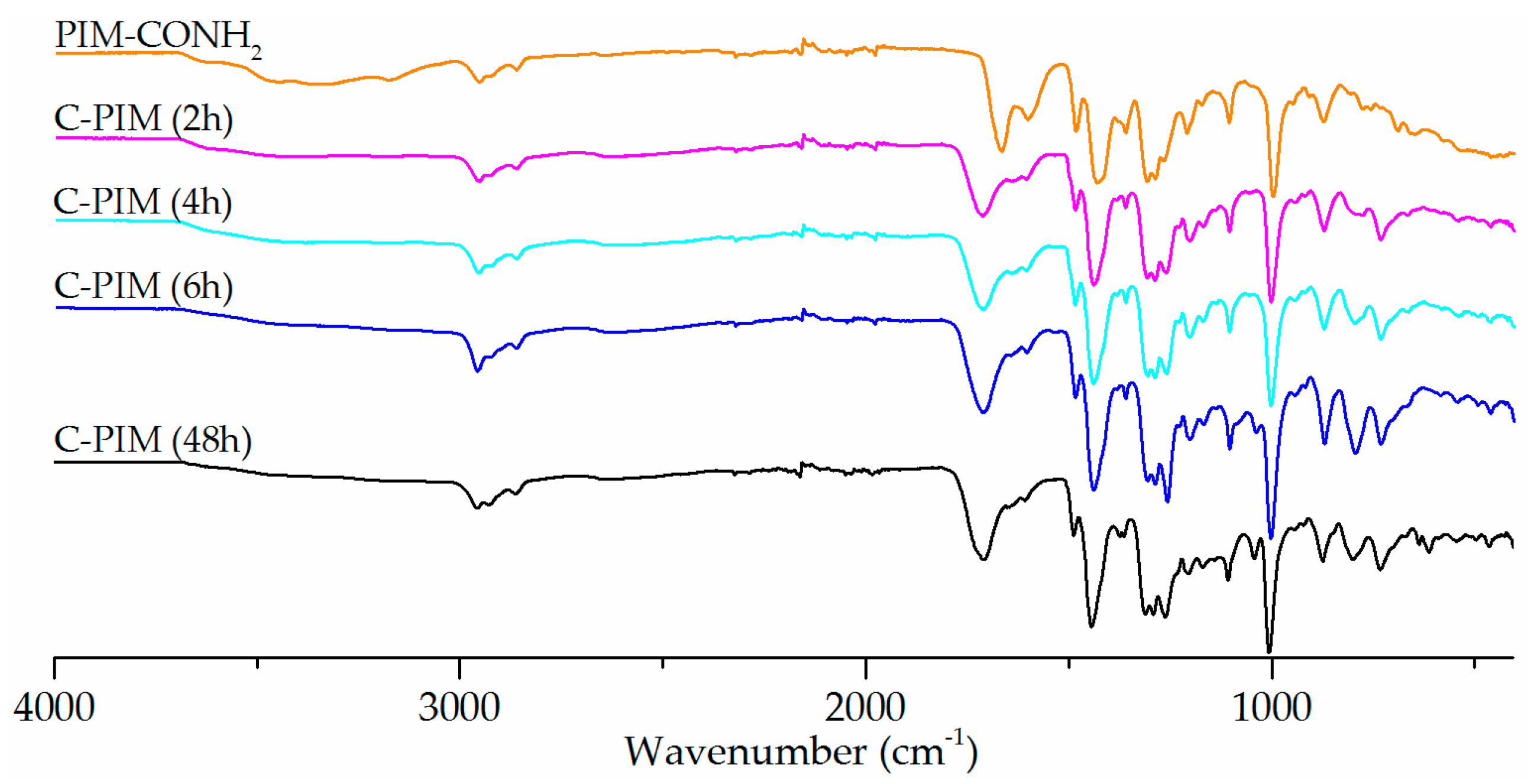
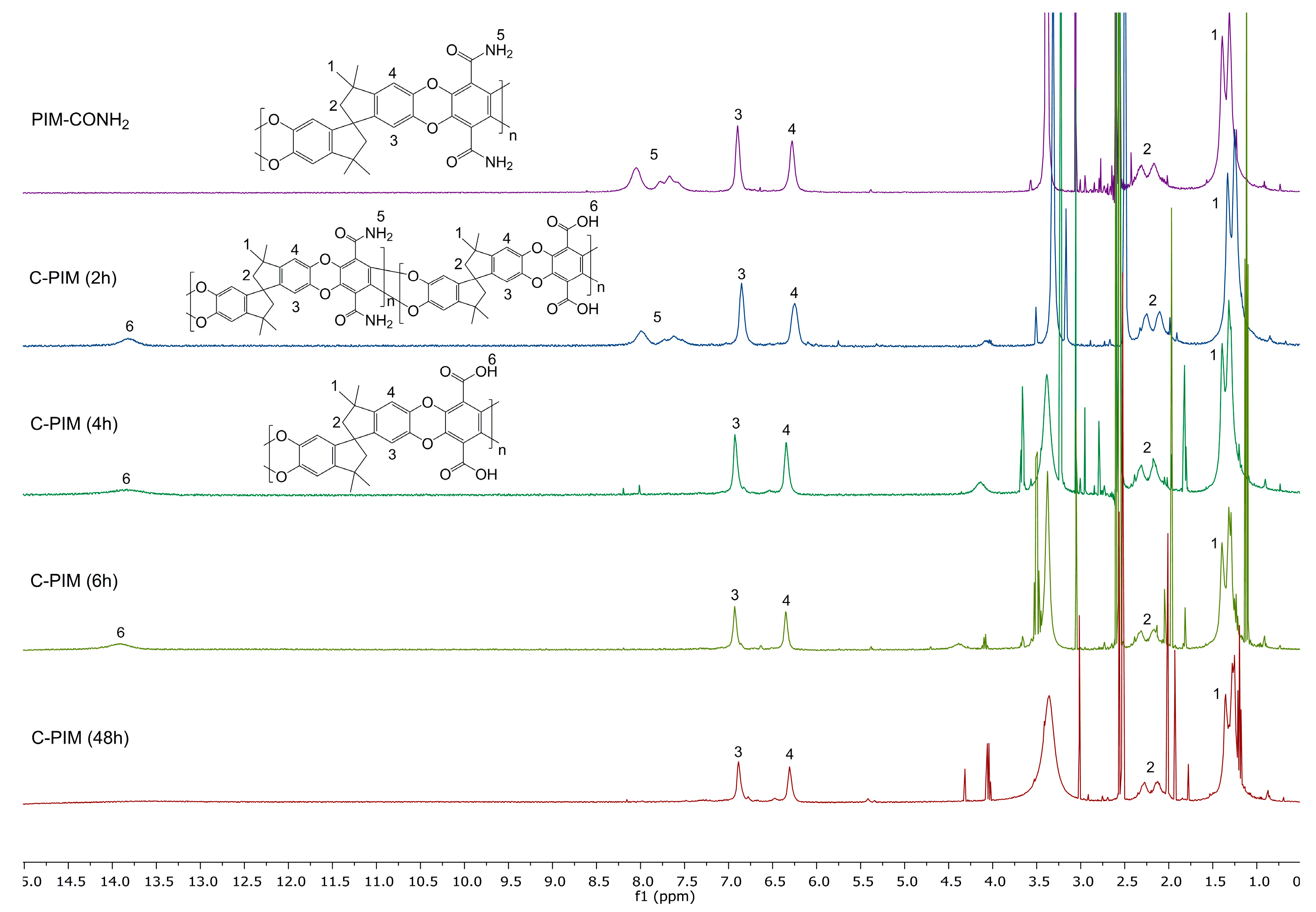
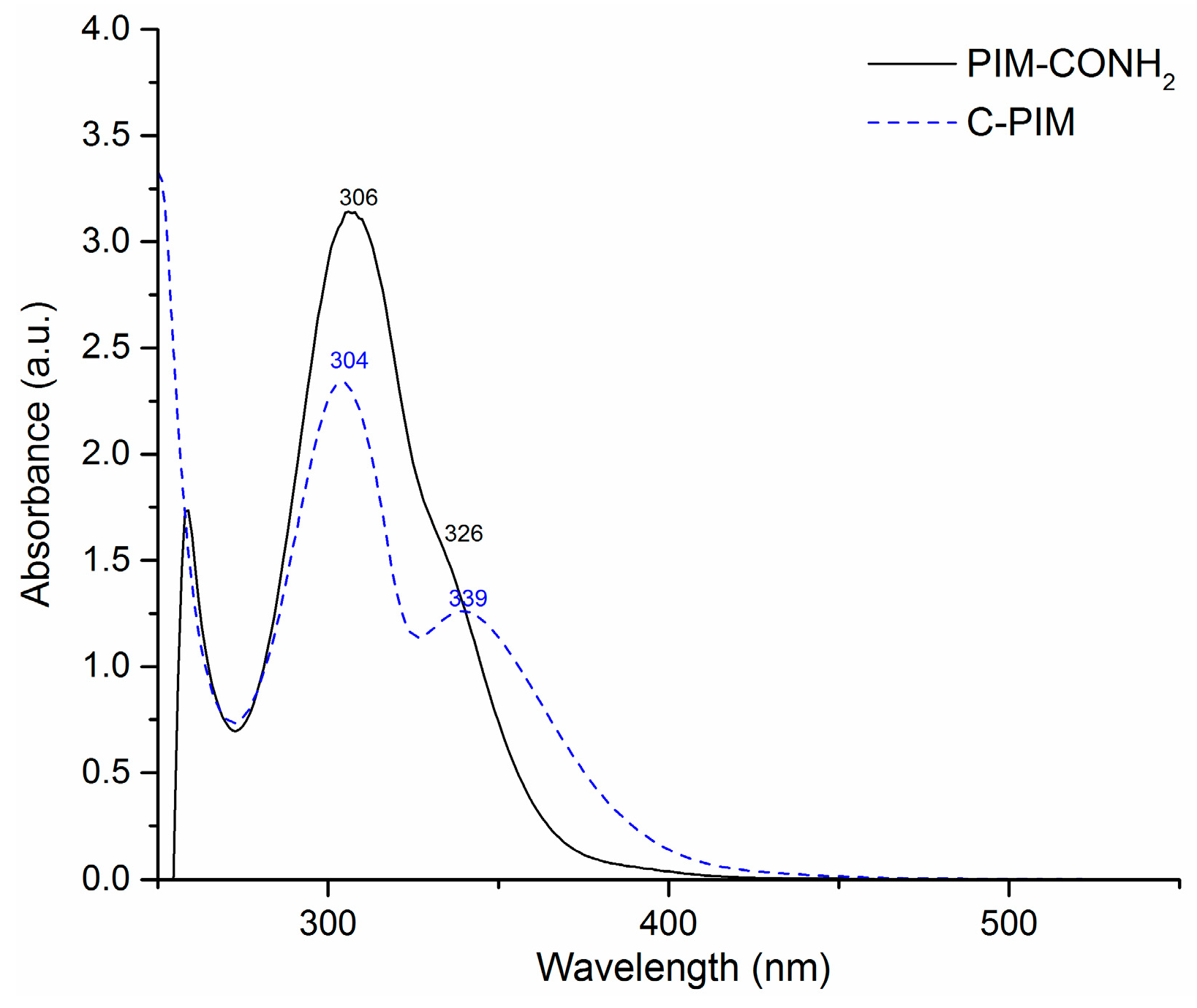
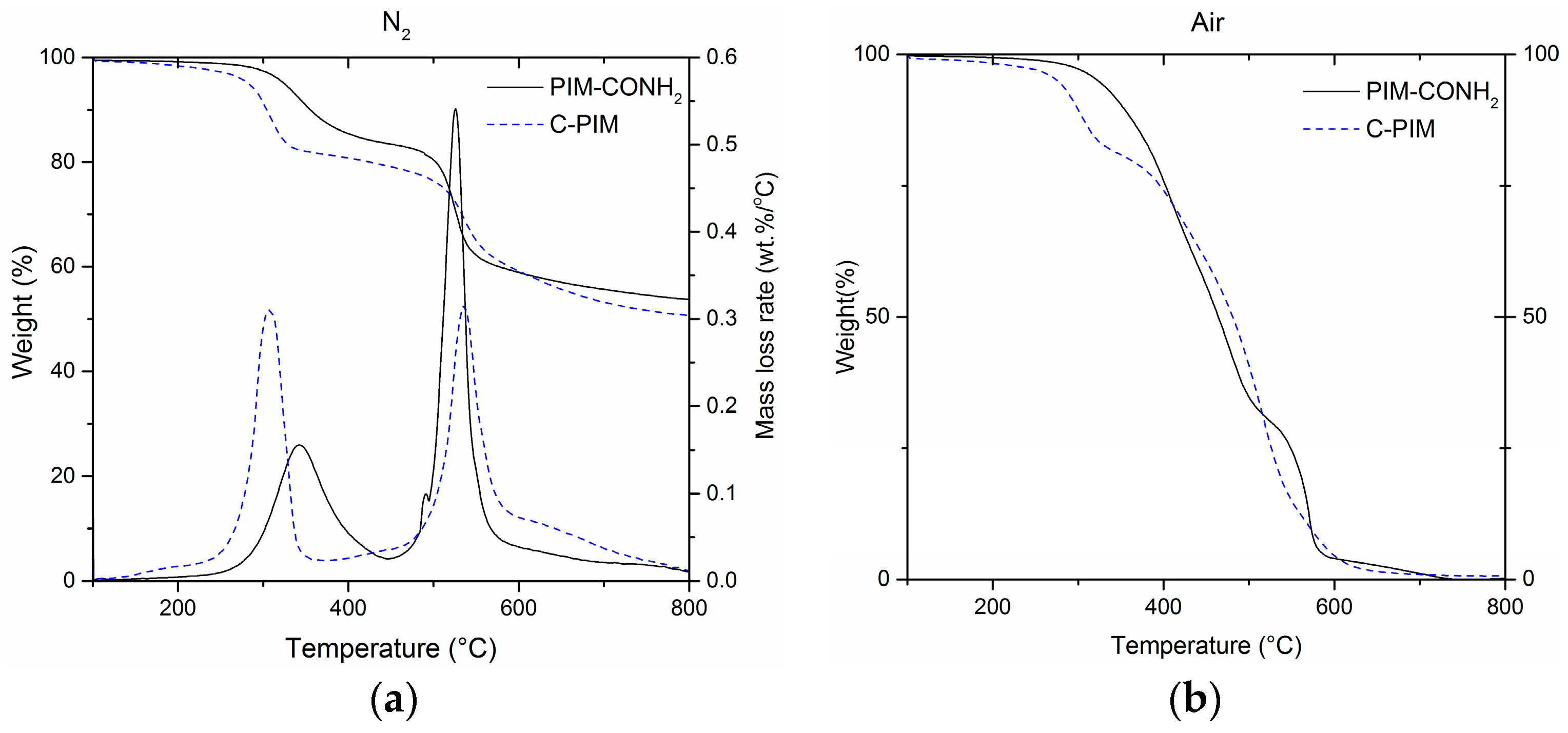
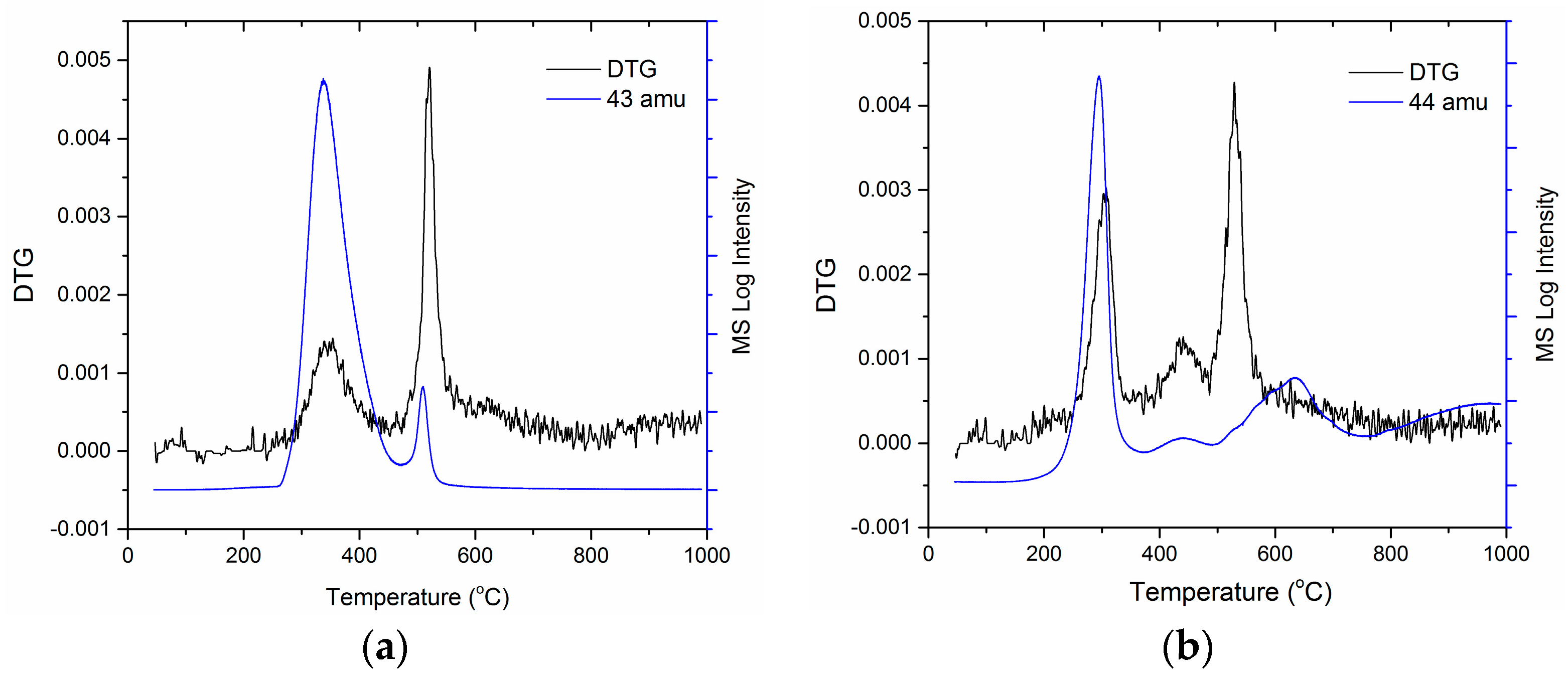
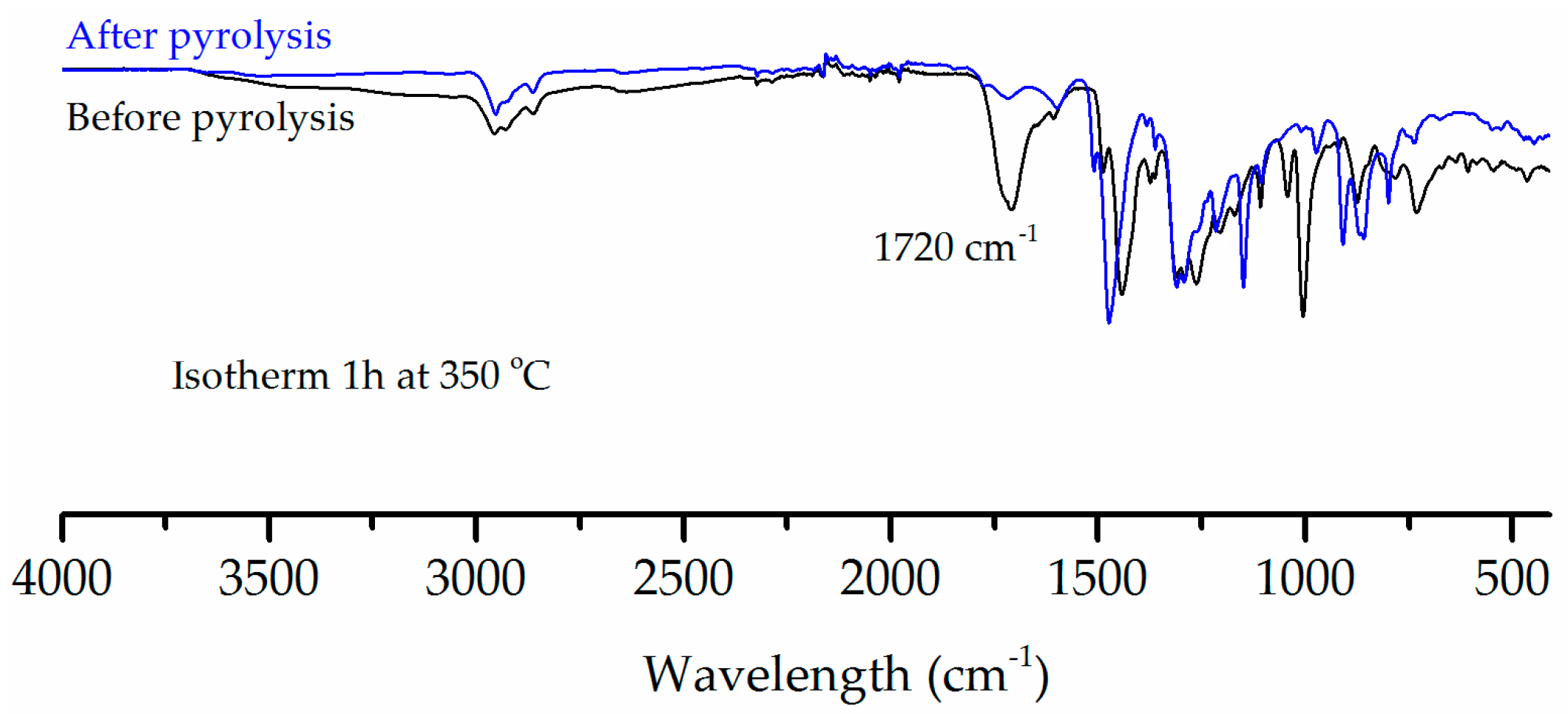
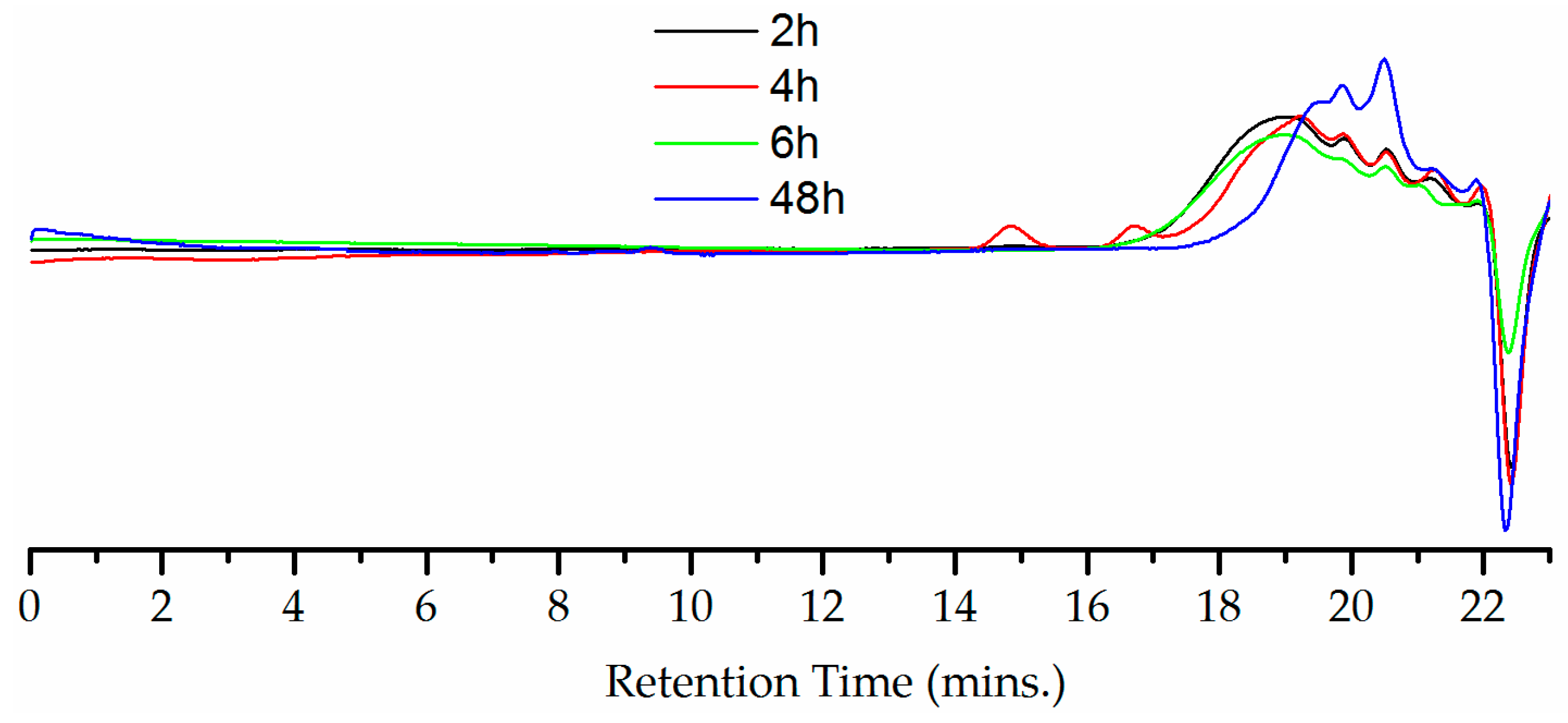
3. Results
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pachauri, R.K.; Allen, M.R.; Barros, V.R.; Broome, J.; Cramer, W.; Christ, R.; Church, J.A.; Clarke, L.; Dahe, Q.; Dasgupta, P.; et al. Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; IPCC: Geneva, Switzerland, 2014. [Google Scholar]
- Sokolov, A.P.; Stone, P.H.; Forest, C.E.; Prinn, R.; Sarofim, M.C.; Webster, M.; Paltsev, S.; Schlosser, C.A.; Kicklighter, D.; Dutkiewicz, S.; et al. Probabilistic forecast for twenty-first-century climate based on uncertainties in emissions (without policy) and climate parameters. J. Clim. 2009, 22, 5175–5204. [Google Scholar] [CrossRef]
- Agiaye, E.O.; Othman, M. CO2 Capture & Utilization: Harnessing the CO2 Content in Natural Gas for Environmental and Economic Gains. In Proceedings of the SPE Nigeria Annual International Conference and Exhibition, Lagos, Nigeria, 4–6 August 2015; Society of Petroleum Engineers: Richardson, TX, USA, 2015. [Google Scholar]
- Chakravarti, S.; Gupta, A.; Hunek, B. Advanced Technology for the Capture of Carbon Dioxide from Flue Gases. In Proceedings of the First National Conference on Carbon Sequestration, Washington, DC, USA, 15–17 May 2001; Praxair, Inc.: Tonawanda, NY, USA, 2001; pp. 15–17. [Google Scholar]
