
Permeation of a Homologous Series of NBD-Labeled Fatty Amines through Lipid Bilayers: A Molecular Dynamics Study




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Unrestrained MD Simulations for Membrane Characterization
2.2. Calculation of Free Energy Profiles of Long Amphiphiles Interacting with Lipid Membranes
2.2.1. Umbrella Sampling Simulations in Cholesterol-Containing Bilayers
2.2.2. Convergence of Free Energy Profiles
- (i)
- Assuming (incorrectly) that no equilibration is needed, in each sampling window using increasing amounts of simulation time for the sampling of the PMF.
- (ii)
- In each sampling window, systematically increase the slice of the simulation time used for equilibration, and use the rest of the simulation data for analysis.
- (iii)
- Again in each sampling window, systematically increase the amount of data used for equilibration, being the analysis for the PMF profile (in each window) carried out over a fixed period of 20 ns.
2.2.3. Permeability Calculations Using the Inhomogeneous Solubility-Diffusion Model (ISDM)
3. Results and Discussion
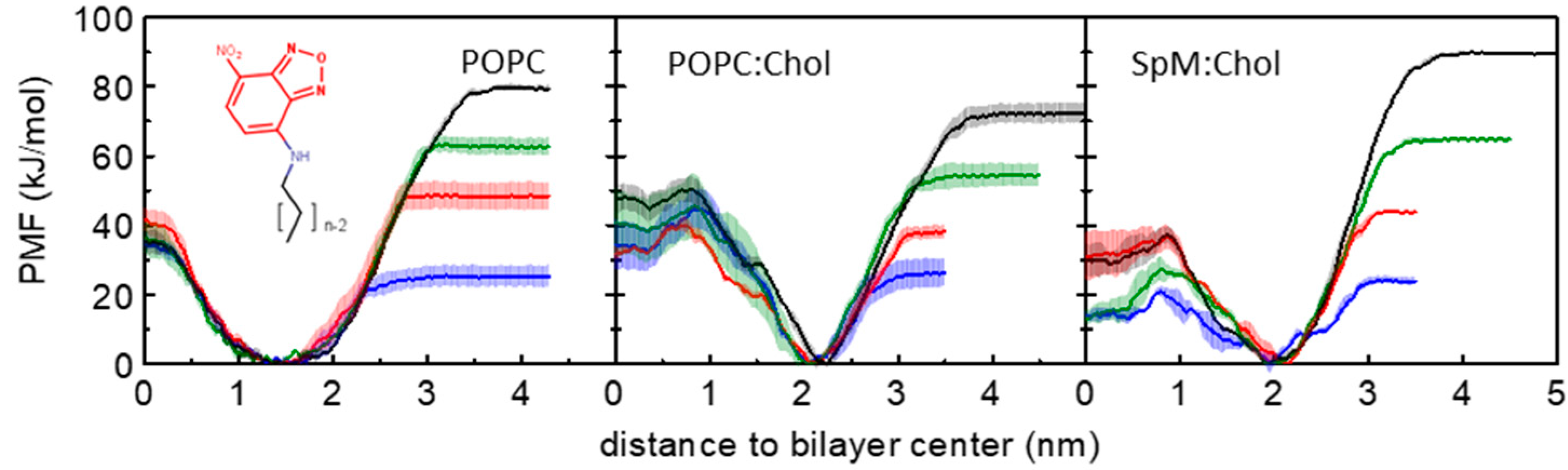
