


Development of a New Affinity Gold Polymer Membrane with Immobilized Protein A

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental
2.2.1. Functionalization of the Membranes
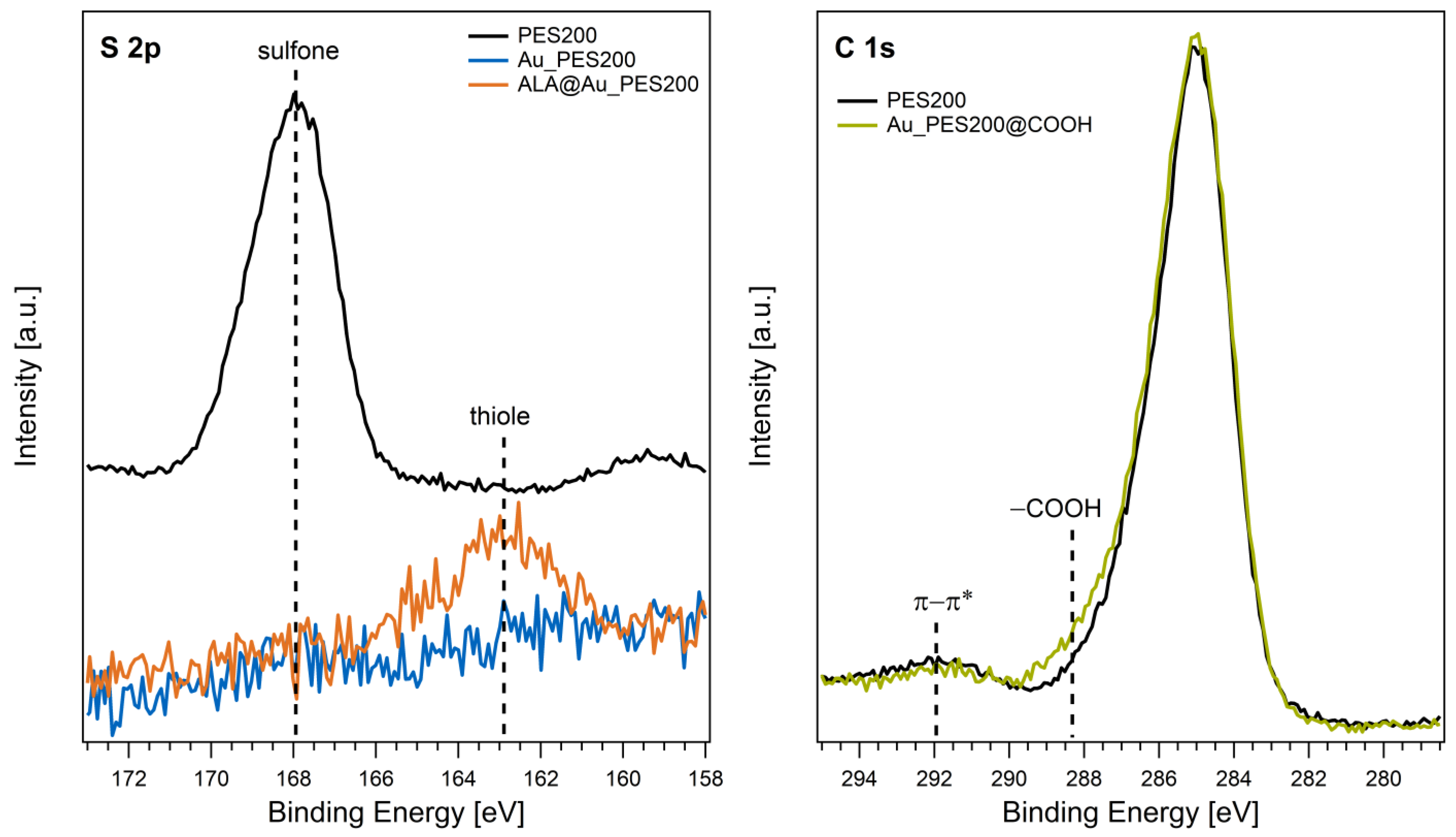
- Functionalization of the gold surface (see Figure 1A) via alphalipoic acid (ALA): Alphalipoic acid undergoes a process of self-assembled monolayer formation [24] due to a thiol-gold bond. Therefore, any gold surface can be carboxylated. For the functionalization, a 2% (w/v) solution of alphalipoic acid in ethanol was prepared. The respective membrane pieces were then incubated at 22 °C for 24 h at 500 rpm in a tabletop shaker. After incubation, the membranes were washed three times with ethanol and dried with compressed air.