- Pires, J.; Martins, F.; Alvim-Ferraz, M.; Simões, M. Recent developments on carbon capture and storage: An overview. Chem. Eng. Res. Des. 2011, 89, 1446–1460. [Google Scholar] [CrossRef]
- Riemer, P.W. The Capture of Carbon Dioxide from Fossil Fuel Fired Power Stations; IEA Greenhouse Gas R & D Programme: Cheltenham, UK, 1993. [Google Scholar]
- Pfaff, I.; Kather, A. Comparative thermodynamic analysis and integration issues of CCS steam power plants based on oxy-combustion with cryogenic or membrane based air separation. Energy Procedia 2009, 1, 495–502. [Google Scholar] [CrossRef]
- Scholes, C.A.; Kentish, S.E.; Stevens, G.W. Effects of minor components in carbon dioxide capture using polymeric gas separation membranes. Sep. Purif. Rev. 2009, 38, 1–44. [Google Scholar] [CrossRef]
- Budd, P.M.; Ghanem, B.S.; Makhseed, S.; McKeown, N.B.; Msayib, K.J.; Tattershall, C.E. Polymers of intrinsic microporosity (PIMs): Robust, solution-processable, organic nanoporous materials. Chem. Commun. 2004, 230–231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jin, J.; Cooney, R.; Fu, Q.; Qiao, G.G.; Thomas, S.; Merkel, T.C. Synthesis of perfectly alternating copolymers for polymers of intrinsic microporosity. Polym. Chem. 2015, 6, 5003–5008. [Google Scholar] [CrossRef]
- Zhang, J.; Jin, J.; Cooney, R.; Zhang, S. Synthesis of polymers of intrinsic microporosity using an ab-type monomer. Polymer 2015, 57, 45–50. [Google Scholar] [CrossRef]
- Zhang, J.; Jin, J.; Cooney, R.; Zhang, S. Fluoride-mediated polycondensation for the synthesis of polymers of intrinsic microporosity. Polymer 2015, 76, 168–172. [Google Scholar] [CrossRef]
