1. Introduction
Biological membranes are laterally heterogeneous (formation of domains with distinct lipid and protein compositions and hence unique physicochemical properties) and vertically asymmetric (different global and local compositions in the two bilayer leaflets). A longstanding question is to what extent domains present in one bilayer leaflet affect the physical properties of the membrane patch opposing that domain. In the plasma membrane, phosphatidylserine (PS) lipids are found exclusively in the inner leaflet of the lipid bilayer and its asymmetric distribution is crucial for many physiological functions [
1,
2]. Flippases maintain PS asymmetry by transporting the lipid from the outside to the inside of the cell [
3], while scramblases diminish the asymmetric PS distribution [
4,
5]. It has been proposed that transient loss of membrane asymmetry might have significant biological functions [
6,
7]. In terms of lateral heterogeneities (domain formation), phosphatidylserine has been shown to form nanodomains [
8] and is of major importance for the assembly and dynamics of caveolae [
9]. The London group recently showed that loss of membrane asymmetry might promote formation of detergent resistant membranes [
10].
Considering the many physiological functions of PS, it is important to understand the physiochemical underpinnings of PS function in asymmetric lipid bilayer systems. Giant unilamellar vesicles (GUVs) have been utilized for decades to investigate lipid properties and domain formation [
11] however, their use was limited to symmetric GUVs (sGUVs) where the two bilayer leaflets have the same lipid composition. This changed with the introduction of the β-cyclodextrin lipid exchange method by the London group [
12], which enabled the generation of asymmetric large unilamellar vesicles. In the following, a range of methods were introduced to fabricate large and giant unilamellar vesicles that exhibit different lipid compositions in the two bilayer leaflets [
13,
14,
15,
16]. Membrane mimics with an asymmetric distribution of PS have been obtained via enzymatic conversion [
17], resorting of lipids between leaflets [
18], droplet transfer method [
19], inkjet printing [
20] and lipid exchange with mβCD [
12,
21] (for a recent review of asymmetric lipid vesicle fabrication methods see Krompers and Heerklotz [
14]). However, several of these techniques furnished membrane mimics with an incomplete membrane asymmetry and potential follow up experiments are difficult to design.
The hemifusion method, developed by Enoki and Feigenson [
22], is generally a very elegant approach to obtain asymmetric GUVs. The method involves the exchange of the outer leaflet of sGUVs via calcium cation induced fusion with a supported lipid bilayer (SLB). The inner leaflet of the resulting aGUV exhibits the lipid composition of the sGUV used for the fusion, while the outer leaflet has the lipid composition of the SLB. This method has been successfully used to study lipid phase diagrams, domain formation and driving forces for lipid order [
23,
24,
25,
26]. At present, this technique has not been applied to lipid systems with anionic lipids.
The goal of this paper is to establish the hemifusion method for the fabrication of aGUVs with an anionic lipid composition in one leaflet of the lipid bilayer. Our experimental design results in aGUVs that exhibit PS in the outer leaflet so that interactions of peptides or proteins with the anionic lipid can be studied. In addition to adopting the Ca
2+ based hemifusion method developed by Enoki and Feigenson [
22], we expand the scope of the method to Mg
2+ as the hemifusion initiating cation. The reason is that Ca
2+ is known to cluster anionic lipids, including PS, and the use of Mg
2+ might avoid complications due to domain formation in the supported lipid bilayer that is being fused with the sGUV. We obtain aGUVs with a high degree of lipid asymmetry and propose a data filtering method that allows for the selection of aGUVs with complete exchange.
2. Materials and Methods
2.1. Materials
1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), sodium 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS), and ammonium 1-palmitoyl-2-(dipyrrometheneboron difluoride)undecanoyl-sn-glycero-3-phospho-L-serine (TF-PS) were purchased from Avanti Polar Lipids (Alabaster, AL, USA), while 1,1-dioctadecyl-3,3,3,3-tetramethylindodicarbocyanine 4-chlorobenzenesulfonate (DiD) was purchased from Invitrogen (Waltham, MA, USA). Each reagent was obtained in powder form and used as received. A DiD stock solution was prepared by dissolving DiD powder in ethanol, while lipid stock solutions were dissolved in a solution of 2:1 chloroform:methanol (by volume). All stock solution was stored at −20 °C. The concentration of the fluorophore (DiD and TF-PS) stock solutions was determined using the Beer–Lambert relationship, approximated from absorbance measurements in methanol at 646 nm and 496 nm, respectively, and the corresponding extinction coefficients (ε). The ε of DiD was obtained from the reagent lot’s certificate of analysis (ε = 250,000 M−1cm−1), while the ε of TF-PS was obtained from Avanti Polar Lipids (ε = 97,000 M−1cm−1).
Nunc Lab-Tech II 4-well chambered coverglasses were purchased from Thermo Fisher (Waltham, MA, USA). Commercial artist-grade tracing paper was purchased from Amazon (Seattle, WA, USA). Liquinox was obtained from Alconox (White Planes, NY, USA). Sodium chloride, potassium chloride, calcium chloride, magnesium chloride, disodium ethylenediamine tetraacetic acid (Na2EDTA), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), sucrose, and glucose were obtained in high purity grades either through Fisher Scientific (Fairlawn, NJ, USA) or Sigma Aldrich (St. Louis, MO, USA). HPLC grade chloroform, ethanol, isopropanol, and methanol were also obtained through Fisher Scientific (Fairlawn, NJ, USA). Deionized water (18.2 MΩ-cm) was obtained using a RODI (Aztec, NM, USA) high-purity water system.
2.2. sGUV Preparation
sGUVs were prepared using the PAPYRUS method with tracing paper as described by Pazzi et al. [
27]. In brief, a rectangular piece of tracing paper was first cleaned by submerging it in chloroform for 30 min, gently stirring the mixture every 10 min. This process was then repeated with a fresh volume of chloroform, after which it was repeated again with DI water. Next, the paper was dried in a vacuum oven. Once dry, a 6.35 mm diameter circular cutout was removed from the paper and coated with a 10 μL solution containing 0.5 to 1 mM lipids dissolved in 2:1 chloroform:methanol (by volume) at the same mole ratio as the desired sGUVs. After applying this mixture, the cutout was dried in a vacuum oven. Next, it was placed in the bottom of a microwell plate. If the sGUVs were to be prepared in low-salt buffer, 150 μL 200 mM sucrose, 5 mM HEPES, pH 7.4 buffer was added to the well, causing vesicles to bud from the dried lipid film on the paper and into solution. The resulting sGUVs were collected 2 h later. For sGUVs being prepared in normal (physiological) ionic strength buffer, 142.5 μL 105 mM sucrose was first added to the well containing the paper cutout, followed by 7.5 μL 100 mM sucrose, 100 mM NaCl, 50 mM KCl, 25 mM HEPES, pH 7.4 buffer 10 min later. The sGUVs were then collected after 110 min.