- Functionalization of carboxylated PES membranes (see Figure 1B): The gold-sputtered PES membranes were carboxylated via an electron beam (e beam) process.
2.2.2. Analysis of Functionalization via XPS
2.2.3. Protein Immobilization via NHS-EDC
- (I)
- For the immobilization, the membranes were washed four times with an activation buffer (50 mM of 2-(N-morpholino)ethanesulfonic acid (MES) buffer at pH 6.0), following an immobilization protocol outlined by Merck Millipore [26]. After washing, 24 µL of an aqueous 200 mM 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide solution (EDC) was supplemented per mL of activation buffer. Immediately after the EDC-solution, 240 µL per mL of activation buffer of a 200 mM N-hydroxysulfosuccinimide (sulfo-NHS) solution (dissolved in activation buffer) was added. The mixture was incubated for 30 min at room temperature at 600 rpm. After incubation, the supernatant was removed, and the membranes washed three times with the activation buffer. The activated membranes were then immersed in a 1 mg mL−1 Protein A in the activation buffer solution and incubated at 25 °C for 2.5 h at 600 rpm. After incubation, 50 µL ethanolamine were added and incubated for 30 min to stop the reaction. The supernatant was removed, and the membranes were immersed in a 50 mM Tris(hydroxymethyl)aminomethane (Tris) 0.5% (w/v) casein blocking buffer at pH 8.0 and incubated at room temperature overnight at 600 rpm. The supernatant was removed, and the membranes washed twice with blocking buffer.
- (II)
- During a second immobilization approach following a protocol from G-Bioscience [27], the membranes were immersed in a 0.1 M MES buffer at pH 4.6 and supplemented with an aqueous solution of 0.4 mg mL−1 EDC and 0.6 mg sulfo-NHS. The mixture was incubated for 15 min at 600 rpm at room temperature. To stop the reaction, 1.5 µL of 3-mercaptoethanol was added before immersing the membranes in a 1 mg mL−1 Protein A in 50 mM NaH2PO4, 150 mM NaCl solution at pH 7.4. The immersed membranes were incubated for 2 h in the solution before quenching the reaction with 0.5 M hydroxylamine.
2.2.4. Fluorescence Labeling
2.2.5. Static Experiments
2.2.6. Dynamic Experiments
3. Results and Discussion
