

Fractionation of a Procyanidin-Rich Grape Seed Extract by a Preparative Integrated Ultrafiltration/Reverse Osmosis/Solid-Phase Extraction Procedure

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Chemicals
2.2. Experimental Equipment
2.3. Analytical Equipment
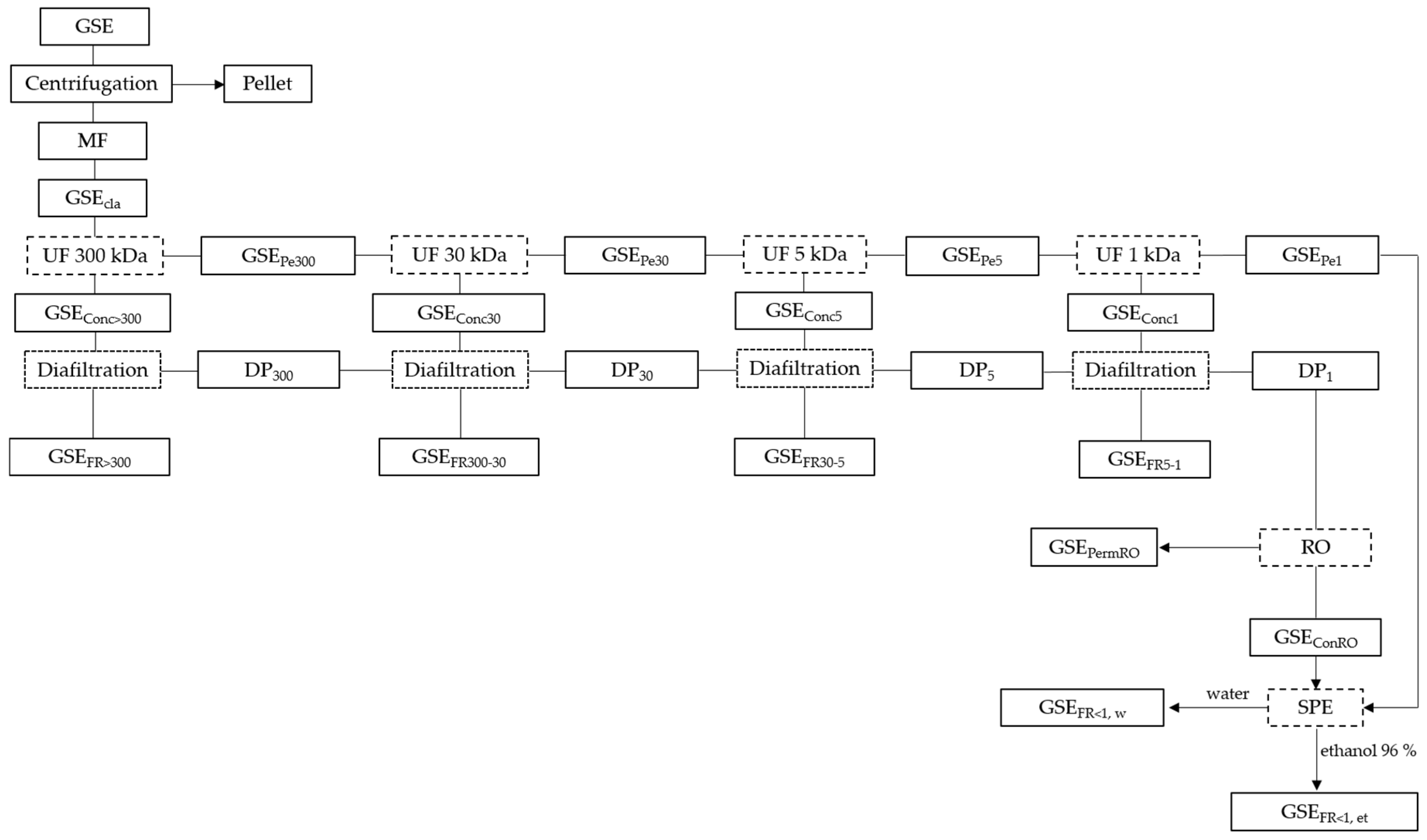
2.4. Preparative Separation Methodologies
2.4.1. Clarification of Grape Seed Extract
2.4.2. Integrated UF Fractionation of Grape Seed Extract
2.4.2.1. Separation of a Very-High-Molecular-Mass Fraction (>300 kDa) by Ultra/Diafiltration of the GSE with a 300 kDa MMCO Membrane
2.4.2.2. Separation of a High-Molecular-Mass Fraction (300–30 kDa) by Ultra/Diafiltration of the GSE Permeate from the 300 kDa Membrane Using a 30 kDa Membrane
2.4.2.3. Separation of an Intermediate-Molecular-Mass Fraction (30–5 kDa) by Ultra/Diafiltration of the GSE Permeate from the 30 kDa Membrane Using a 5 kDa Membrane