To collect sGUVs after their formation, a pipette with a wide orifice tip was used to mix the solution in the well by aspirating and dispensing 100 μL of it 6 times. The solution was then removed. If the sGUVs were prepared in the higher salt buffer, the osmolality of their collected solution was then measured with an Advanced Instruments model 3300 Micro Osmometer (Norwood, MA, USA) Any buffers later added to these sGUVs were adjusted to within ±2 mOsm/kg this reading using the same protocol as described for the low-salt buffers.
2.3. Coating of Cover Glasses
sGUV controls were imaged in chamber well plates with coverglass bottoms, which were coated with bovine serum albumin (BSA) to prevent the vesicles from sticking to the glass. To perform this coating on a chambered coverglass, it was first cleaned by rinsing its chambers with DI water three times, followed by ethanol two times, isopropanol once, then ethanol once. This rinsing was then repeated, ending with a final 3 rinses of DI water. Next, the coverglass was sonicated for 15 min in dilute Liquinox detergent (around 2% (w/v) in DI water) heated to 69 °C in a Branson 1510 water bath sonicator (Brookfield, CT). Once done, the coverglass was rinsed with DI water 12 times, followed by ethanol two times, isopropanol once, then ethanol once. This rinsing was then repeated with a final 12 rinses of DI water. The coverglass was dried under N2 gas and primed for BSA coating by filling each chamber with 0.1 M NaCl. After 40 min, 0.1 M NaCl was replaced with 1 mg/mL BSA in 0.1 M NaCl at pH = 5.0 to apply the BSA coating. After 3 or more hours, the coverglass was rinsed with DI water 12 times to remove excess BSA not bound to its surface.
2.4. Solid Supported Lipid Bilayer (SLB) Preparation
SLBs were fabricated in chambered coverglasses using the vesicle fusion method first described by Brian and McConnell [
28]. In this procedure, SUVs were first prepared by creating a 1 mM lipid mixture in 2:1 chloroform:methanol (by volume) with the lipids at the same mole ratios as the desired SLB. This solution was then dried down under N
2 and resuspended in a 500 mM NaCl, 20 mM citrate, pH = 4.0 buffer to form multilamellar vesicles. Next, the multilamellar vesicles were subjected to six freeze/thaw cycles to obtain SUVs. Then, the suspension was sonicated using a Sonics Vibra-Cell VCX130 Ultrasonic Processor tip sonicator (Newton, CT, USA), where an amplitude of 50% was applied to the vesicles in 15 s pulses (15 s on, 15 s off) for 30 min. The resulting SUVs were then diluted fivefold in the buffer described above.