3.1. Qualitative Functionalization Analysis
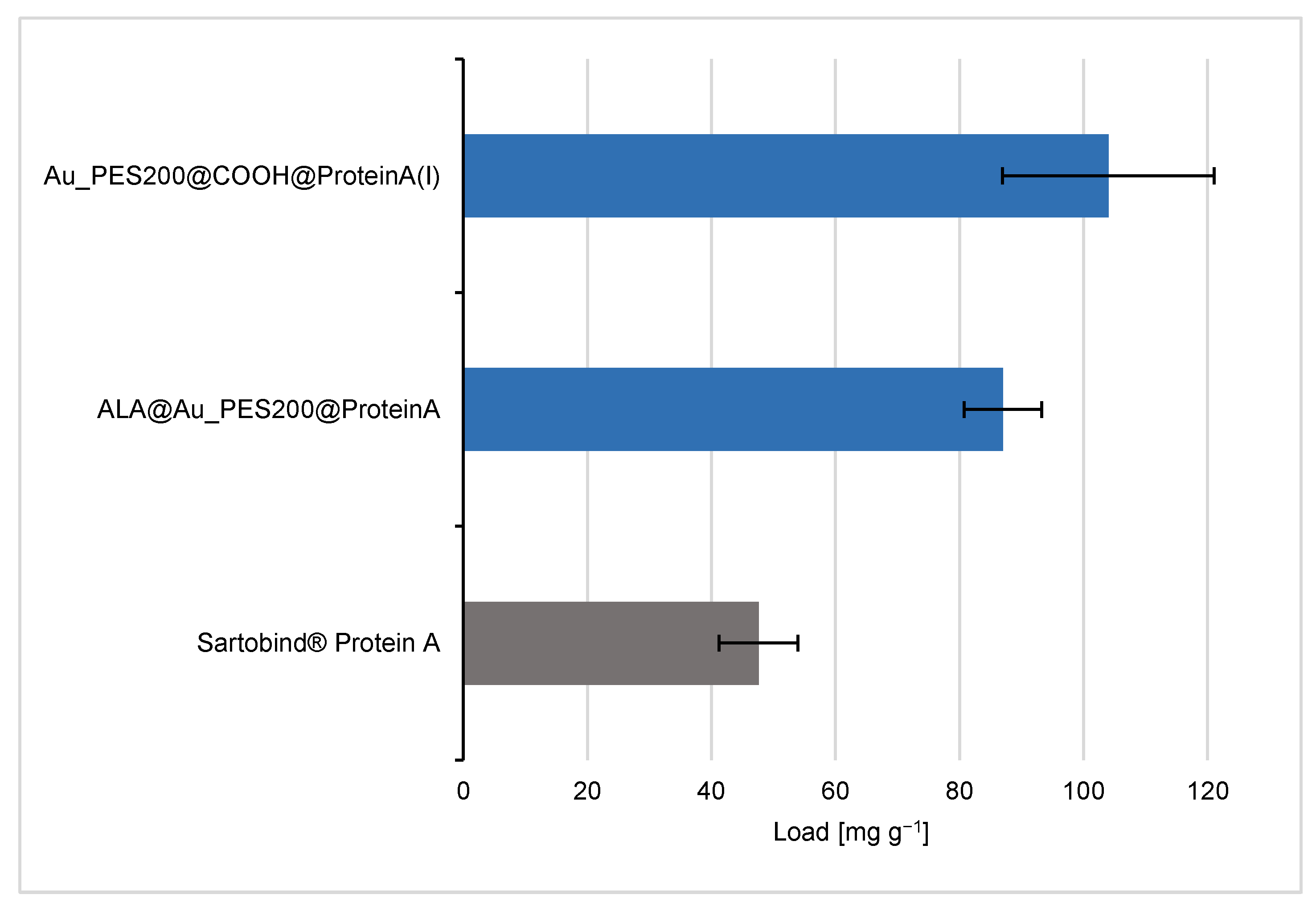
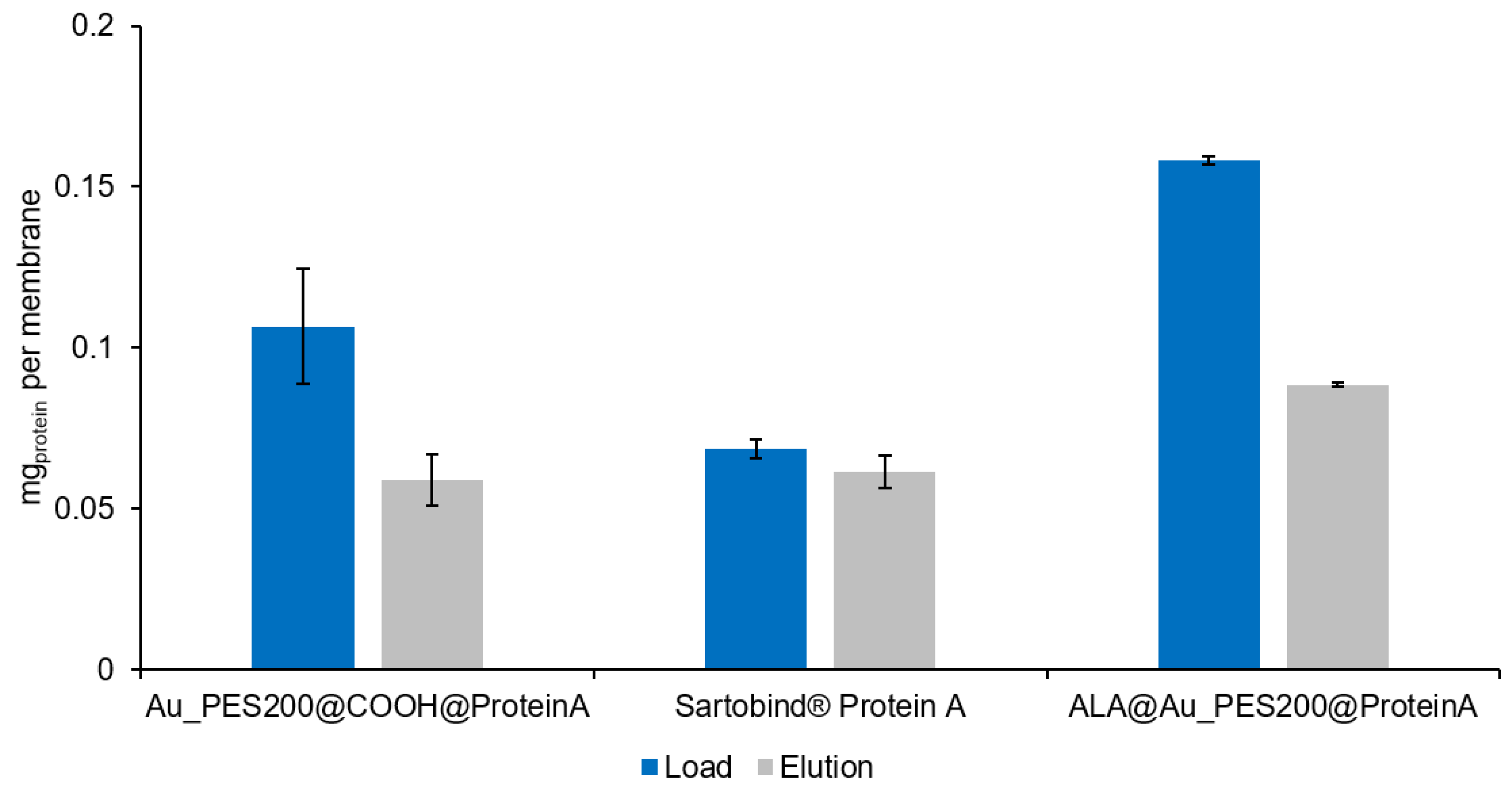
3.2. Evaluation of the Binding and Elution Efficiencies of Ligand-Immobilized Membranes
3.3. Dynamic Processing of New Membranes with Purified Trastuzumab
3.4. Stability of the Functionalized Membranes (Reusability and Storage)
3.5. Application to a Real Process: Antibody Purification from Fresh Frozen Human Blood Plasma
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghosh, R. Protein separation using membrane chromatography: Opportunities and challenges. J. Chromatogr. A 2002, 952, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Gerstner, J.A.; Hamilton, R.; Cramer, S.M. Membrane chromatographic systems for high-throughput protein separations. J. Chromatogr. A 1992, 596, 173–180. [Google Scholar] [CrossRef]
- Lalli, E.; Silva, J.S.; Boi, C.; Sarti, G. Affinity Membranes and Monoliths for Protein Purification. Membranes 2019, 10, 1. [Google Scholar] [CrossRef]
- Brandt, S.; Goffe, R.A.; Kessler, S.B.; O’Connor, J.; Zale, S.E. Membrane-Based Affinity Technology for Commercial Scale Purifications. Nat. Biotechnol. 1988, 6, 779–782. [Google Scholar] [CrossRef]
- Orr, V.; Zhong, L.; Moo-Young, M.; Chou, C. Recent advances in bioprocessing application of membrane chromatography. Biotechnol. Adv. 2013, 31, 450–465. [Google Scholar] [CrossRef] [PubMed]
- Ramos-de-la-Peña, A.M.; González-Valdez, J.; Aguilar, O. Protein A chromatography: Challenges and progress in the purification of monoclonal antibodies. J. Sep. Sci. 2019, 42, 1816–1827. [Google Scholar] [CrossRef] [PubMed]
- Boi, C.; Dimartino, S.; Sarti, G.C. Performance of a new protein A affinity membrane for the primary recovery of antibodies. Biotechnol. Prog. 2008, 24, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Özbek, M.A.; Çimen, D.; Bereli, N.; Denizli, A. Metal-chelated polyamide hollow fiber membranes for ovalbumin purification from egg white. J. Chromatogr. B 2022, 1203, 123293. [Google Scholar] [CrossRef]