- Zhang, J.; Kang, H.; Martin, J.; Zhang, S.; Thomas, S.; Merkel, T.C.; Jin, J. The enhancement of chain rigidity and gas transport performance of polymers of intrinsic microporosity via intramolecular locking of the spiro-carbon. Chem. Commun. 2016, 52, 6553–6556. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.R.; Jin, J.; Freeman, B.D.; Paul, D.R. Physical aging, CO2 sorption and plasticization in thin films of polymer with intrinsic microporosity (PIM-1). J. Membr. Sci. 2017, 537, 362–371. [Google Scholar] [CrossRef]
- Starannikova, L.; Belov, N.; Shantarovich, V.; Zhang, J.; Jin, J.; Yampolskii, Y. Effective increase in permeability and free volume of PIM copolymers containing ethanoanthracene unit and comparison between the alternating and random copolymers. J. Membr. Sci. 2018, 548, 593–597. [Google Scholar] [CrossRef]
- Du, N.; Park, H.B.; Robertson, G.P.; Dal-Cin, M.M.; Visser, T.; Scoles, L.; Guiver, M.D. Polymer nanosieve membranes for CO2-capture applications. Nat. Mater. 2011, 10, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Du, N.; Robertson, G.P.; Dal-Cin, M.M.; Scoles, L.; Guiver, M.D. Polymers of intrinsic microporosity (PIMs) substituted with methyl tetrazole. Polymer 2012, 53, 4367–4372. [Google Scholar] [CrossRef]
- Patel, H.A.; Yavuz, C.T. Noninvasive functionalization of polymers of intrinsic microporosity for enhanced CO2 capture. Chem. Commun. 2012, 48, 9989–9991. [Google Scholar] [CrossRef] [PubMed]
- Swaidan, R.; Ghanem, B.S.; Litwiller, E.; Pinnau, I. Pure-and mixed-gas CO2/CH4 separation properties of PIM-1 and an amidoxime-functionalized PIM-1. J. Membr. Sci. 2014, 457, 95–102. [Google Scholar] [CrossRef]