Before using the SUVs to form the SLBs, an unused chambered coverglass was first cleaned using the same method as described for the BSA-coated coverglasses, but in this case, after drying the cleaned coverglass with N2, it was plasma-cleaned for 1 m 20 s using a Mercator Control Systems LF-5 Plasma System with O2 gas. Immediately after plasma cleaning, 1 mL of the diluted SUVs was added to each chamber to begin SLB formation. In this process, the SUVs in each chamber settle to the bottom and fuse to form a planar bilayer. The high ionic strength and acidity of the buffer helps to overcome electrostatic repulsions that would inhibit fusing.
After 40 or more minutes, the unfused vesicle remnants were washed out by submerging the coverglass in 2.5 L of DI water and using a syringe to gently rinse each chamber two times with 10 mL DI water. Next, the coverglass was removed from the water, and liquid was gently removed from the top of each chamber to return their volumes to 1 mL. Finally, the water in each chamber was exchanged for either a 65 mM NaCl, 35 mM KCl, 25 mM HEPES, pH 7.4 buffer or a 100 mM NaCl, 100 mM KCl, 25 mM HEPES, pH 7.4 buffer depending on if the SLB was being prepared in low or high-salt buffer respectively. To do so, 1 mL of the buffer was added to a chamber dropwise, then 1 mL was gently removed from a location in the chamber different than where the buffer was added. This process was repeated for each chamber 16–24 times, stopping once the chamber’s osmolality was equal to the buffer used to prepare the sGUVs within ±2 mOsm/kg, measured using The Advanced Micro Osmometer, Model 3300. SLBs were used immediately after being prepared.
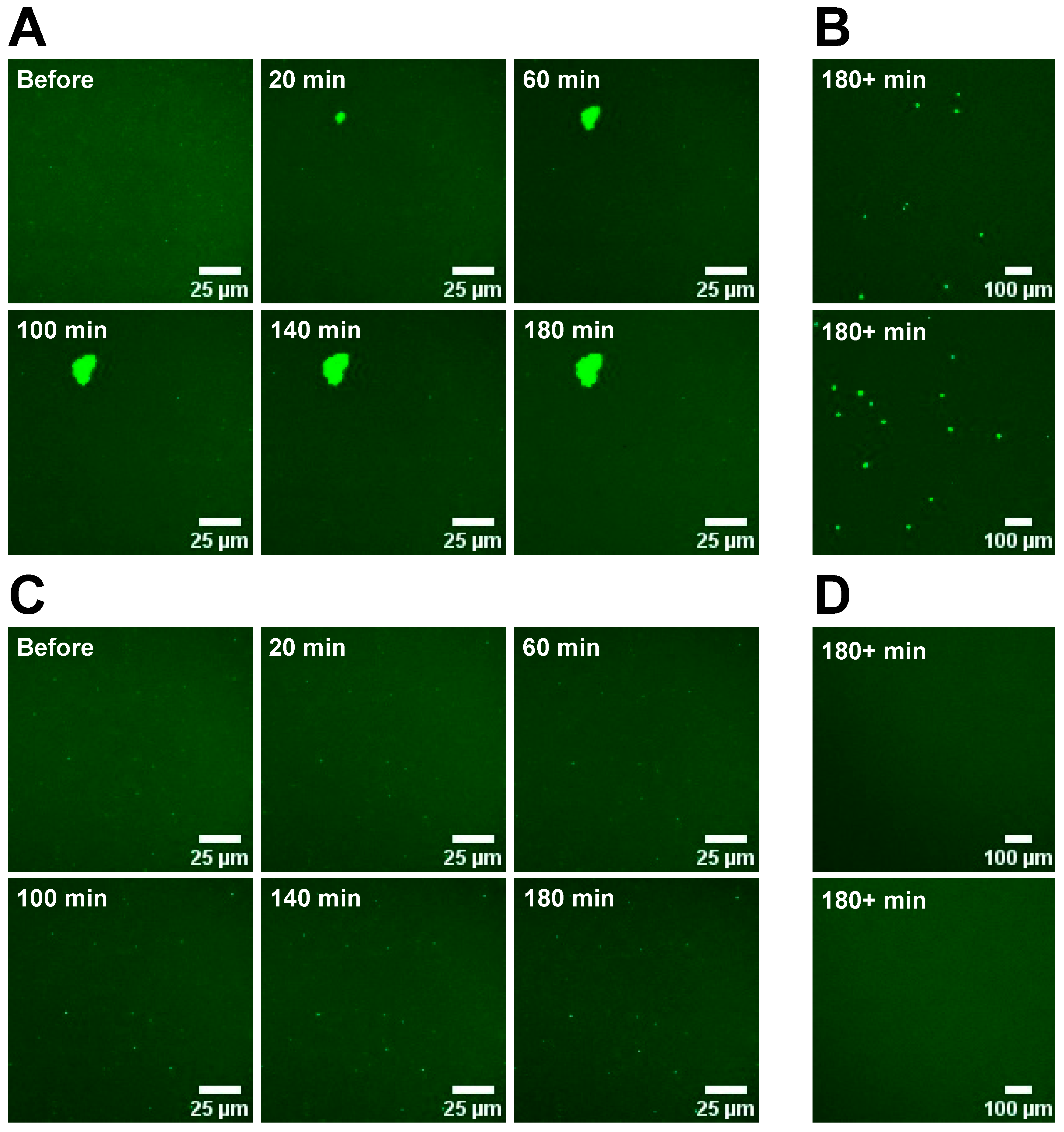
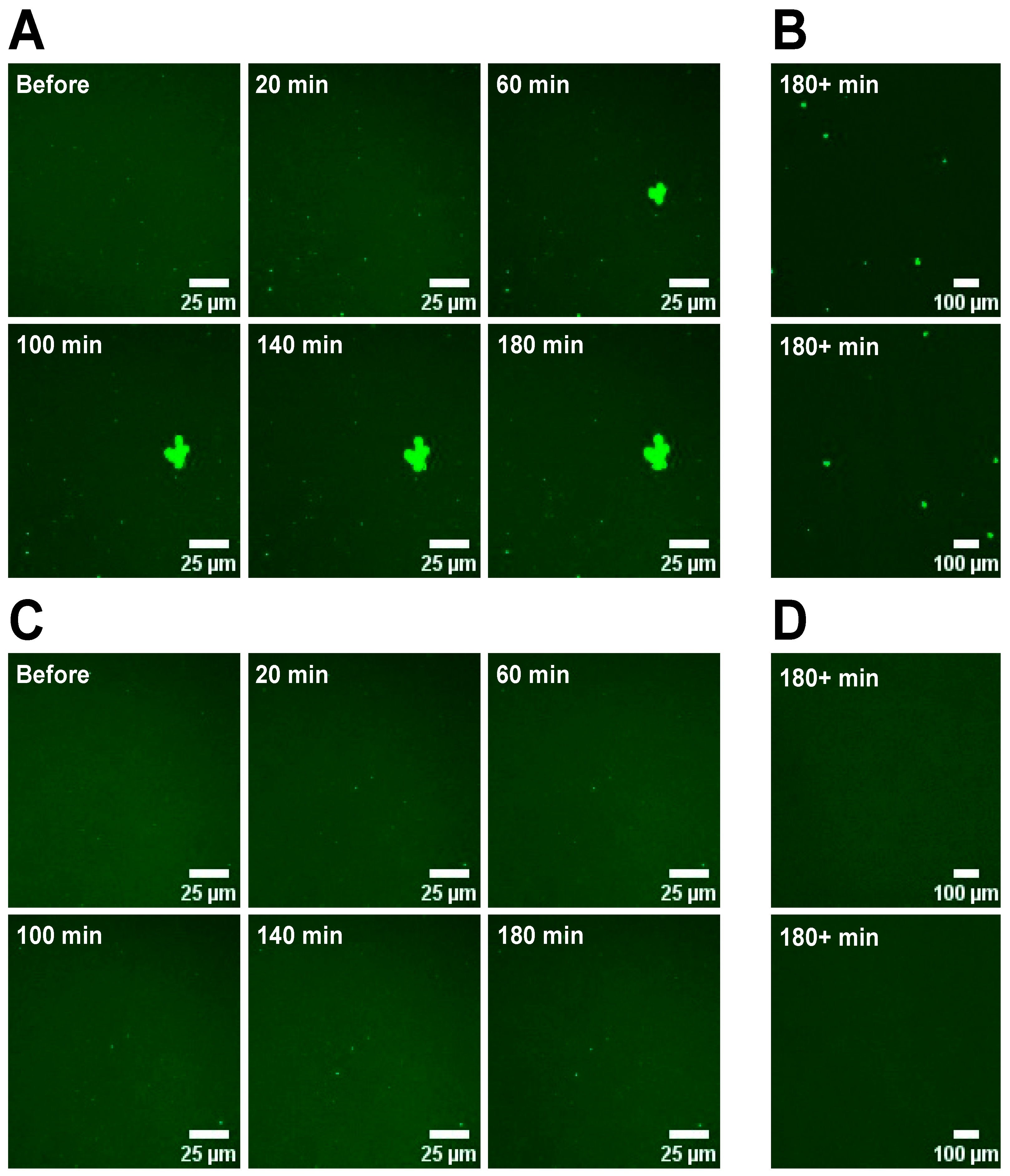
2.5. Monitoring Domain Formation in SLBs
SLBs were prepared in low or higher salt buffer with DOPC/POPS/TF-PS (69.9/30/0.1 mol%). These were then imaged before and after the introduction of 3 mM Ca2+ or Mg2+ to the aqueous phase by the addition of 400 μL 11 mM MCl2 (M = Ca2+ or Mg2+), 42 mM NaCl, 42 mM KCl, 25 mM HEPES, pH 7.4 buffer (“low-salt” buffer) or 400 μL 11 mM MCl2 (M = Ca2+ or Mg2+), 108 mM NaCl, 76 mM KCl, 25 mM HEPES, pH 7.4 buffer (“high-salt” buffer). 50 μL 200 mM sucrose, 5 mM HEPES, pH 7.4 buffer (if in low-salt buffer) or 100 mM sucrose, 100 mM NaCl, 50 mM KCl, 25 mM HEPES, pH 7.4 buffer (if in high-salt buffer) was added to the SLBs at the same time as the cations to mimic the conditions of hemifusion. For each SLB, images were taken every 20 min following the additions at the same locations on the SLB.