- Xu, Z.; Wan, L.; Huang, X. (Eds.) Surface Engineering of Polymer Membranes: Functionalization Methods for Membrane Surfaces; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Mahmoudifard, M.; Soudi, S.; Soleimani, M.; Hosseinzadeh, S.; Esmaeili, E.; Vossoughi, M. Efficient protein immobilization on polyethersolfone electrospun nanofibrous membrane via covalent binding for biosensing applications. Mater. Sci. Eng. C 2016, 58, 586–594. [Google Scholar] [CrossRef]
- Shaimi, R.; Low, S.C. Prolonged protein immobilization of biosensor by chemically cross-linked glutaraldehyde on mixed cellulose membrane. J. Polym. Eng. 2016, 36, 655–661. [Google Scholar] [CrossRef]
- Berensmeier, S.; Rauwolf, S.; Zanker, A.; Steegmüller, T.; Schwaminger, P. One Multifunctional Affinity Peptide Tag for Different Non-Functionalized Materials and Applications. Chem. Ing. Tech. 2022, 94, 1253. [Google Scholar] [CrossRef]
- Rauwolf, S.; Steegmüller, T.; Schwaminger, S.P.; Berensmeier, S. Purification of a peptide tagged protein via an affinity chromatographic process with underivatized silica. Eng. Life Sci. 2021, 21, 549–557. [Google Scholar] [CrossRef] [PubMed]
- von Roman, M.F.; Berensmeier, S. Improving the binding capacities of protein A chromatographic materials by means of ligand polymerization. J. Chromatogr. A 2014, 1347, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Kołodziejczak-Radzimska, A.; Jesionowski, T. Engineering of Immobilized Enzymes: pH, Thermal Stability and Kinetic Aspects. In Practical Aspects of Chemical Engineering; Ochowiak, M., Woziwodzki, S., Mitkowski, P.T., Doligalski, M., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 161–170. [Google Scholar]
- Cheryan, M. Ultrafiltration and Microfiltration Handbook; CRC Press: Boca Raton, FL, USA, 1998. [Google Scholar]
- Zhao, C.; Xue, J.; Ran, F.; Sun, S. Modification of polyethersulfone membranes—A review of methods. Prog. Mater. Sci. 2013, 58, 76–150. [Google Scholar] [CrossRef]
- Merchant, B. Gold, the noble metal and the paradoxes of its toxicology. Biologicals 1998, 26, 49–59. [Google Scholar] [CrossRef]
- Carnovale, C.; Bryant, G.; Shukla, R.; Bansal, V. Identifying Trends in Gold Nanoparticle Toxicity and Uptake: Size, Shape, Capping Ligand, and Biological Corona. ACS Omega 2019, 4, 242–256. [Google Scholar] [CrossRef]