2.4.2.4. Separation of a of Low-Molecular-Mass Fraction (5–1 kDa) by Ultra/Diafiltration of the GSE Permeate from the 5 kDa Membrane Using a 1 kDa Membrane
2.4.3. Partial Concentration of a Very-Low-Molecular-Mass Fraction (<1 kDa) of the GSE by Reverse Osmosis
2.4.4. Membrane Regeneration
2.4.5. Purification of Catechins and Procyanidin Oligomers by Preparative SPE
2.5. Statistical Analysis
3. Results and Discussion
3.1. Separation of a Very-High-Molecular-Mass Fraction (>300 kDa) by Ultra/Diafiltration of GSEcla Using a 300 kDa MMCO Membrane
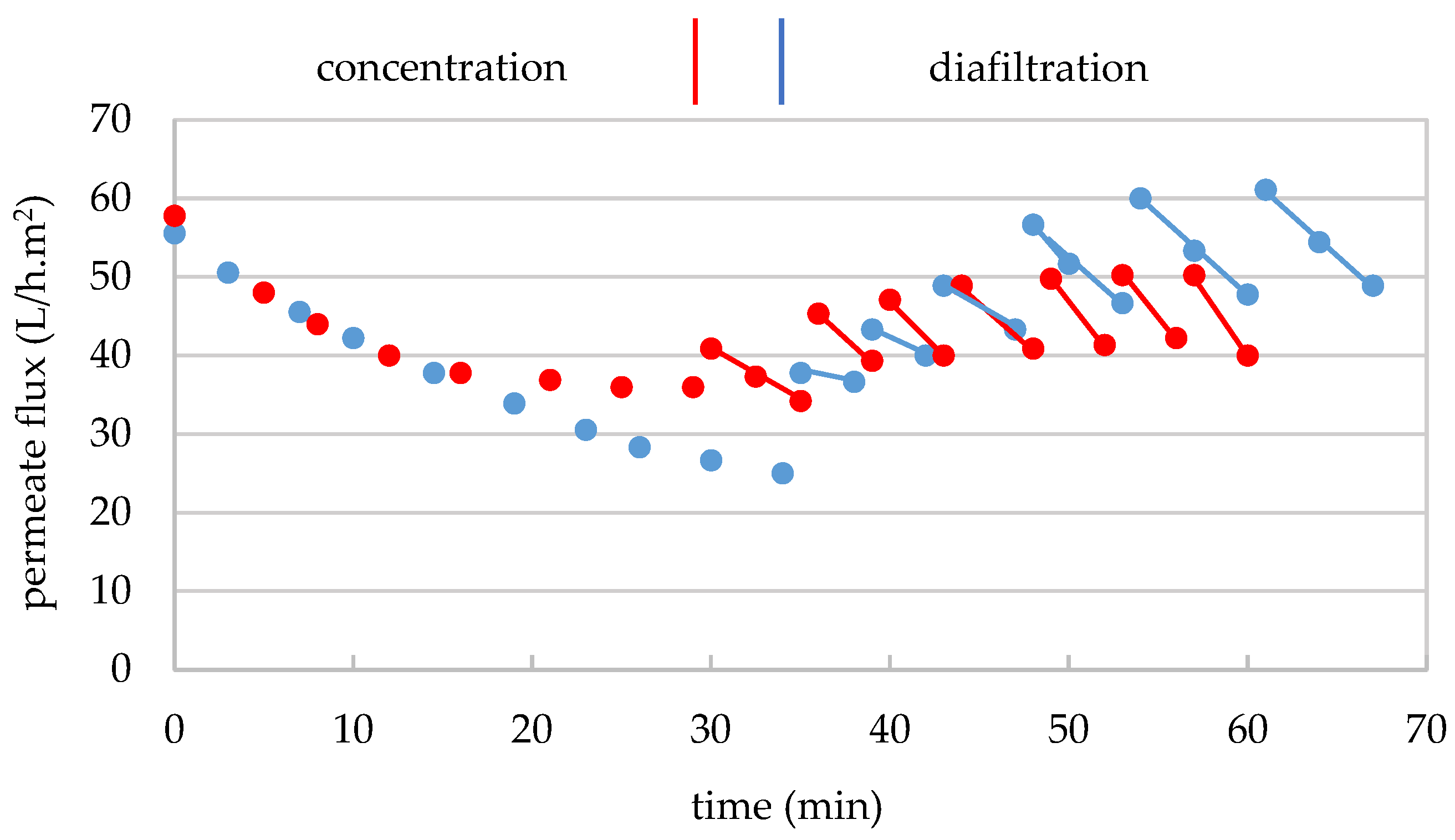
3.1.1. Operating Conditions and Material Balance
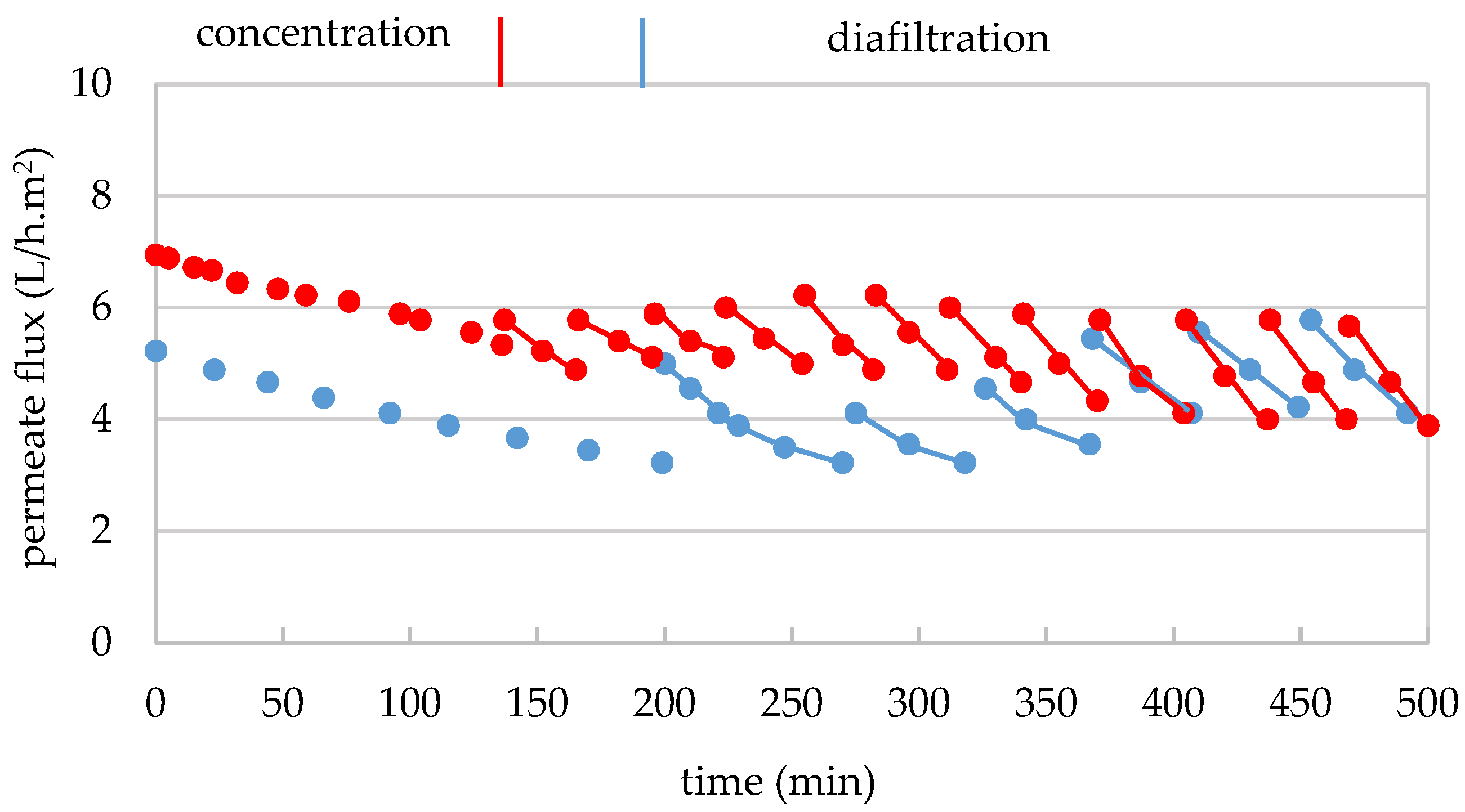
3.1.2. Mass Transfer Through the 300 kDa Membrane
3.1.2.1. TDS Transfer
3.1.2.2. Electrolyte Transfer
3.1.2.3. Effect on pH
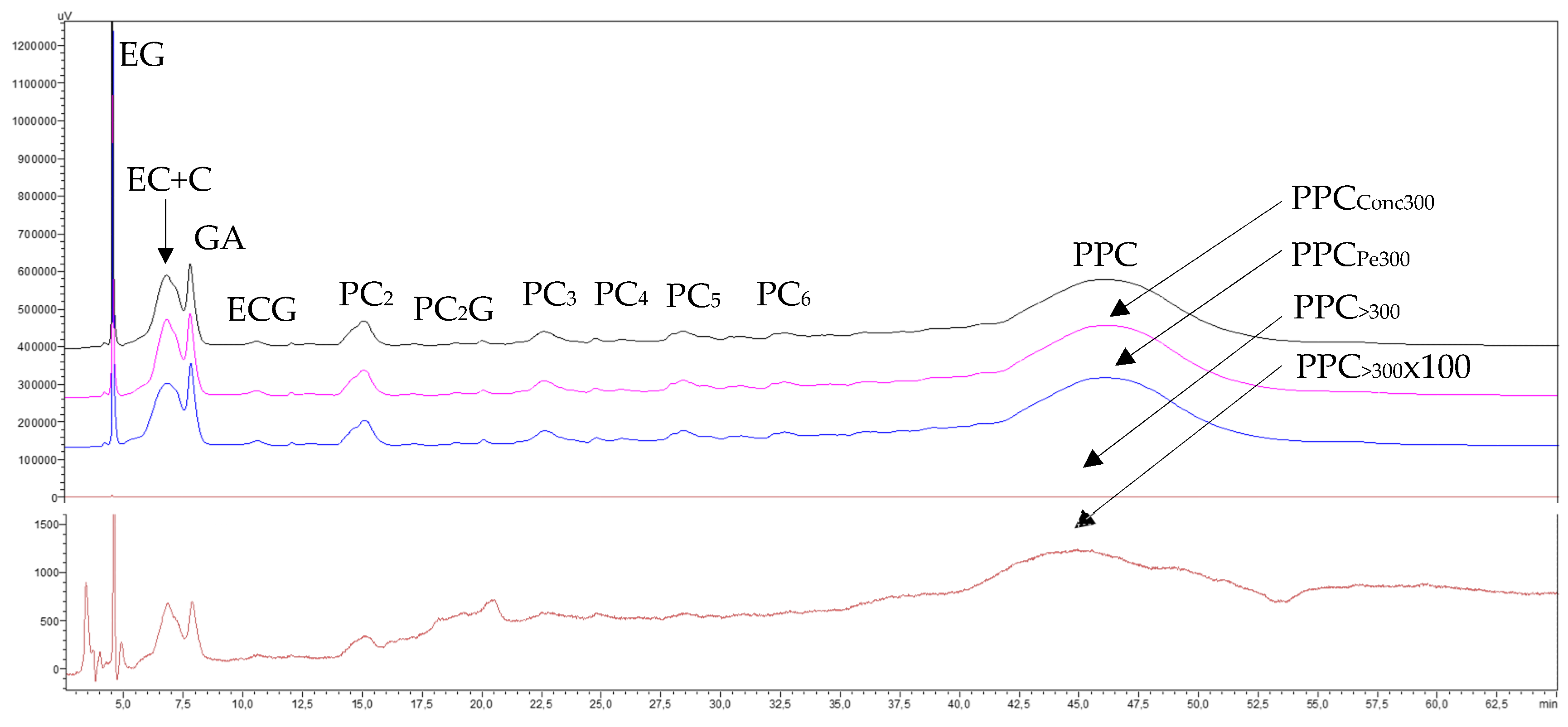
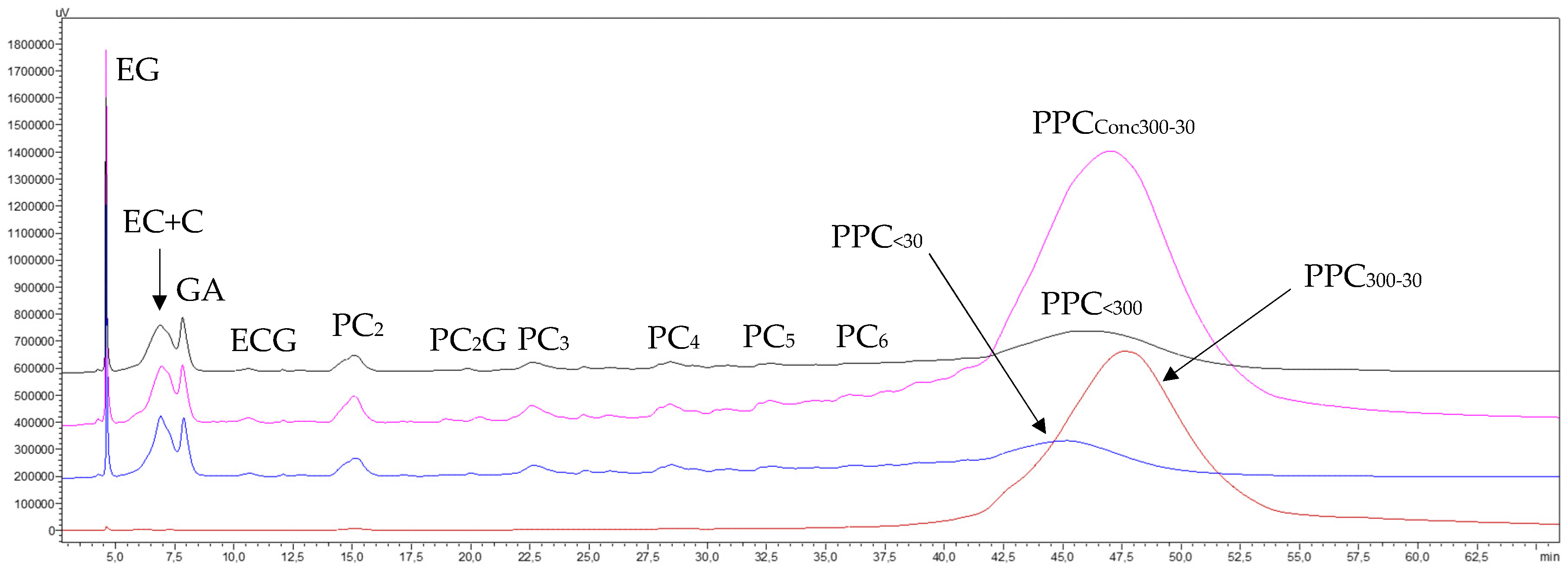
3.1.2.4. Phenolic Compounds Transfer
3.1.3. Evaluation of the Hydraulic Permeability of the 300 kDa Membrane After Filtration and Its Chemical Regeneration