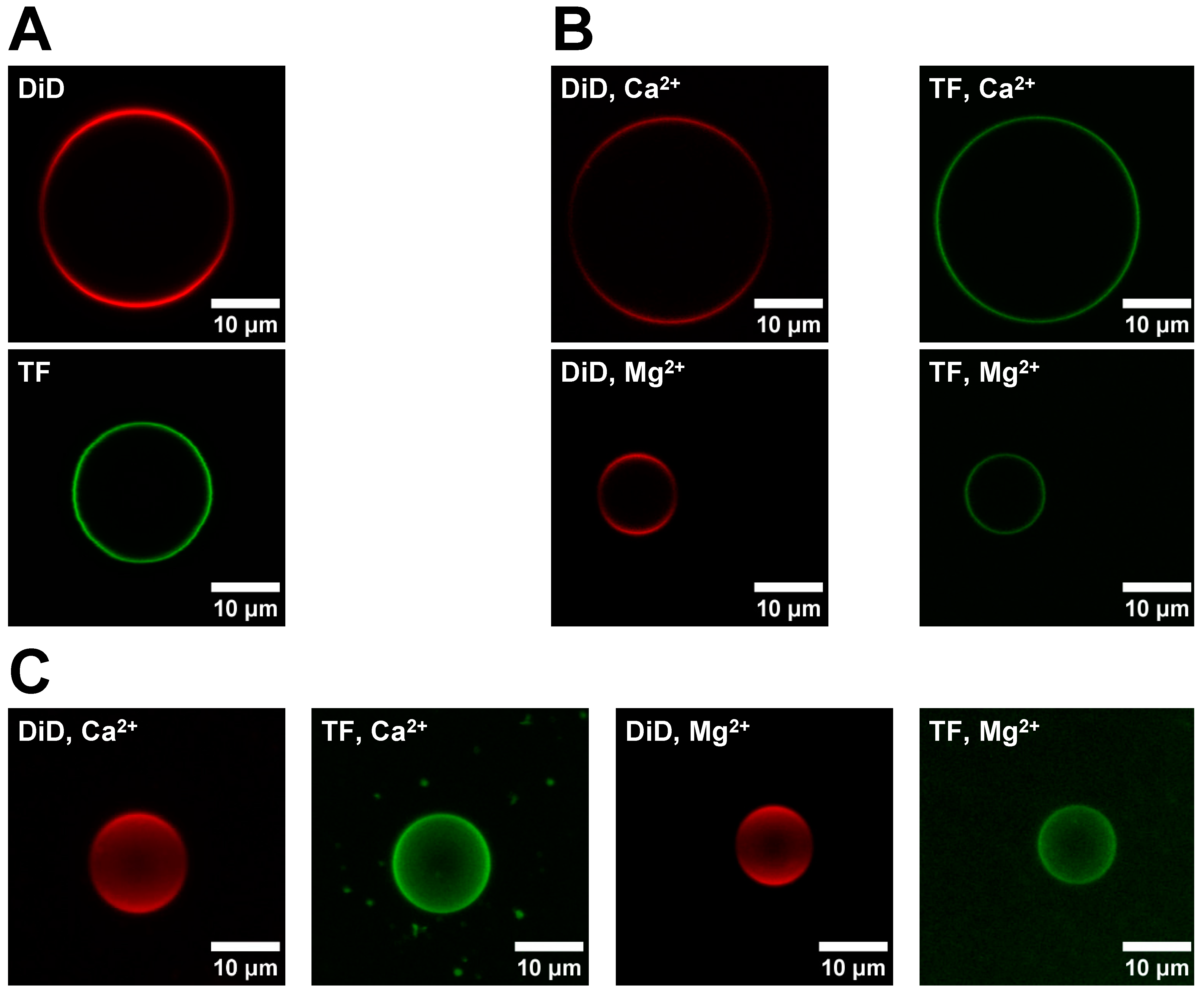
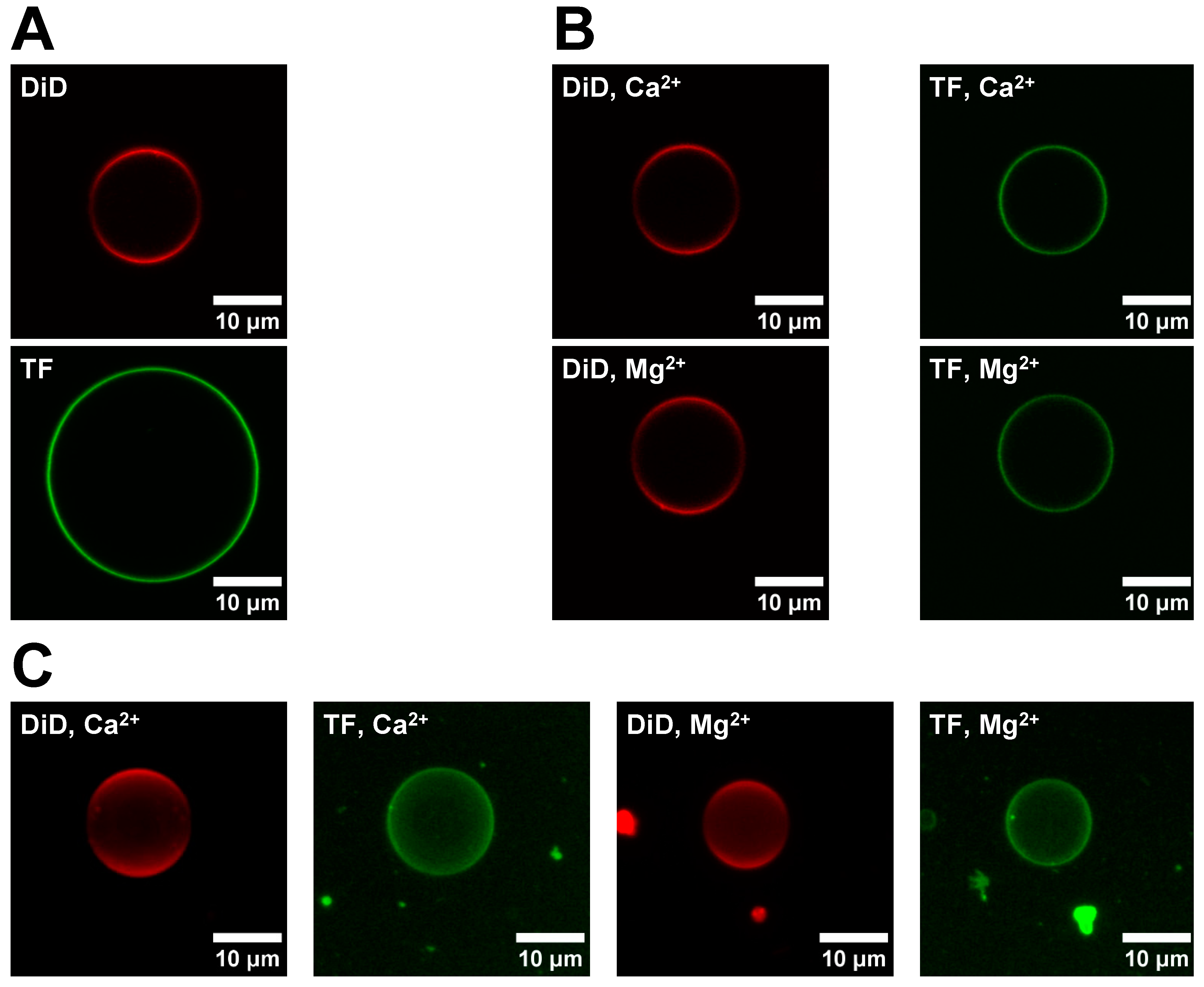
2.6. Asymmetric GUV (aGUV) Preparation
Asymmetric GUVs were prepared using an adapted form of the hemifusion method described by Enoki et al. [
22] In this procedure, 50 μL DOPC/DiD (99.9/0.1 mol%) sGUVs in low or high-salt buffer was added to a chamber containing a DOPC/POPS/TF-PS (69.9/30/0.1 mol%) SLB in low or high-salt buffer, respectively. After waiting 10 min for the sGUVs to settle to the bottom of the chamber, hemifusion between the sGUVs and SLB was induced by adding 5.5 mM Ca
2+ or Mg
2+ to the chamber via the addition of 400 μL 20 mM MCl
2 (M = Ca
2+ or Mg
2+), 35 mM NaCl, 35 mM KCl, 25 mM HEPES, pH 7.4 (if in low-salt buffer) or buffer 20 mM MCl
2 (M = Ca
2+ or Mg
2+), 100 mM NaCl, 70 mM KCl, 25 mM HEPES, pH 7.4 (if in high-salt buffer). After waiting 23 min for hemifusion and lipid exchange to occur between the membranes, fission of the GUVs from the SLB was induced by adding 6 mM EDTA to the chamber via the addition of 600 μL 20 mM Na
2EDTA, 35 mM NaCl, 35 mM KCl, 25 mM HEPES, pH 7.4 buffer (for low-salt buffer) or 20 mM Na
2EDTA, 100 mM NaCl, 76 mM KCl, 25 mM HEPES, pH 7.4 buffer (for high-salt buffer). After waiting 10 min for fission to complete, the resulting aGUVs were harvested.
2.7. Fluorescence Imaging