- García, A.; Rodríguez, B.; Giraldo, H.; Quintero, Y.; Quezada, R.; Hassan, N.; Estay, H. Copper-Modified Polymeric Membranes for Water Treatment: A Comprehensive Review. Membranes 2021, 11, 93. [Google Scholar] [CrossRef]
- Alenazi, N.A.; Hussein, M.A.; Alamry, K.A.; Asiri, A. Modified polyether-sulfone membrane: A mini review. Des. Monomers Polym. 2017, 20, 532–546. [Google Scholar] [CrossRef]
- Weisse, H.; Keul, H.; Höcker, H. A new route to carboxylated poly(ether sulfone)s: Synthesis and characterization. Polymer 2001, 42, 5973–5978. [Google Scholar] [CrossRef]
- Kaveh-Baghbaderani, Y.; Allgayer, R.; Schwaminger, S.P.; Fraga-García, P.; Berensmeier, S. Magnetic Separation of Antibodies with High Binding Capacity by Site-Directed Immobilization of Protein A-Domains to Bare Iron Oxide Nanoparticles. ACS Appl. Nano Mater. 2021, 4, 4956–4963. [Google Scholar] [CrossRef]
- Duan, C.; Meyerhoff, M.E. Immobilization of proteins on gold coated porous membranes via an activated self-assembled monolayer of thioctic acid. Mikrochim. Acta 1995, 117, 195–206. [Google Scholar] [CrossRef][Green Version]
- Hopkins, J.; Badyal, J.P.S. XPS and atomic force microscopy of plasma-treated polysulfone. J. Polym. Sci. Part A 1996, 34, 1385–1393. [Google Scholar] [CrossRef]
- Meck Millipore. Microsphere Coupling—Two-Step EDC/Sulfo NHS Covalent Coupling Procedure for Estapor Carboxy-Modified Dyed Microspheres; Meck Millipore: Burlington, MA, USA, 2015. [Google Scholar]
- Protein Cross-Linking & Protein Modification. 11 August 2022. Available online: https://www.gbiosciences.com/Cross-Linking-Modification (accessed on 20 November 2023).
- Jena Bioscience GmbH. Atto 488 Protein Labeling Kit, Fluorescent Amine Protein Labeling—Jena Bioscience. 11 August 2022. Available online: https://www.jenabioscience.com/probes-epigenetics/protein-labeling/labeling-of-purified-proteins/fluorescent-amine-labeling/fp-201-488-atto-488-protein-labeling-kit (accessed on 20 November 2023).
- Cytiva. Amersham Typhoon Operating Instructions. Available online: https://d3.cytivalifesciences.com/prod/IFU/29193226.pdf (accessed on 20 November 2023).
- Moulder, J.F. (Ed.) Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data; Perkin-Elmer Corporation: Eden Prairie, MN, USA, 1992. [Google Scholar]