- Mason, C.R.; Maynard-Atem, L.; Al-Harbi, N.M.; Budd, P.M.; Bernardo, P.; Bazzarelli, F.; Clarizia, G.; Jansen, J.C. Polymer of intrinsic microporosity incorporating thioamide functionality: Preparation and gas transport properties. Macromolecules 2011, 44, 6471–6479. [Google Scholar] [CrossRef]
- Mason, C.R.; Maynard-Atem, L.; Heard, K.W.; Satilmis, B.; Budd, P.M.; Friess, K.; Lanč, M.; Bernardo, P.; Clarizia, G.; Jansen, J.C. Enhancement of CO2 affinity in a polymer of intrinsic microporosity by amine modification. Macromolecules 2014, 47, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Satilmis, B.; Alnajrani, M.N.; Budd, P.M. Hydroxyalkylaminoalkylamide PIMs: Selective adsorption by ethanolamine-and diethanolamine-modified PIM-1. Macromolecules 2015, 48, 5663–5669. [Google Scholar] [CrossRef]
- Weng, X.; Baez, J.E.; Khiterer, M.; Hoe, M.Y.; Bao, Z.; Shea, K.J. Chiral polymers of intrinsic microporosity: Selective membrane permeation of enantiomers. Angew. Chem. Int. Ed. 2015, 54, 11214–11218. [Google Scholar] [CrossRef] [PubMed]
- Du, N.; Robertson, G.P.; Song, J.; Pinnau, I.; Guiver, M.D. High-performance carboxylated polymers of intrinsic microporosity (PIMs) with tunable gas transport properties. Macromolecules 2009, 42, 6038–6043. [Google Scholar] [CrossRef]
- Du, N.; Dal-Cin, M.M.; Robertson, G.P.; Guiver, M.D. Decarboxylation-induced cross-linking of polymers of intrinsic microporosity (PIMs) for membrane gas separation. Macromolecules 2012, 45, 5134–5139. [Google Scholar] [CrossRef]