All GUVs and SLBs were imaged using a Zeiss LSM 510 confocal microscope (Oberkochen, Germany). SLBs and aGUVs were imaged in the coverglass wellplate they were prepared in, while sGUVs were imaged in BSA-coated coverglasses by adding 50 μL of the sGUVs to the chamber, followed by 550 μL 200 mM glucose, 5 mM HEPES, pH 7.4 buffer or to 550 μL 100 mM glucose, 100 mM NaCl, 50 mM KCl, 25 mM HEPES, pH 7.4 buffer if they were prepared in low or high-salt buffer, respectively. Adding the glucose buffer after adding the GUVs helps the vesicles settle on the bottom and provides a better imaging contrast. When imaging vesicles, only those with no deformities (e.g., multilamellarity, tubules, non-spherical morphology) were imaged. All GUV images were obtained by using a plan-apochromat 63× oil immersion objective, while SLB images were taken either with this lens, or a plan-apochromat 10× air objective. Unless otherwise specified, all GUV images were taken by capturing a single frame at the equator of the vesicle, and later processed by applying a Gaussian blur of radius 1. Meanwhile, all 63× lens images of SLBs were taken as Z-stacks, with the Z-slices spanning from right above to right below the membrane. All Z-stacks in this study were converted into composite 2D images, where each pixel in the image has the average intensity of all the Z-slices at that pixel. Certain Z-stacks of SLB domains were taken at a low detector gain, and later, digitally brightened to avoid image artifacts. All 10× lens images of SLBs were single frames that were digitally brightened as well.