3.2. Separation of a High-Molecular-Mass Fraction (300–30 kDa) by Ultra/Diafiltration of the GSE Permeate from the 300 kDa MMCO Membrane Treatment Using a 30 kDa MMCO Membrane
3.2.1. Operating Conditions and Material Balance
3.2.2. Mass Transfer Through the 30 kDa Membrane
3.2.2.1. TDS Transfer
3.2.2.2. Electrolyte Transfer
3.2.2.3. Effect on pH
3.2.2.4. Phenolic Compounds Transfer
3.2.3. Evaluation of the Hydraulic Permeability of the 30 kDa Membrane After Filtration and Its Chemical Regeneration
3.3. Separation of an Intermediate-Molecular-Mass Fraction (30—5 kDa) by Ultra/Diafiltration of the GSE Permeate from the 30 kDa MMCO Membrane Treatment Using a 5 kDa MMCO Membrane
3.3.1. Operating Conditions and Material Balance
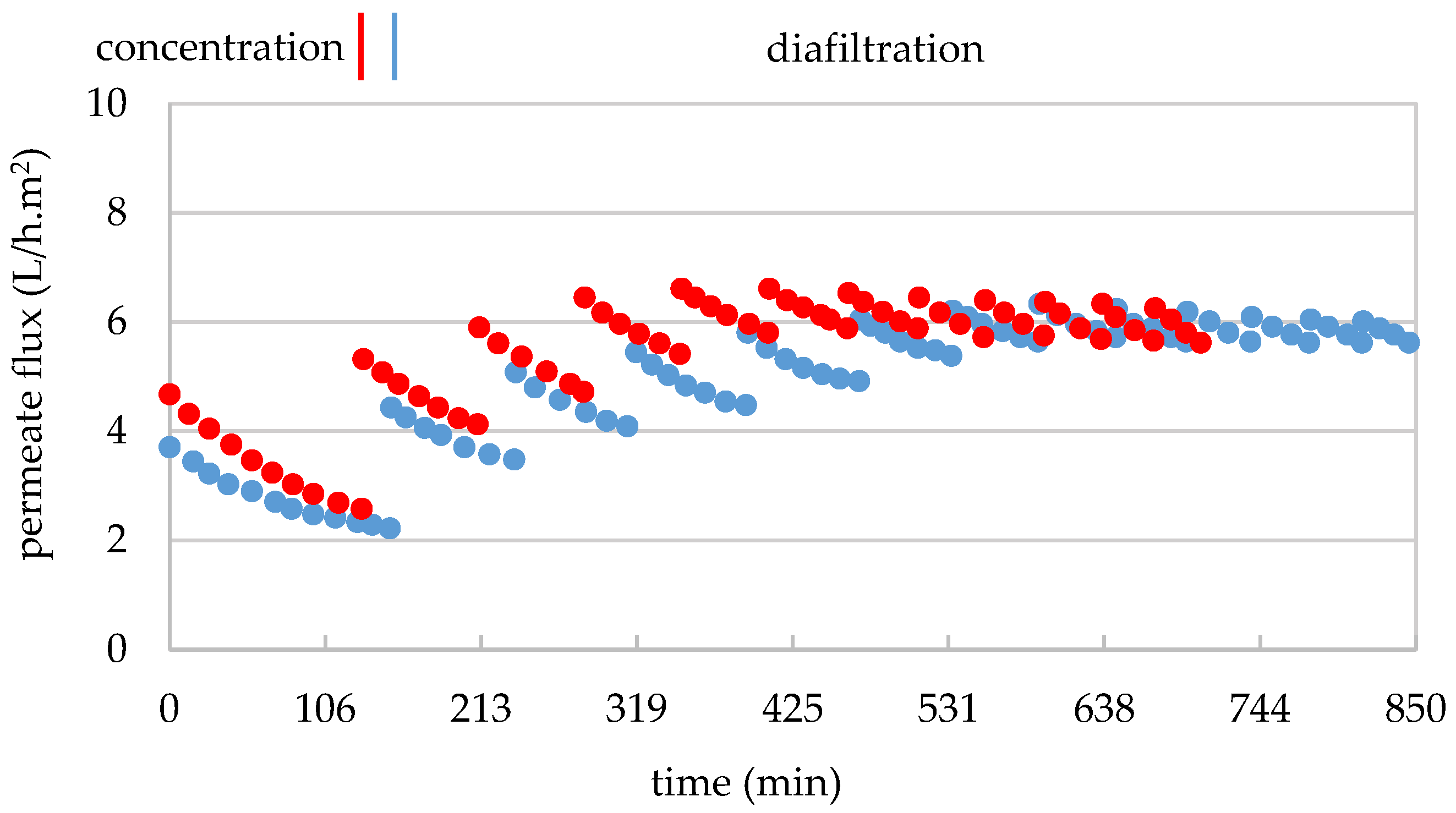
3.3.2. Mass Transfer Through the 5 kDa Membrane
3.3.2.1. TDS Transfer
3.3.2.2. Electrolyte Transfer
3.3.2.3. Effect on pH
3.3.2.4. Phenolic Compounds Transfer