- Yong, W.F.; Chung, T.-S. Miscible blends of carboxylated polymers of intrinsic microporosity (cPIM-1) and Matrimid. Polymer 2015, 59, 290–297. [Google Scholar] [CrossRef]
- Yong, W.F.; Li, F.Y.; Chung, T.S.; Tong, Y.W. Molecular interaction, gas transport properties and plasticization behavior of cPIM-1/Torlon blend membranes. J. Membr. Sci. 2014, 462, 119–130. [Google Scholar] [CrossRef]
- Salehian, P.; Yong, W.F.; Chung, T.-S. Development of high performance carboxylated PIM-1/P84 blend membranes for pervaporation dehydration of isopropanol and CO2/CH4 separation. J. Membr. Sci. 2016, 518, 110–119. [Google Scholar] [CrossRef]
- Liao, K.-S.; Lai, J.-Y.; Chung, T.-S. Metal ion modified PIM-1 and its application for propylene/propane separation. J. Membr. Sci. 2016, 515, 36–44. [Google Scholar] [CrossRef]
- Zhao, H.; Xie, Q.; Ding, X.; Chen, J.; Hua, M.; Tan, X.; Zhang, Y. High performance post-modified polymers of intrinsic microporosity (PIM-1) membranes based on multivalent metal ions for gas separation. J. Membr. Sci. 2016, 514, 305–312. [Google Scholar] [CrossRef]
- Wind, J.D.; Staudt-Bickel, C.; Paul, D.R.; Koros, W.J. The effects of crosslinking chemistry on CO2 plasticization of polyimide gas separation membranes. Ind. Eng. Chem. Res. 2002, 41, 6139–6148. [Google Scholar] [CrossRef]
- Satilmis, B.; Budd, P.M. Base-catalysed hydrolysis of PIM-1: Amide versus carboxylate formation. RSC Adv. 2014, 4, 52189–52198. [Google Scholar] [CrossRef]
- Santoso, B.; Yanaranop, P.; Kang, H.; Leung, I.K.; Jin, J. A Critical Update on the Synthesis of Carboxylated Polymers of Intrinsic Microporosity (C-PIMs). Macromolecules 2017, 50, 3043–3050. [Google Scholar] [CrossRef]