2.8. Analysis of the Fluorescence Intensities
Using the analysis method developed by Enoki et al. [
22], the fluorescence intensities of aGUVs were compared to those of sGUVs to evaluate the amount of lipid exchange each aGUV underwent during hemifusion. This analysis first involved deriving an intensity value for the DiD and TF-PS emissions of each aGUV. To do so, ImageJ macros generously provided by Thais Enoki and Gerald Feigenson (personal communication; used in [
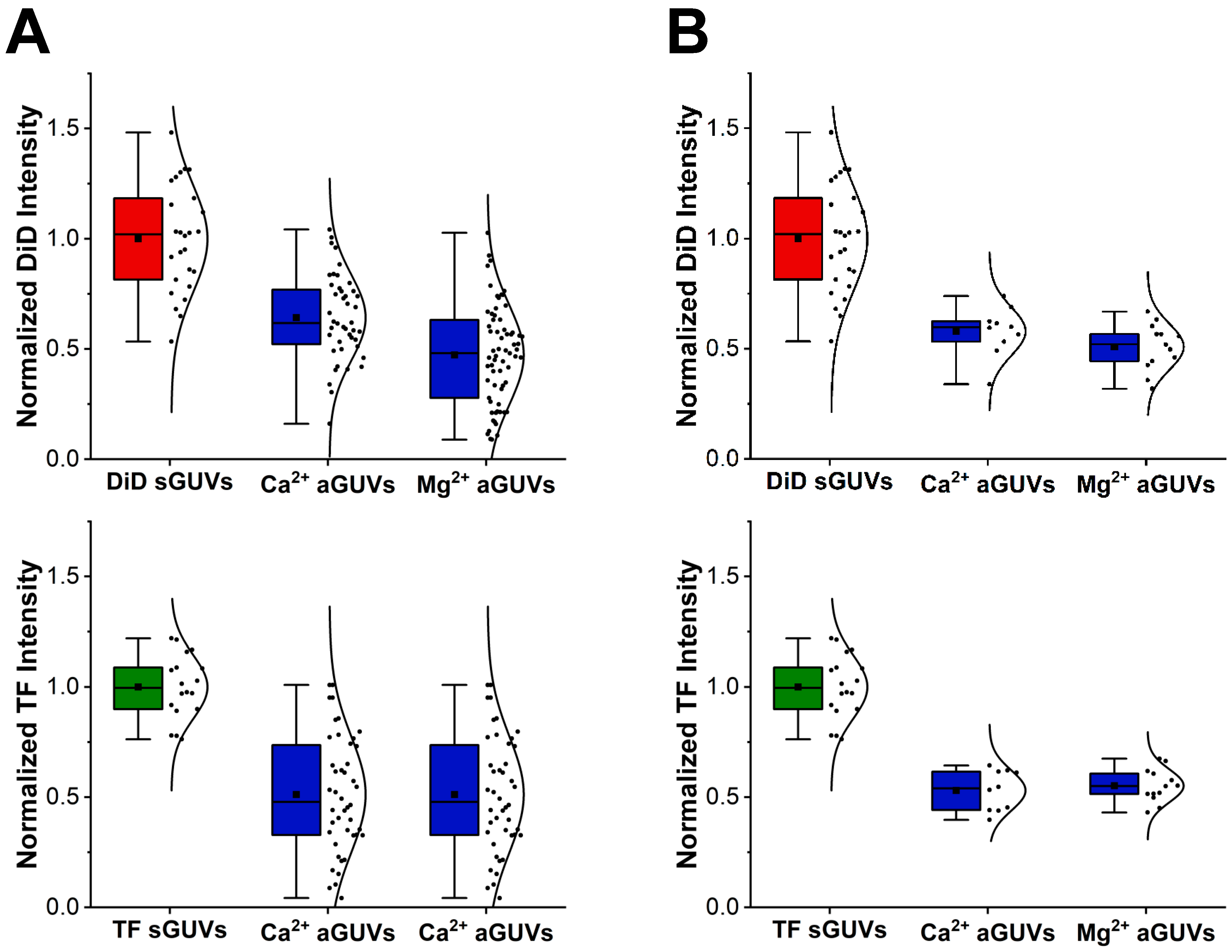
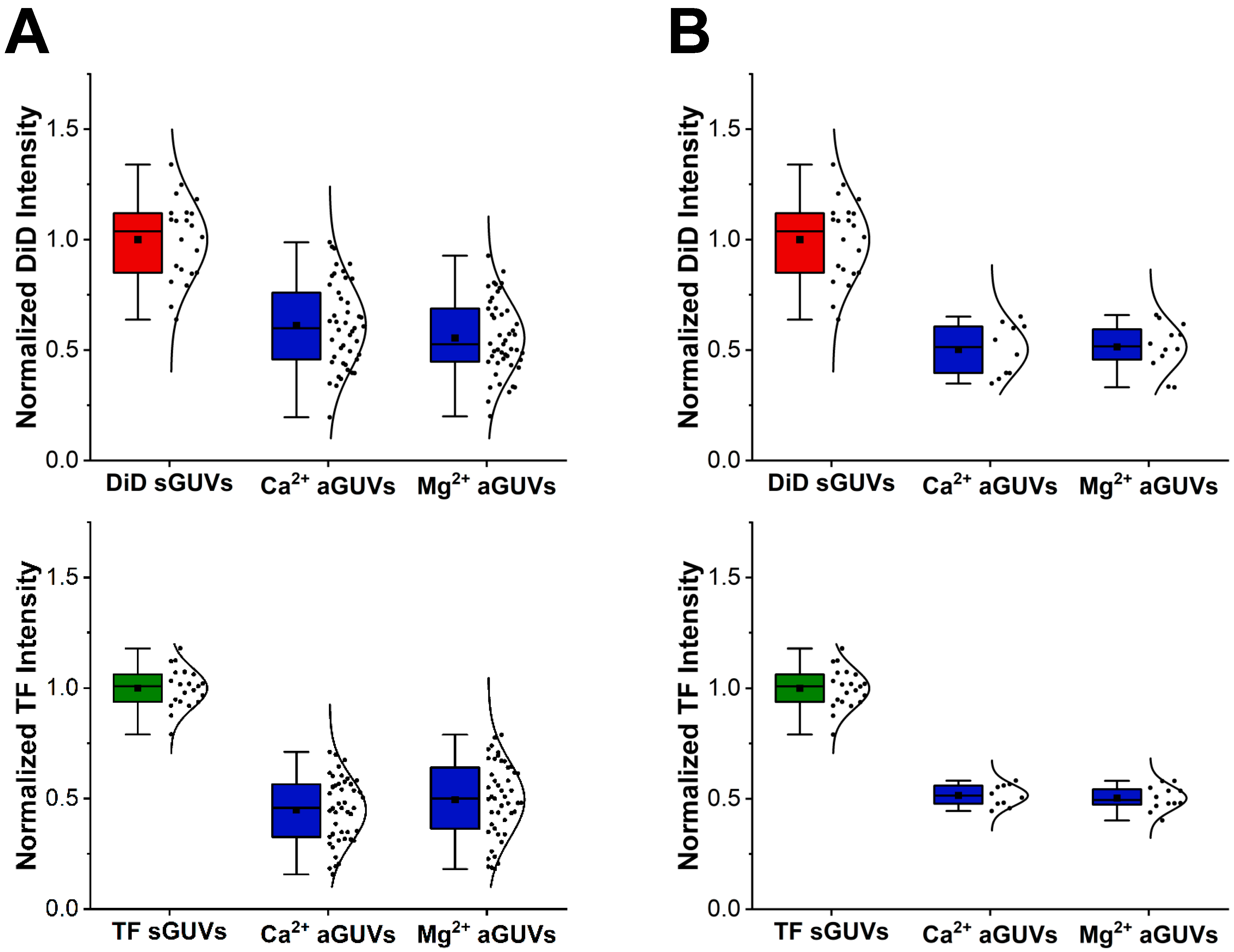
22]) were used to find the maximum DiD and TF-PS emission intensity along a ray going from the center of the vesicle outwards. This measurement was repeated for 359 more rays, with each angled in a different manner such to capture all areas of the vesicle. The maximum intensities captured by each ray were then averaged together to get an average maximum DiD intensity and average maximum TF-PS intensity for the vesicle. Going forward, these values will be referred to as “DiD intensity” and “TF intensity” respectively. In addition to being found for each aGUV, the DiD or TF intensity was also determined for sGUVs with a composition matching the theoretical inner or outer leaflet composition of the aGUVs.