3.3.3. Evaluation of the Hydraulic Permeability of the 5 kDa Membrane After Filtration and Its Chemical Regeneration
3.4. Separation of a Low-Molecular-Mass Fraction (5–1 kDa) by Ultra/Diafiltration of the GSE Permeate from the 5 kDa MMCO Membrane Treatment Using a 1 kDa MMCO Membrane
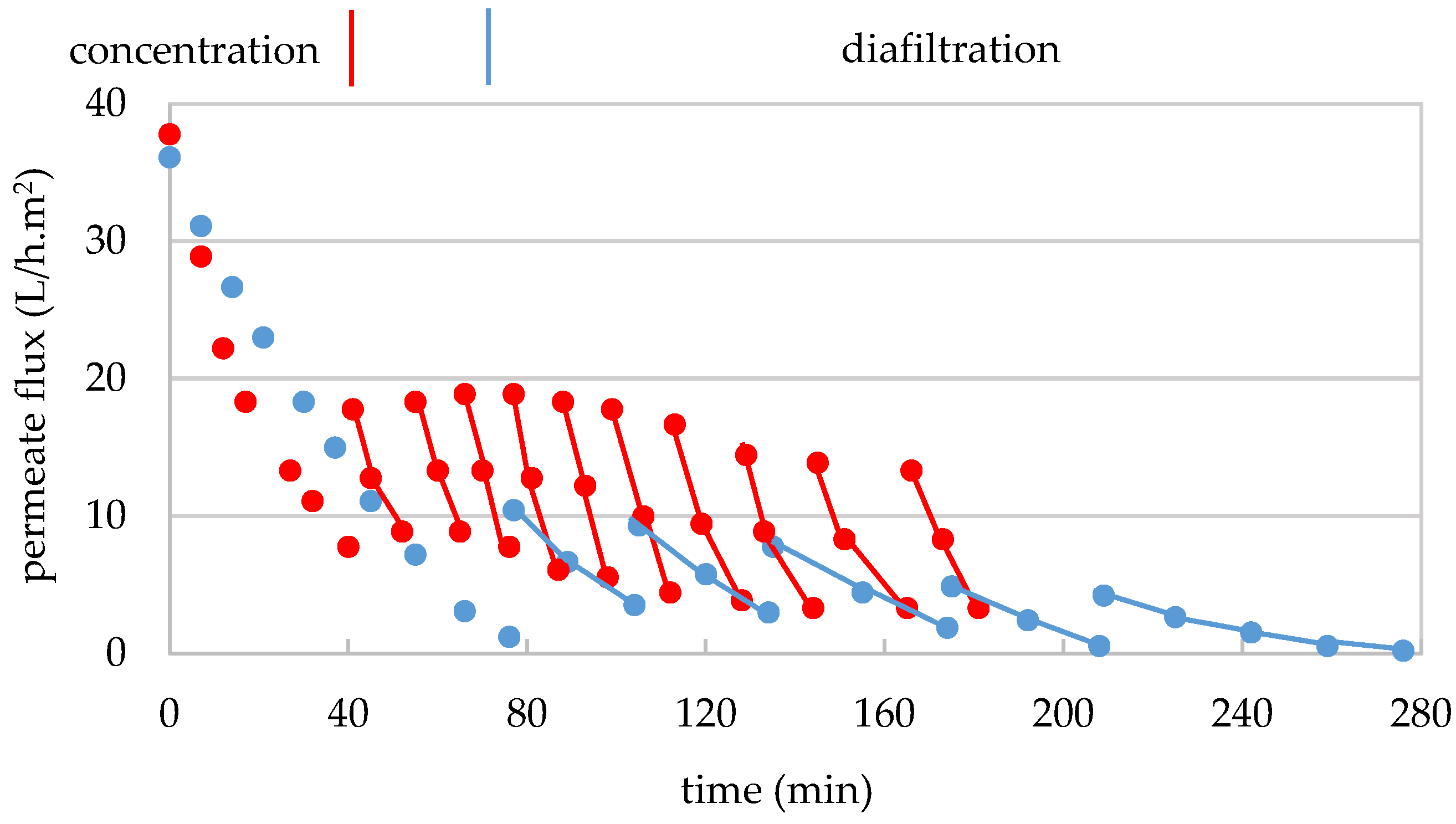
3.4.1. Operating Conditions and Material Balance
3.4.2. Mass Transfer Through the 1 kDa Membrane
3.4.2.1. TDS Transfer
3.4.2.2. Electrolyte Transfer
3.4.2.3. Effect on pH
3.4.2.4. Phenolic Compounds Transfer
3.4.3. Evaluation of the Hydraulic Permeability of the 1 kDa Membrane After Filtration and Its Chemical Regeneration
3.5. Partial Concentration of the Diafiltration Permeates of the GSE from the 1 kDa Membrane by a RO Membrane with 96% NaCl Rejection
3.5.1. Operating Conditions and Material Balance
3.5.2. Mass Transfer Through the RO Membrane
3.5.2.1. TDS Transfer
3.5.2.2. Electrolyte Transfer
3.5.2.3. Effect on pH