- Ruiz, G.; Ryan, N.; Rutschke, K.; Awotunde, O.; Driskell, J. Antibodies Irreversibly Adsorb to Gold Nanoparticles and Resist Displacement by Common Blood Proteins. Langmuir 2019, 35, 10601–10609. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.; Schwahn, A.B.; Lin, S.; Pohl, C.A.; De Pra, M.; Tremintin, S.M.; Cook, K. New Insights into the Chromatography Mechanisms of Ion-Exchange Charge Variant Analysis: Dispelling Myths and Providing Guidance for Robust Method Optimization. Anal. Chem. 2020, 92, 13411–13419. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Biswas, M.E.; Chen, P. Study of Binding between Protein A and Immunoglobulin G Using a Surface Tension Probe. Biophys. J. 2003, 84, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, K.; Driskell, J.D. Quantifying Bound and Active Antibodies Conjugated to Gold Nanoparticles: A Comprehensive and Robust Approach To Evaluate Immobilization Chemistry. ACS Omega 2018, 3, 8253–8259. [Google Scholar] [CrossRef] [PubMed]
- Carta, G.; Jungbauer, A. Protein Chromatography: Process Development and Scale-Up; Wiley-VCH: Weinheim, Germany, 2020. [Google Scholar]
- Osuofa, J.; Husson, S.M. Comparative Evaluation of Commercial Protein A Membranes for the Rapid Purification of Antibodies. Membranes 2023, 13, 511. [Google Scholar] [CrossRef] [PubMed]
- Boi, C.; Malavasi, A.; Carbonell, R.G.; Gilleskie, G. A direct comparison between membrane adsorber and packed column chromatography performance. J. Chromatogr. A 2020, 1612, 460629. [Google Scholar] [CrossRef]
- Sartorius Lab Instruments GmBH & Co. KG. Sartobind® Protein A Manual; Sartorius Lab Instruments GmBH & Co. KG: Goettingen, Germany, 2022. [Google Scholar]
- Cho, N.-S.; Cho, H.-Y.; Shin, S.-J.; Choi, Y.-J.; Leonowicz, A.; Ohga, S. Production of Fungal Laccase and Its Immobilization and Stability. J. Fac. Agric. Kyushu Univ. 2008, 53, 13–18. [Google Scholar] [CrossRef]
- Latip, W.; Rosli, N.E.; Ali, M.S.M.; Kamarudin, N.H.A.; Rahman, R.N.Z.R.A. Stability Enhancement of Aldehyde Dehydrogenase from Anoxybacillus geothermalis Strain D9 Immobilized onto Seplite LX120. Catalysts 2023, 13, 368. [Google Scholar] [CrossRef]