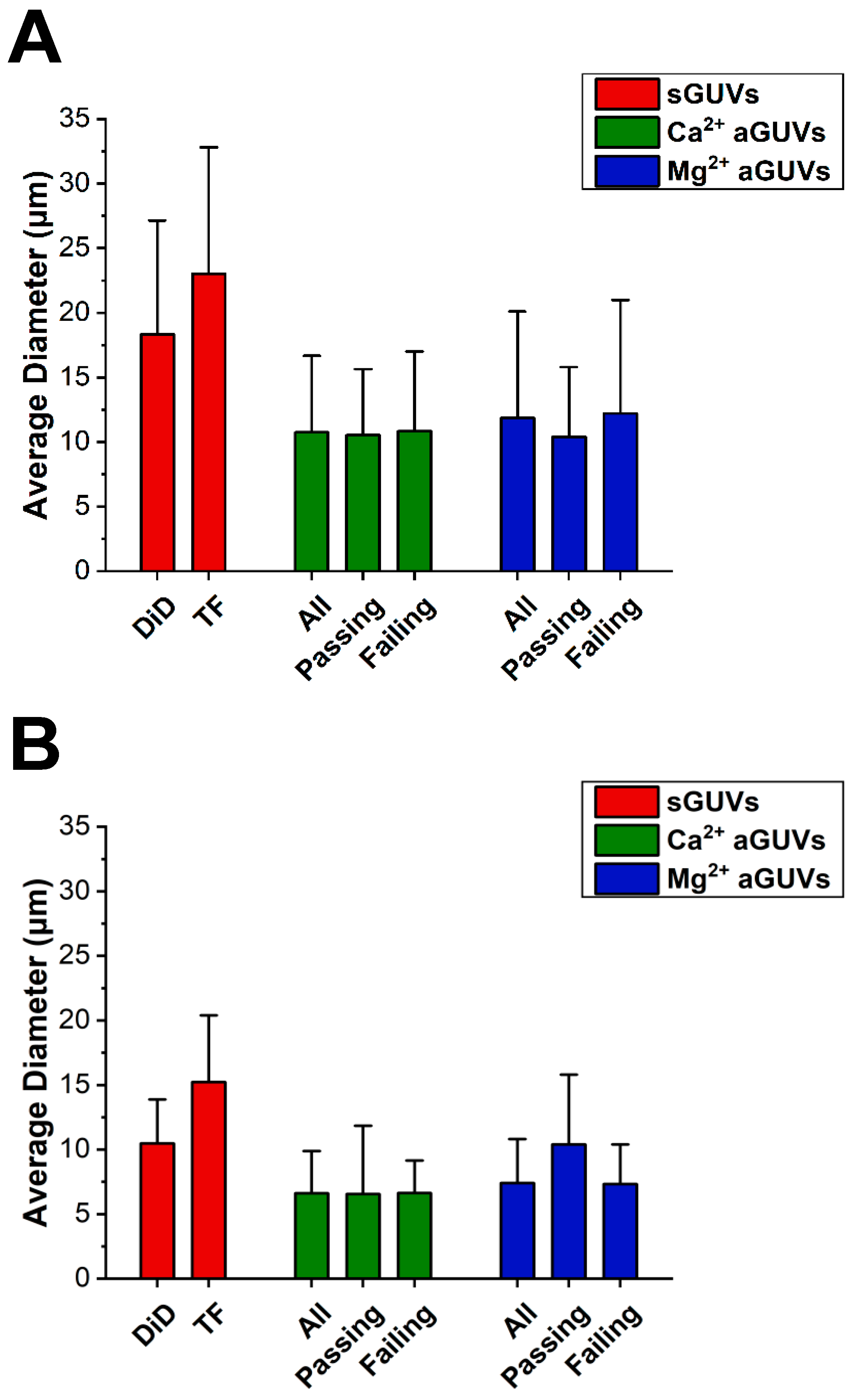
Before performing any further analysis with each vesicle’s DiD or TF intensity, outliers were removed from the data set. An outlier was defined as a vesicle that had a DiD or TF intensity outside the range of their corresponding average DiD or TF intensity ± double the standard deviation. This average and standard deviation were taken from the vesicle’s sample, meaning all data for vesicles of that type, depending on composition (i.e., aGUV, sGUV with DiD, or sGUV with TF-PS) and buffer composition (i.e., low or high-salt buffer). An aGUV found to be an outlier for one intensity (DiD or TF) value but not the other was considered an outlier, and both intensities were completely discarded. As with this outlier removal, all future analyses were performed separately for vesicles in low and high-salt buffers, meaning intensities from vesicles in low-salt buffer were never compared to those from vesicles in high-salt buffer and vice versa.
A vesicle’s DiD or TF intensity essentially represents the average emission intensity of DiD or TF-PS across it, which is chiefly dependent on the fraction of DiD or TF-PS in the vesicle. aGUVs that experienced full outer leaflet exchange with the SLB should have half the fraction of DiD and TF-PS as their sGUV counterparts, meaning their DiD and TF intensities should be half that of the sGUVs. This reasoning was used to determine which aGUVs experienced complete outer leaflet exchange. In this analysis, an aGUV was considered to have undergone this complete exchange if both of the following were true:
Its DiD intensity fell within the range of the halved maximum and halved minimum DiD intensity of the corresponding sGUVs.
Its TF intensity fell within the range of the halved maximum and halved minimum TF intensity of the corresponding sGUVs.
When both of these conditions were met, it means the aGUV gave a DiD and TF intensity that was in line with what is expected from a population of vesicles with half the DiD and TF-PS content of the sGUVs. An aGUV that met these criteria is referred to as a “range-passing” aGUV, while those that did not are “range-failing”.