3.5.2.4. Phenolic Compounds Transfer
3.5.3. Evaluation of the Hydraulic Permeability of 96% NaCl Rejection Reverse Osmosis Membrane After Filtration and Its Chemical Regeneration
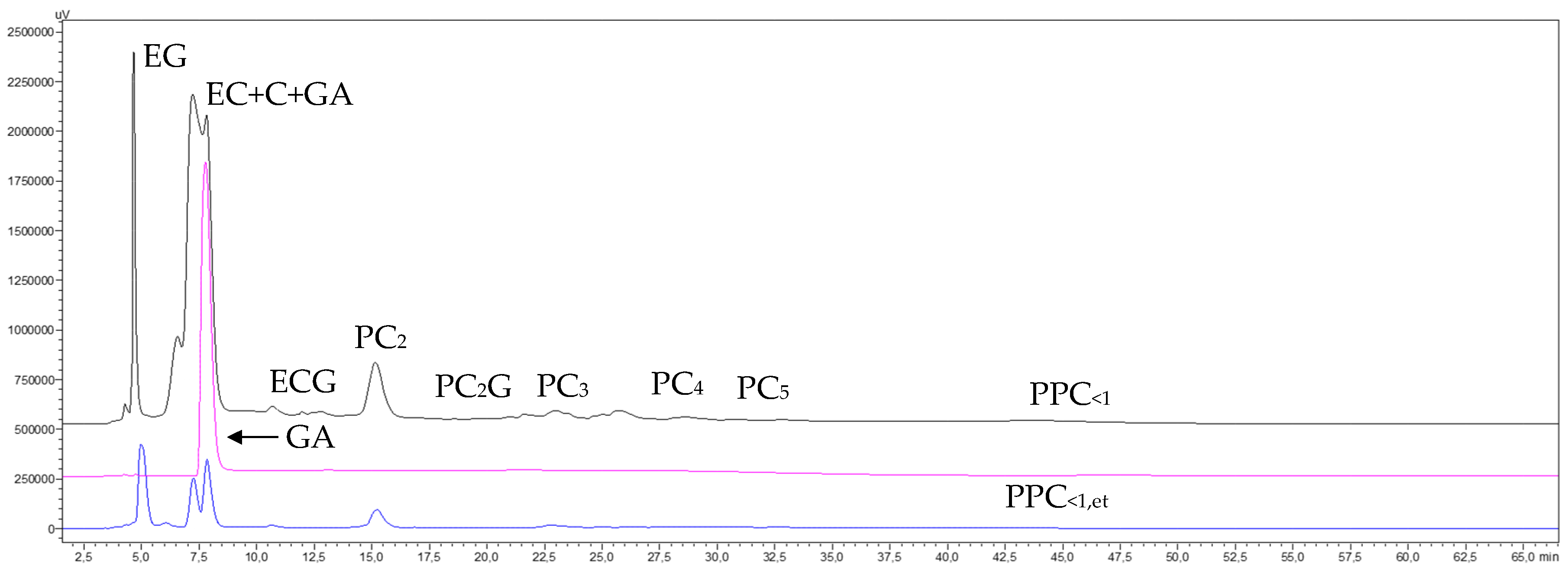
3.6. Purification of Catechins and Procyanidin Oligomers of GSEFR<1 by SPE
3.6.1. Operating Conditions and Material Balance
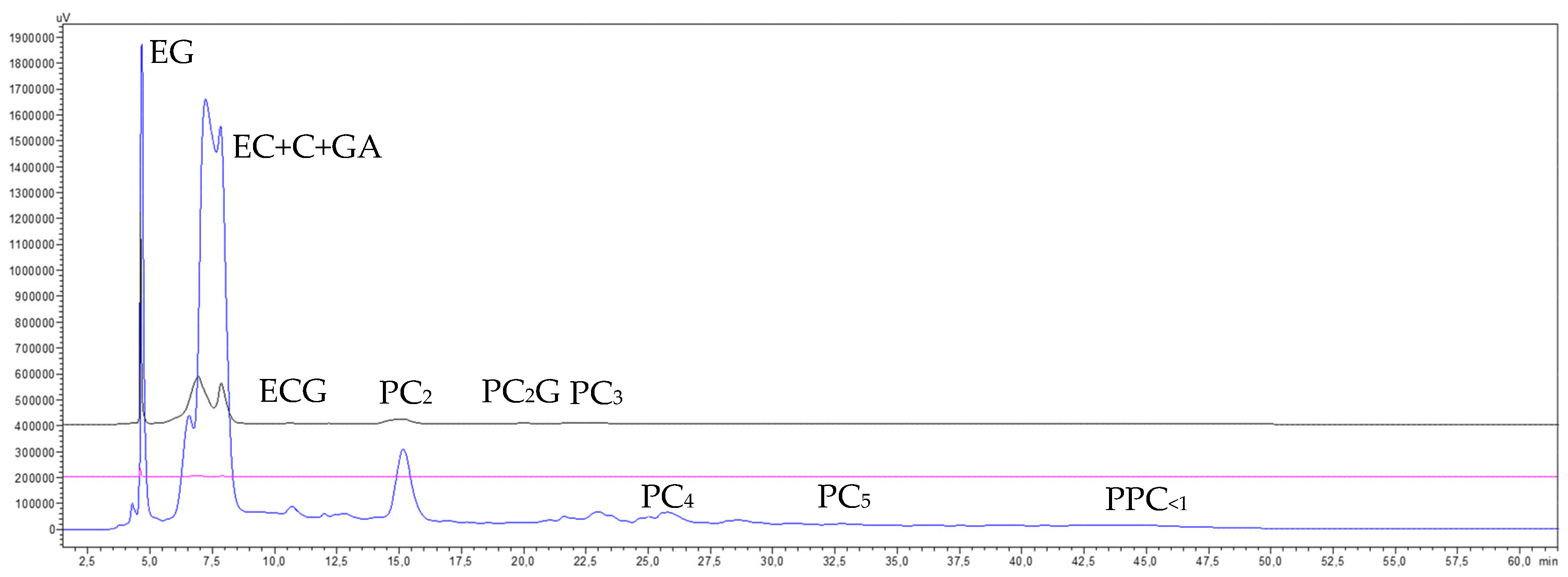
3.6.2. Phenolic Compounds Transfer
3.7. Estimation of the GSE Composition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anonymous. Grape seed extracts market size & trends. In Grape Seed Extracts Market Size, Share & Trends Analysis Report by Product (Powder, Liquid) by Application (Food & Beverage, Personal Care; Dietary Supplements), by Region, and Segment Forecasts; 2024–2030; Grand View Research, Inc.: San Francisco, CA, USA, 2024; Available online: https://www.grandviewresearch.com/industry-analysis/grape-seed-extracts-market (accessed on 28 January 2025).