- Kirley, T.L.; Norman, A.B. Unfolding of IgG domains detected by non-reducing SDS-PAGE. Biochem. Biophys. Res. Commun. 2018, 503, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Wiley (Ed.) Protein Purification: Principles, High Resolution Methods and Applications; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- Schwark, S.; Sun, W.; Stute, J.; Lütkemeyer, D.; Ulbricht, M.; Sellergren, B. Monoclonal antibody capture from cell culture supernatants using epitope imprinted macroporous membranes. RSC Adv. 2016, 6, 53162–53169. [Google Scholar] [CrossRef]
- Barroso, T.; Temtem, M.; Hussain, A.; Aguiar-Ricardo, A.; Roque, A. Preparation and characterization of a cellulose affinity membrane for human immunoglobulin G (IgG) purification. J. Membr. Sci. 2010, 348, 224–230. [Google Scholar] [CrossRef]
- Winzen, S.; Schoettler, S.; Baier, G.; Rosenauer, C.; Mailaender, V.; Landfester, K.; Mohr, K. Complementary analysis of the hard and soft protein corona: Sample preparation critically effects corona composition. Nanoscale 2015, 7, 2992–3001. [Google Scholar] [CrossRef] [PubMed]
- Szekeres, G.P.; Kneipp, J. Different binding sites of serum albumins in the protein corona of gold nanoparticles. Analyst 2018, 143, 6061–6068. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steegmüller, T.; Kratky, T.; Gollwitzer, L.; Schwaminger, S.P.; Berensmeier, S. Development of a New Affinity Gold Polymer Membrane with Immobilized Protein A. Membranes 2024, 14, 31. https://doi.org/10.3390/membranes14020031
Steegmüller T, Kratky T, Gollwitzer L, Schwaminger SP, Berensmeier S. Development of a New Affinity Gold Polymer Membrane with Immobilized Protein A. Membranes. 2024; 14(2):31. https://doi.org/10.3390/membranes14020031
Chicago/Turabian StyleSteegmüller, Tobias, Tim Kratky, Lena Gollwitzer, Sebastian Patrick Schwaminger, and Sonja Berensmeier. 2024. "Development of a New Affinity Gold Polymer Membrane with Immobilized Protein A" Membranes 14, no. 2: 31. https://doi.org/10.3390/membranes14020031
APA StyleSteegmüller, T., Kratky, T., Gollwitzer, L., Schwaminger, S. P., & Berensmeier, S. (2024). Development of a New Affinity Gold Polymer Membrane with Immobilized Protein A. Membranes, 14(2), 31. https://doi.org/10.3390/membranes14020031