- Jeon, J.W.; Kim, D.-G.; Sohn, E.-H.; Yoo, Y.; Kim, Y.S.; Kim, B.G.; Lee, J.-C. Highly Carboxylate-Functionalized Polymers of Intrinsic Microporosity for CO2-Selective Polymer Membranes. Macromolecules 2017, 50, 8019–8027. [Google Scholar] [CrossRef]
- McMurry, J. Organic Chemistry, 7th ed.; Thomson Brooks Cole: Pacific Grove, CA, USA, 2008. [Google Scholar]
- Yanaranop, P.; Santoso, B.; Etzion, R.; Jin, J. Facile conversion of nitrile to amide on polymers of intrinsic microporosity (PIM-1). Polymer 2016, 98, 244–251. [Google Scholar] [CrossRef]
- Satilmis, B.; Budd, P.M.; Uyar, T. Systematic hydrolysis of PIM-1 and electrospinning of hydrolyzed PIM-1 ultrafine fibers for an efficient removal of dye from water. React. Funct. Polym. 2017, 121, 67–75. [Google Scholar] [CrossRef]
- Olah, G.A.; Olah, J.A. Reactions of Amides and Sulfonamides with Nitrosonium Salts. J. Org. Chem. 1965, 30, 2386–2387. [Google Scholar] [CrossRef]
- Bhole, Y.S. Investigations on Gas Permeation and Related Physical Properties of structurally Architectured Aromatic Polymers (Polyphenylene Oxides and Polyarylates), Polyarlate-Clay Nanocomposites and Poly (Ionic Liquid). Ph.D. Thesis, CSIR-National Chemical Laboratory, Pune, India, 2007. [Google Scholar]
- Sakaguchi, T.; Yumoto, K.; Shiotsuki, M.; Sanda, F.; Yoshikawa, M.; Masuda, T. Synthesis of poly (diphenylacetylene) membranes by desilylation of various precursor polymers and their properties. Macromolecules 2005, 38, 2704–2709. [Google Scholar] [CrossRef]