- Web of Science Citation Report. 2025. Available online: https://www.webofscience.com (accessed on 25 January 2025).
- Sáyago-Ayerdi, S.G.; Brenes, A.; Viveros, A.; Goñi, I. Antioxidative effect of dietary grape pomace concentrate on lipid oxidation of chilled and long-term frozen stored chicken patties. Meat Sci. 2009, 83, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.M.; Alfaia, C.M.; Lopes, P.A.; Pestana, J.M.; Prates, J.A.M. Grape by-products as feedstuff for pig and poultry production. Animals 2022, 12, 2239. [Google Scholar] [CrossRef]
- Gutierrez-Docio, A.; Almodóvar, P.; Moreno-Fernández, S.; Silvan, J.M.; Martínez-Rodríguez, A.J.; Alonso, G.L.; Prodanov, M. Evaluation of an integrated ultrafiltration/solid phase extraction process for purification of oligomeric grape seed procyanidins. Membranes 2020, 10, 147. [Google Scholar] [CrossRef]
- Weseler, A.R.; Bast, A. Masquelier’s grape seed extract: From basic flavonoid research to a well-characterized food supplement with health benefits. Nutr. J. 2017, 16, 5. [Google Scholar] [CrossRef]
- Grases, F.; Prieto, R.M.; Fernández-Cabot, R.A.; Costa-Bauzá, A.; Sánchez, A.M.; Prodanov, M. Effect of consuming a grape seed supplement with abundant phenolic compounds on the oxidative status of healthy human volunteers. Nutr. J. 2015, 14, 94–101. [Google Scholar] [CrossRef]
- Wahyuningtyas, E.; Triwardhani, A.; Ardani, I.G.A.W.; Surboyo, M.D.C. The effect of grape seed extract on the alveolar; jaw; and skeletal bone remodeling: A scoping review. Eur. J. Dent. 2024, 18, 73–85. [Google Scholar] [CrossRef]
- Prodanov, M.; Vacas, V.; Hernández, T.; Estrella, I.; Amador, B.; Winterhalter, P. Chemical characterisation of Malvar grape seeds (Vitis vinifera L.) by ultrafiltration and RP-HPLC-PAD-MS. J. Food Compos. Anal. 2013, 31, 284–292. [Google Scholar] [CrossRef]
- Tang, G.Y.; Zhao, C.N.; Liu, Q.; Feng, X.L.; Xu, X.Y.; Cao, S.Y.; Meng, X.; Li, S.; Gan, R.Y.; Li, H.B. Potential of grape wastes as a natural source of bioactive compounds. Molecules 2018, 23, 2598. [Google Scholar] [CrossRef]
- Bedran-Russo, A.K.; Pauli, G.F.; Chen, S.N.; McAlpine, J.; Castellan, C.S.; Phansalkar, R.S.; Aguiar, T.R.; Vidal, C.M.P.; Napotilano, J.G.; Nam, J.W.; et al. Dentin biomodification: Strategies; renewable resources and clinical applications. Dent. Mater. 2014, 30, 62–76. [Google Scholar] [CrossRef]
- Montero, L.; Herrero, M.; Prodanov, M.; Ibañez, E.; Cifuentes, A. Characterization of grape seed procyanidins by comprehensive two-dimensional hydrophilic interaction x reversed phase liquid chromatography coupled to diode array detection and tandem mass spectrometry (HILICxRPLC-DAD-MS/MS). Anal. Bioanal. Chem. 2013, 405, 4627–4638. [Google Scholar] [CrossRef]
- Hayasaka, Y.; Waters, E.J.; Cheynier, V.; Herderich, M.J.; Vidal, S. Characterization of proanthocyanidins in grape seeds using electrospray mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Núñez, V.; Gómez-Cordovés, M.C.; Bartolomé, B.; Hong, Y.J.; Mitchell, A.E. Non-galloylated and galloylated proanthocyanidin oligomers in grape seeds from Vitis vinifera L. cv. Graciano, Tempranillo and Cabernet Sauvignon. J. Sci. Food Agric. 2006, 86, 915–921. [Google Scholar] [CrossRef]
- Muñoz-Labrador, A.; Prodanov, M.; Villamiel, M. Effects of high intensity ultrasound on disaggregation of a macromolecular procyanidin-rich fraction from Vitis vinifera L. seed extract and evaluation of its antioxidant activity. Ultrason. Sonochem. 2019, 50, 74–81. [Google Scholar] [CrossRef]
- Waterhouse, A.L.; Ignelzi, S.; Shirley, J.R. A comparison of methods for quantifying oligomeric proanthocyanidins from grape seed extracts. Am. J. Enol. Viticult. 2000, 51, 383–389. [Google Scholar] [CrossRef]
- Kuhnert, S.; Lehmann, L.; Winterhalter, P. Rapid characterisation of grape seed extracts by a novel HPLC method on a diol stationary phase. J. Funct. Foods 2015, 15, 225–232. [Google Scholar] [CrossRef]
- Kelm, M.A.; Johnson, J.C.; Robbins, R.J.; Hammerstone, J.F.; Schmitz, H.H. High performance liquid chromatography separation and purification of cacao (Theobroma cacao L.) procyanidins according to degree of polymerization using a diol stationary phase. J. Agric. Food Chem. 2006, 54, 1571–1576. [Google Scholar] [CrossRef]
- Prasain, J.K.; Peng, N.; Dai, Y.; Moore, R.; Arabshahi, A.; Wilson, L.; Barnes, S.; Michael Wyss, J.; Kim, H.; Watts, R.L. Liquid chromatography tandem mass spectrometry identification of proanthocyanidins in rat plasma after oral administration of grape seed extract. Phytomedicine 2009, 16, 233–243. [Google Scholar] [CrossRef]
- Dorenkott, M.; Griffin, L.; Goodrich, K.; Thompson-Witrick, K.; Fundaro, G.; Li, L.; Neilson, A. Oligomeric cocoa procyanidins possess enhanced bioactivity compared to monomeric and polymeric cocoa procyanidins for preventing the development of obesity, insulin resistance, and impaired glucose tolerance during highfat feeding. J. Agric. Food Chem. 2014, 62, 2216–2227. [Google Scholar] [CrossRef]
- Choy, Y.Y.; Waterhouse, A.L. Proanthocyanidin metabolism, a mini review. Nutr. Aging 2014, 2, 111–116. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Li, D.; Ho, C.; Li, J.; Wan, X. The absorption; distribution, metabolism and excretion of procyanidins. Food Funct. 2016, 7, 1273–1281. [Google Scholar] [CrossRef]
- Plumb, G.W.; de Pascual-Teresa, S.; Santos-Buelga, C.; Cheynier, V.; Williamson, G. Antioxidant properties of catechins and proanthocyanidins: Effect of polymerization, galloylation and glycosilation. Free Radic. Res. 1998, 29, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.S.; Belchior, G.P.; Ricardo-da-Silva, J.M.; Spranger, M.I. Isolation and purification of dimeric and trimeric procyanidins from grape seeds. J. Chromatogr. A 1999, 841, 115–121. [Google Scholar] [CrossRef]
- Sharma, V.; Zhang, C.; Pasinetti, G.M.; Dixon, R.A. Fractionation of grape seed proanthocyanidins for bioactivity assessment. In The Biological Activity of Phytochemicals, Recent Advances in Phytochemicals; Gang, D.R., Ed.; Springer: New York, NY, USA, 2011; pp. 33–46. [Google Scholar]
- Pasini, F.; Chinnici, F.; Caboni, M.F.; Verardo, V. Recovery of oligomeric proanthocyanidins and other phenolic compounds with established bioactivity from grape seed by-products. Molecules 2019, 24, 677. [Google Scholar] [CrossRef]
- Giori, A.; Anneli, A. Grape Seeds Extracts Obtainable by Fractioning on a Resin Patent WO 2007/0.7037 A1, 15 February 2007.