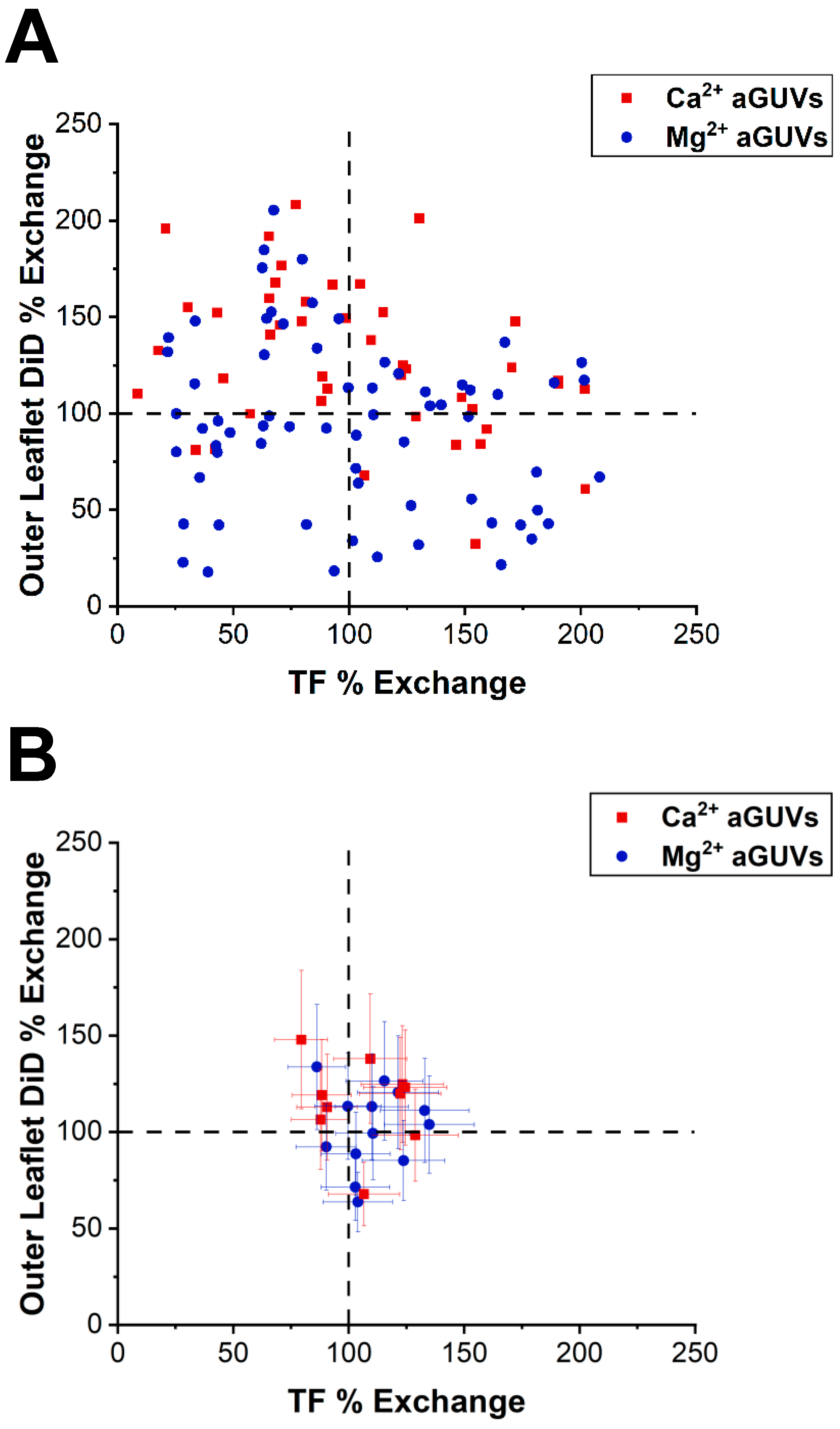
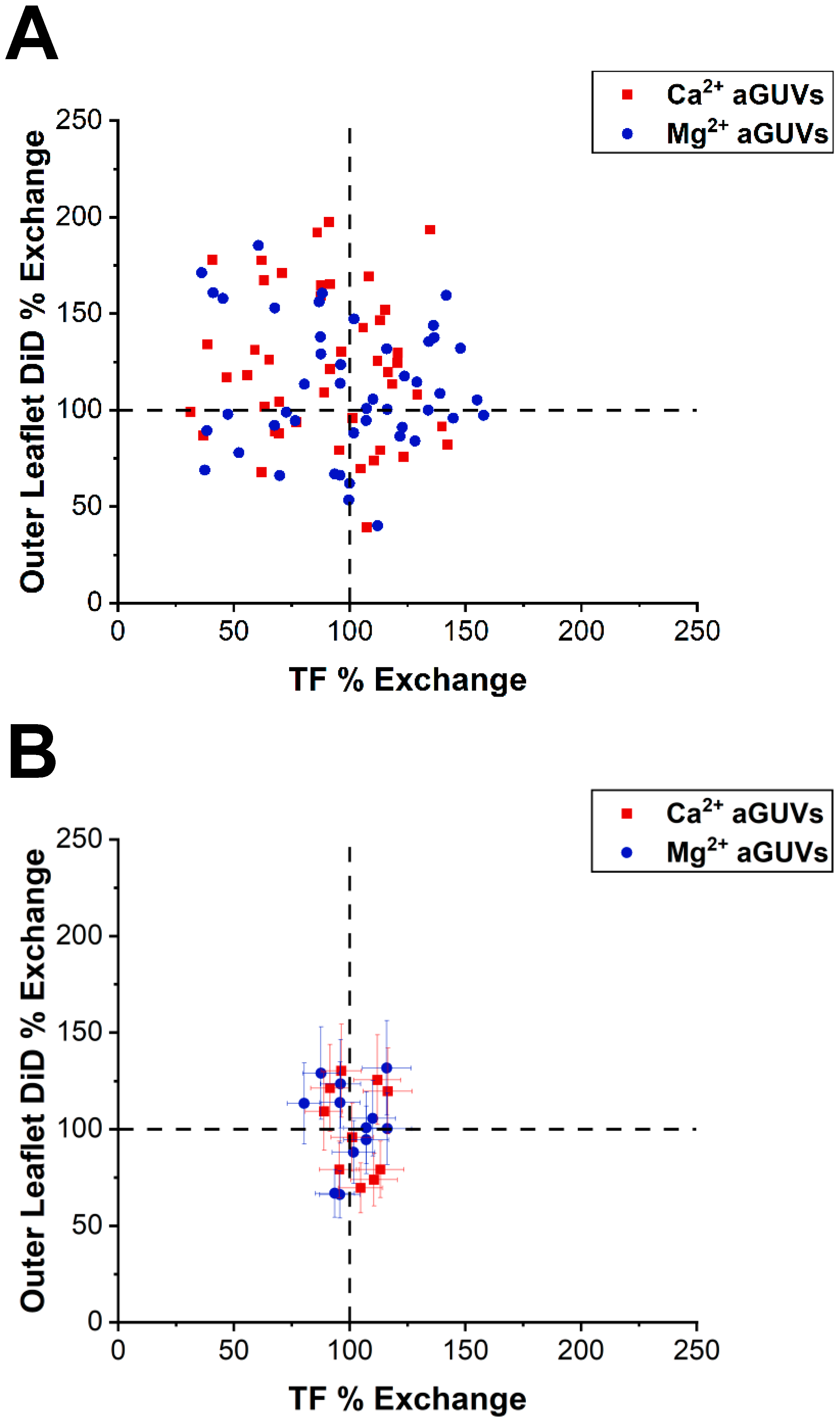
The DiD and TF intensities of the aGUVs were also used to find each vesicle’s outer leaflet DiD % exchange and TF % exchange. These values equal the aGUV’s DiD or TF intensity (I
a) divided by the halved average DiD or TF intensity of the corresponding sGUVs (I
c), expressed as a percentage:
An aGUV having close to a 100% outer leaflet DiD % exchange and TF % exchange indicates that its DiD and TF intensities closely matched the halved average intensities for the sGUVs. This situation suggests that the aGUV underwent a mostly complete outer leaflet exchange.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}