- Krasnokutskaya, E.A.; Semenischeva, N.I.; Filimonov, V.D.; Knochel, P. A new, one-step, effective protocol for the iodination of aromatic and heterocyclic compounds via aprotic diazotization of amines. Synthesis 2007, 2007, 81–84. [Google Scholar] [CrossRef]
- Filimonov, V.D.; Trusova, M.; Postnikov, P.; Krasnokutskaya, E.A.; Lee, Y.M.; Hwang, H.Y.; Kim, H.; Chi, K.-W. Unusually stable, versatile, and pure arenediazonium tosylates: Their preparation, structures, and synthetic applicability. Org. Lett. 2008, 10, 3961–3964. [Google Scholar] [CrossRef] [PubMed]
- Borikar, S.P.; Paul, V. N-Nitrosation of Secondary Amines Using p-TSA-NaNO2 as a Novel Nitrosating Agent Under Mild Conditions. Synth. Commun. 2010, 40, 654–660. [Google Scholar] [CrossRef]
- Søndergaard, R.R.; Norrman, K.; Krebs, F.C. Low-temperature side-chain cleavage and decarboxylation of polythiophene esters by acid catalysis. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 1127–1132. [Google Scholar] [CrossRef]
- Philippides, A.; Budd, P.M.; Price, C.; Cuncliffe, A.V. The nitration of polystyrene. Polymer 1993, 34, 3509–3513. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Base Hydrolysis | Acid Hydrolysis | This Work |
|---|---|---|---|
| Starting Material | PIM-1 | PIM-1 | PIM-CONH2 |
| Reaction Conditions | 20% NaOH at 120 °C, 5 h | H2SO4, H2O and Acetic acid at 105 °C, 48 h | NaNO2, H2SO4 and MeCN at 90 °C, 6 h |
| Samples | C% | H% | N% | O% * |
|---|---|---|---|---|
| PIM-1 | 74.65 | 4.47 | 6.19 | 14.69 |
| PIM-CONH2 | 65.53 | 5.33 | 5.24 | 23.90 |
| C-PIM | 63.55 | 4.80 | 0.91 | 30.74 |