- Köhler, N.; Wray, V.; Winterhalter, P. Preparative isolation of procyanidins from grape seed extracts by high-speed counter-current chromatography. J. Chromatogr. A 2008, 1177, 114–125. [Google Scholar] [CrossRef]
- Phansalkar, R.S.; Nam, J.G.; Chen, S.N.; McAlpine, J.B.; Napotilano, J.G.; Leme, A.; Vidal, C.M.P.; Aguiar, T.; Bedran-Russo, A.K.; Pauli, G.F. A galloylated dimeric proanthocyanidin from grape seed exhibits dentin biomodification potential. Phytoterapia 2015, 101, 169–178. [Google Scholar] [CrossRef]
- Morales, D.; Soler-Rivas, C.; Ribeiro Smiderle, F.; Jimenez Piris, A.; Prodanov, M. Production of a β-D-glucan-rich extract from Shiitake mushrooms (Lentinula edodes) by an extraction/microfiltration/reverse osmosis pilot process. Innov. Food Sci. Emerg. Technol. 2018, 51, 80–90. [Google Scholar] [CrossRef]
- Guerrero-Hurtado, E.; Gutiérrez-Docio, A.; Fiedorowicz, R.; Mollá, E.; Reglero, G.; Prodanov, M. Why proanthocyanidins elute at increasing order of molecular masses when analysed by normal phase high performance liquid chromatography? Considerations of use. J. Chromatogr. A 2023, 1696, 463957. [Google Scholar] [CrossRef]
- Lewis, M.J. Ultrafiltration. In Separation Processes in the Food and Biotechnology Industries: Principles and Applications; Grandinson, A.S., Lewis, M.J., Eds.; Woodhaed Publishing Ltd.: Cambridge, UK, 1996; pp. 97–139. [Google Scholar]
- Lenntech. 2023. Available online: https://www.lenntech.es/calculadoras/tds/tdsyconductividad-electrica.htm (accessed on 25 January 2025).
- Spranger, I.; Sun, B.; Mateus, A.M.; de Freitas, V.; Ricardo-da-Silva, J.M. Chemical characterization and antioxidant activities of oligomeric and polymeric procyanidin fractions from grape seeds. Food Chem. 2008, 108, 519–532. [Google Scholar] [CrossRef]
- Pepper, D. RO for improved products in the food and chemical industries and water treatment. Desalination 1990, 77, 55–71. [Google Scholar] [CrossRef]
- Jiao, B.; Cassano, A.; Drioli, E. Recent advances on membrane processes for the concentration of fruit juices: A review. J. Food Eng. 2004, 63, 303–324. [Google Scholar] [CrossRef]
- Rodriguez, R.B.; Menezes, H.C.; Cabral, L.M.C.; Dornier, M.; Rios, G.M.; Reynes, M. Evaluation of reverse osmosis and osmotic evaporation to concentrate camu–camu juice (Myrciaria dubia). J. Food Eng. 2004, 63, 97–102. [Google Scholar] [CrossRef]


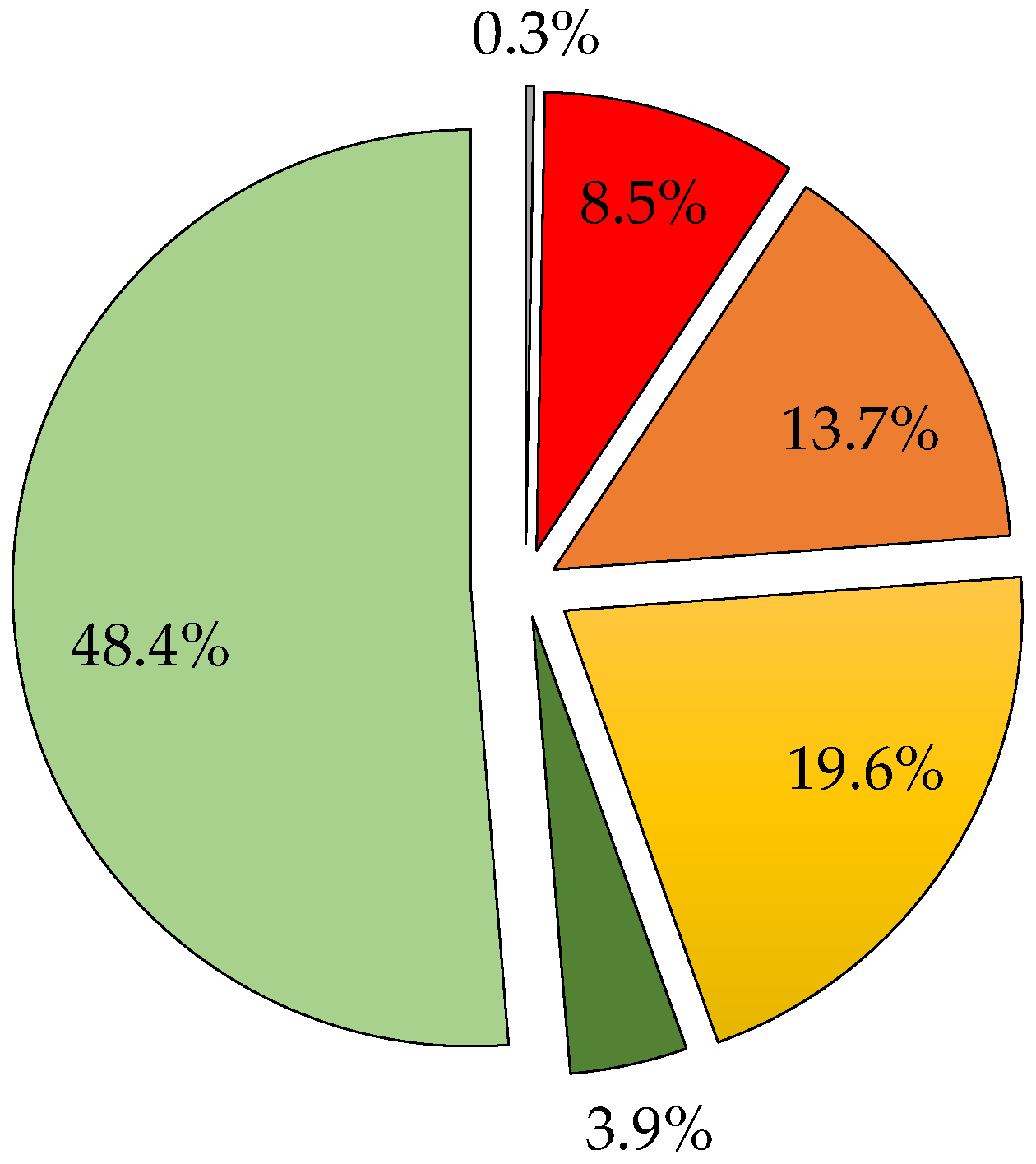

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GSEcla | VGSE | TDSGSEcla | ms,GSEcla | ms,FR>300 | Pro GSEcla | VPe300 | TDSPe300 | ms,Pe300 |
|---|---|---|---|---|---|---|---|---|
| (L) | (g/100 mL) | (g) | (g) | (%) | (L) | (g/100 mL) | (g) | |
| Rep 1 | 12 | 7.2 | 864 | 1.83 | 0.21 | 10.8 | 7.0 | 756 |
| Rep 2 | 12 | 6.4 | 768 | 2.57 | 0.33 | 10.6 | 6.2 | 657 |
| Diafiltration GSEFR>300kDa | Diafiltration Intensity (Number of Washings) | Electrical Conductivity (μS/cm) |
|---|---|---|
| Replicate 1 | 6 | 48 |
| Replicate 2 | 7 | 38 |
| Pe300 | VPe300 | TDSPe300 | ms,Pe300 | ms,FR300–30 | Pro GSEcla | VPe30 | TDSPe30 | ms,Pe30 |
|---|---|---|---|---|---|---|---|---|
| (L) | (g/100 mL) | (g) | (g) | (%) | (L) | (g/100 mL) | (g) | |
| Rep 1 | 10.8 | 7.0 | 756 | 71.5 | 8.5 | 10.0 | 6.8 | 680 |
| Rep 2 | 10.6 | 6.2 | 657 | 65.1 | 9.3 | 9.0 | 6.0 | 540 |
| Diafiltration GSEFR300–30 | Diafiltration Intensity (Number of Washings) | Electrical Conductivity (μS/cm) |
|---|---|---|
| Replicate 1 | 5 | 424 |
| Replicate 2 | 10 | 39 |
| Pe30 | VPem30 | TDSPe30 | ms,Pe30 | ms,FR30–5 | Pro GSEcla | VPe5 | TDSPe5 | ms,Pe5 |
|---|---|---|---|---|---|---|---|---|
| (L) | (g/100 mL) | (g) | (g) | (%) | (L) | (g/100 mL) | (g) | |
| Rep 1 | 10.0 | 6.8 | 680 | 135 | 15.6 | 8.7 | 6.0 | 522 |
| Rep 2 | 9.0 | 6.0 | 540 | 105 | 13.7 | 7.5 | 5.4 | 405 |
| Diafiltration GSEFR30–5 | Diafiltration Intensity (Number of Washings) | Electrical Conductivity (μS/cm) |
|---|---|---|
| Replicate 1 | 7 | 109 |
| Replicate 2 | 12 | 43 |
| Pe5 | VPe5 | TDSPe5 | Ms,Pe5 | ms,FR5–1 | Pro GSEcla | VPe1 | TDSPe1 | ms,Pe1 |
|---|---|---|---|---|---|---|---|---|
| (L) | (g/100 mL) | (g) | (g) | (%) | (L) | (g/100 mL) | (g) | |
| Sub-rep 1′ | 4.0 | 6.0 | 240 | 83.6 | 2.0 | 4.3 | 86 | |
| Sub-rep 1″ | 4.7 | 6.0 | 282 | 98.7 | 2.4 | 4.4 | 106 | |
| Rep 1 | 8.7 | 6.0 | 522 | 182.3 | 21.1 | 4.4 | 4.4 | 194 |
| Sub-rep 2′ | 4.0 | 5.4 | 216 | 80.1 | 2.0 | 3.9 | 78 | |
| Sub-rep 2″ | 3.5 | 5.4 | 189 | 58.1 | 1.8 | 4.0 | 72 | |
| Rep 2 | 7.5 | 5.4 | 405 | 138.2 | 18.0 | 3.8 | 3.9 | 150 |
| Diafiltration GSEFR5–1 | Diafiltration Intensity (Number of Washings) | Electrical Conductivity (μS/cm) |
|---|---|---|
| Replicate 1′ | 12 | 49.2 |
| Replicate 1″ | 12 | 50.1 |
| Replicate 2′ | 11 | 48.6 |
| Replicate 2″ | 11 | 47.2 |
| Input Matter | Ouput Matter | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pe1 | Sub-Fraction < 1, w | Sub-Fraction < 1, et | ||||||||||
| VPe1 | TDSPe1 | msPe1 | V<1,w | ms<1,w | Rec FR<1,w | Pro GSEcla | V<1,et | ms<1,et | Rec FR<1,et | Pro GSEcla | TRM | |
| (L) | (g/100 mL) | (g) | (L) | (g) | (%) | (%) | (L) | (g) | (%) | (%) | (%) | |
| Rep1 | 4.4 | 4.4 | 194 | 6.4 | 158 | 81.4 | 18.3 | 2.1 | 12.3 | 6.4 | 0.7 | 87.8 |
| Rep2 | 3.8 | 3.9 | 148 | 5.8 | 101 | 68.2 | 13.1 | 2.0 | 9.1 | 6.7 | 0.9 | 74.0 |
| DP | 5.2 | 3.8 | 198 | 7.4 | 139 | 70.2 | 17.0 | 2.0 | 11.1 | 6.0 | 1.3 | 76.2 |
| TMB | 13.1 | 4.0 | 542 | 20.3 | 398 | 73.3 | 48.4 | 6.1 | 32.5 | 6.2 | 3.9 | 79.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerrero-Hurtado, E.; Gutiérrez-Docio, A.; Fiedorowicz, R.; Prodanov, M. Fractionation of a Procyanidin-Rich Grape Seed Extract by a Preparative Integrated Ultrafiltration/Reverse Osmosis/Solid-Phase Extraction Procedure. Membranes 2025, 15, 92. https://doi.org/10.3390/membranes15030092
Guerrero-Hurtado E, Gutiérrez-Docio A, Fiedorowicz R, Prodanov M. Fractionation of a Procyanidin-Rich Grape Seed Extract by a Preparative Integrated Ultrafiltration/Reverse Osmosis/Solid-Phase Extraction Procedure. Membranes. 2025; 15(3):92. https://doi.org/10.3390/membranes15030092
Chicago/Turabian StyleGuerrero-Hurtado, Esperanza, Alba Gutiérrez-Docio, Rebeca Fiedorowicz, and Marin Prodanov. 2025. "Fractionation of a Procyanidin-Rich Grape Seed Extract by a Preparative Integrated Ultrafiltration/Reverse Osmosis/Solid-Phase Extraction Procedure" Membranes 15, no. 3: 92. https://doi.org/10.3390/membranes15030092
APA StyleGuerrero-Hurtado, E., Gutiérrez-Docio, A., Fiedorowicz, R., & Prodanov, M. (2025). Fractionation of a Procyanidin-Rich Grape Seed Extract by a Preparative Integrated Ultrafiltration/Reverse Osmosis/Solid-Phase Extraction Procedure. Membranes, 15(3), 92. https://doi.org/10.3390/membranes15030092