| Solvent | PIM-1 | PIM-CONH2 | C-PIM Obtained in This Work |
|---|---|---|---|
| DCM | soluble | insoluble | insoluble |
| EtOAc | insoluble | insoluble | partially soluble |
| CHCl3 | soluble | insoluble | insoluble |
| THF | soluble | partially soluble | soluble |
| DMF | insoluble | soluble | soluble |
| DMAc | insoluble | soluble | soluble |
| NMP | insoluble | soluble | soluble |
| MeOH | insoluble | partially soluble | partially soluble |
| MeCN | insoluble | insoluble | partially soluble |
| EtOH | insoluble | insoluble | partially soluble |
| Acetone | insoluble | insoluble | soluble |
| DMSO | insoluble | soluble | soluble |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, W.-H.; Thomas, P.; Hume, P.; Jin, J. Effective Conversion of Amide to Carboxylic Acid on Polymers of Intrinsic Microporosity (PIM-1) with Nitrous Acid. Membranes 2018, 8, 20. https://doi.org/10.3390/membranes8020020
Wu W-H, Thomas P, Hume P, Jin J. Effective Conversion of Amide to Carboxylic Acid on Polymers of Intrinsic Microporosity (PIM-1) with Nitrous Acid. Membranes. 2018; 8(2):20. https://doi.org/10.3390/membranes8020020
Chicago/Turabian StyleWu, Wei-Hsuan, Paul Thomas, Paul Hume, and Jianyong Jin. 2018. "Effective Conversion of Amide to Carboxylic Acid on Polymers of Intrinsic Microporosity (PIM-1) with Nitrous Acid" Membranes 8, no. 2: 20. https://doi.org/10.3390/membranes8020020
APA StyleWu, W.-H., Thomas, P., Hume, P., & Jin, J. (2018). Effective Conversion of Amide to Carboxylic Acid on Polymers of Intrinsic Microporosity (PIM-1) with Nitrous Acid. Membranes, 8(2), 20. https://doi.org/10.3390/membranes8020020