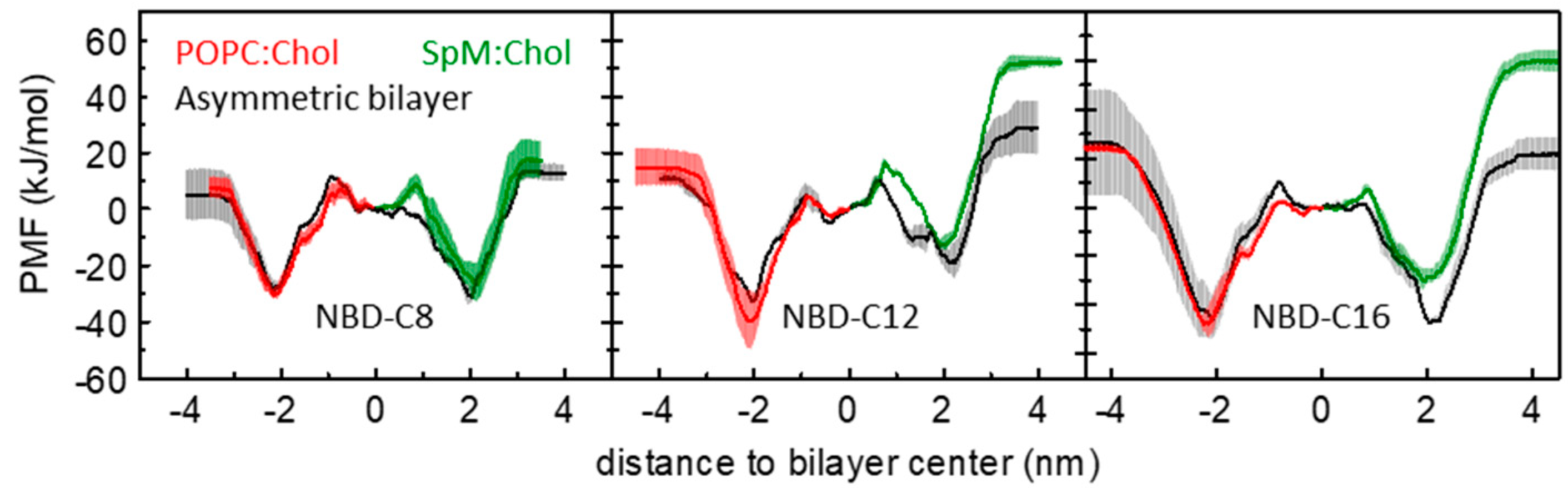
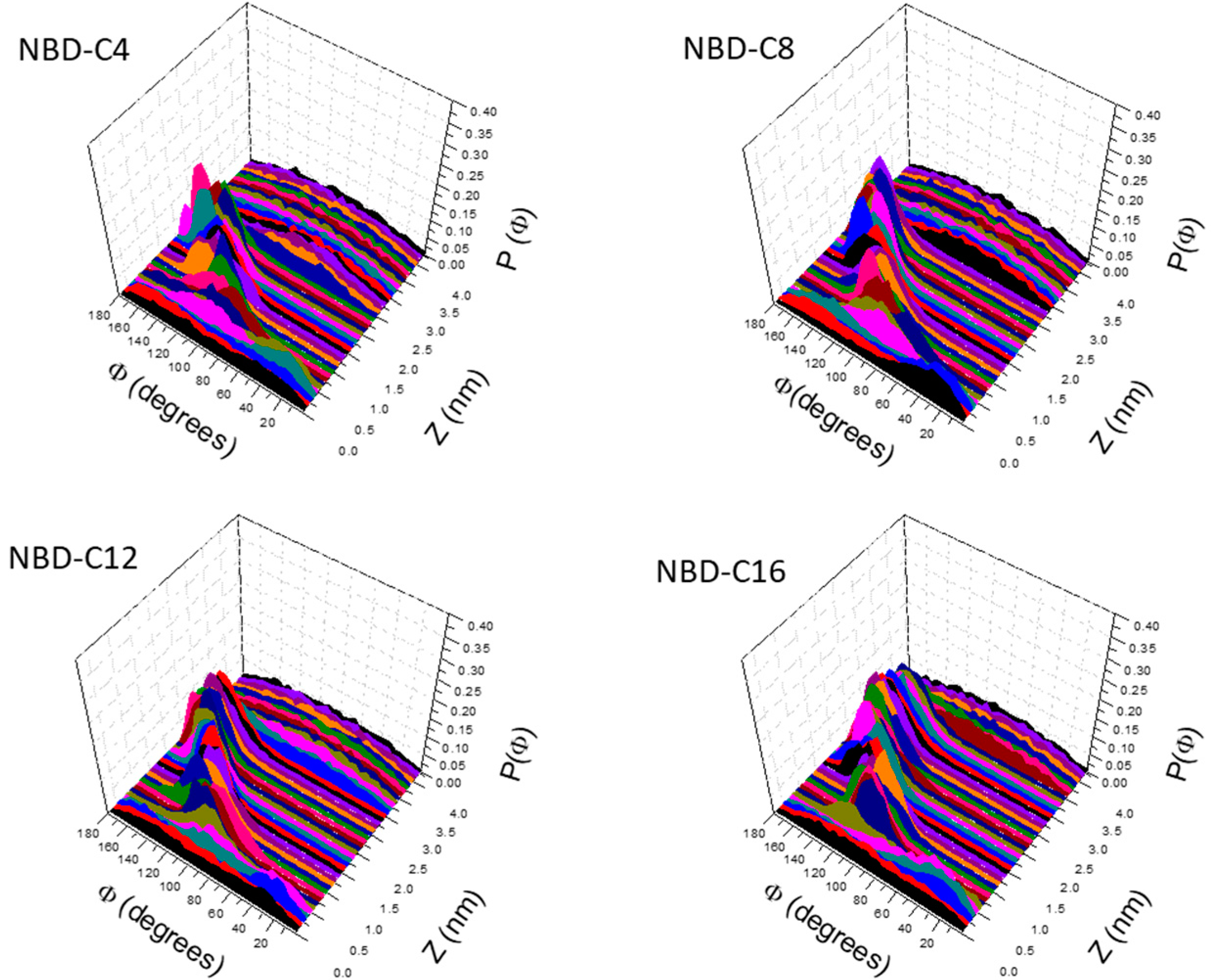
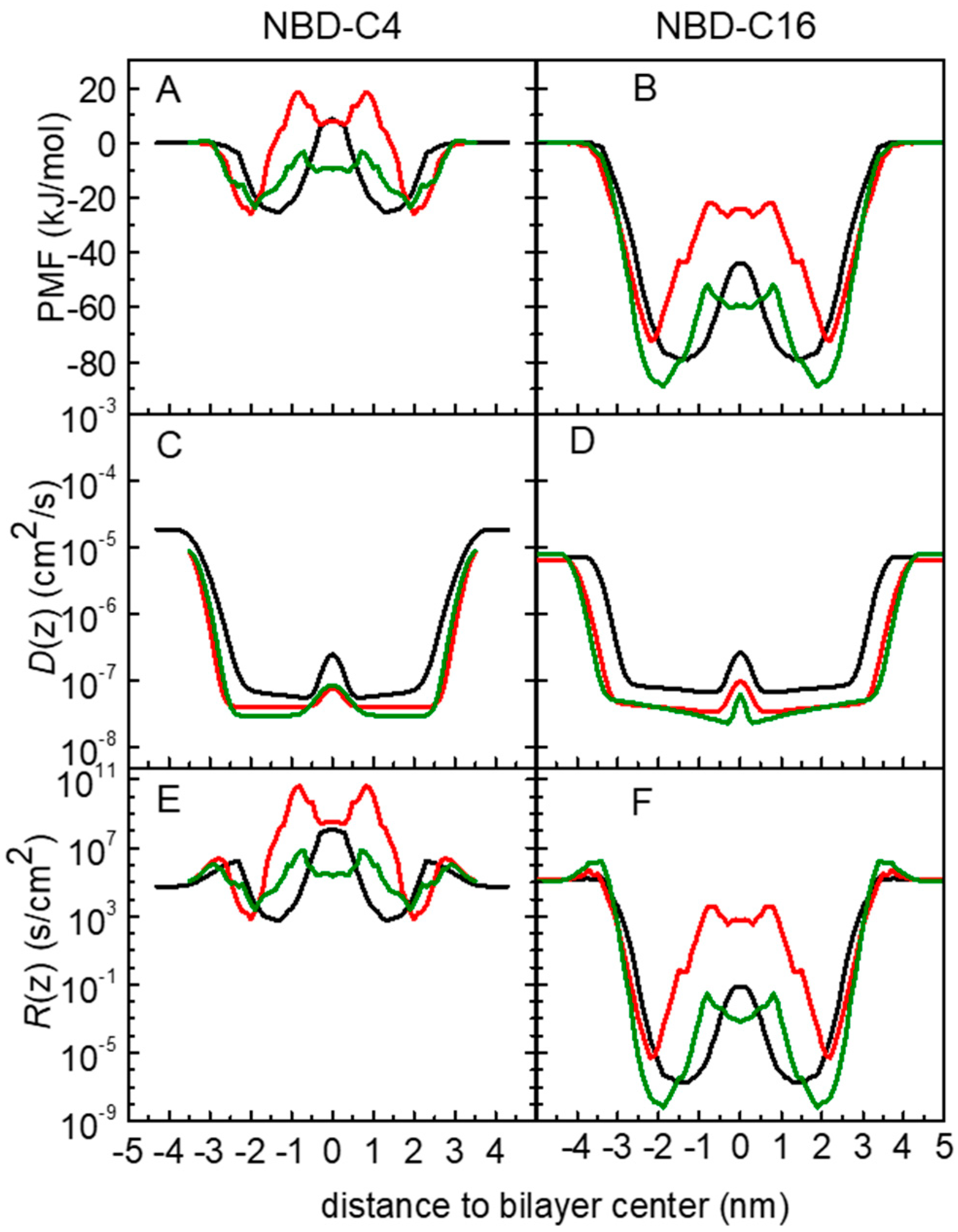
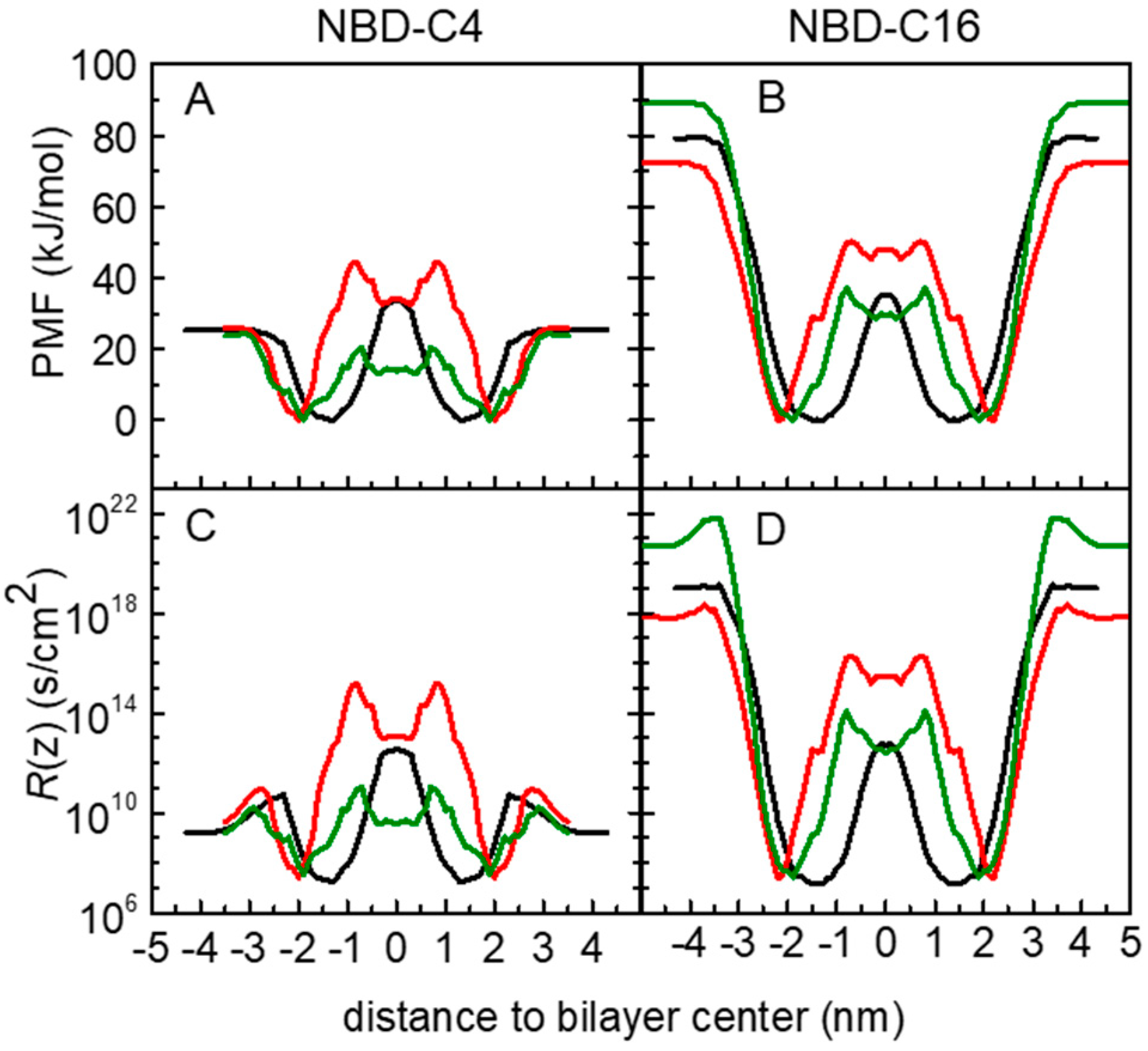
3.1. PMF Profiles for the NBD-Cn Homologous Series through Lipid Membranes
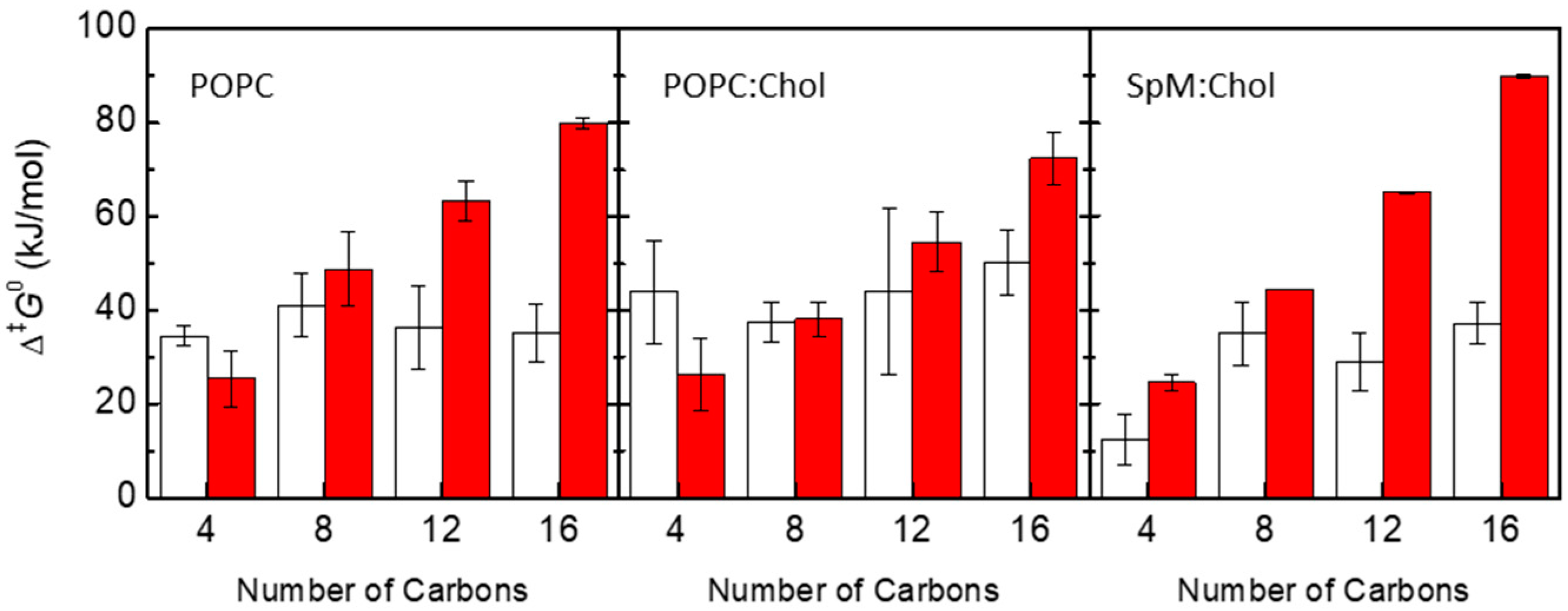
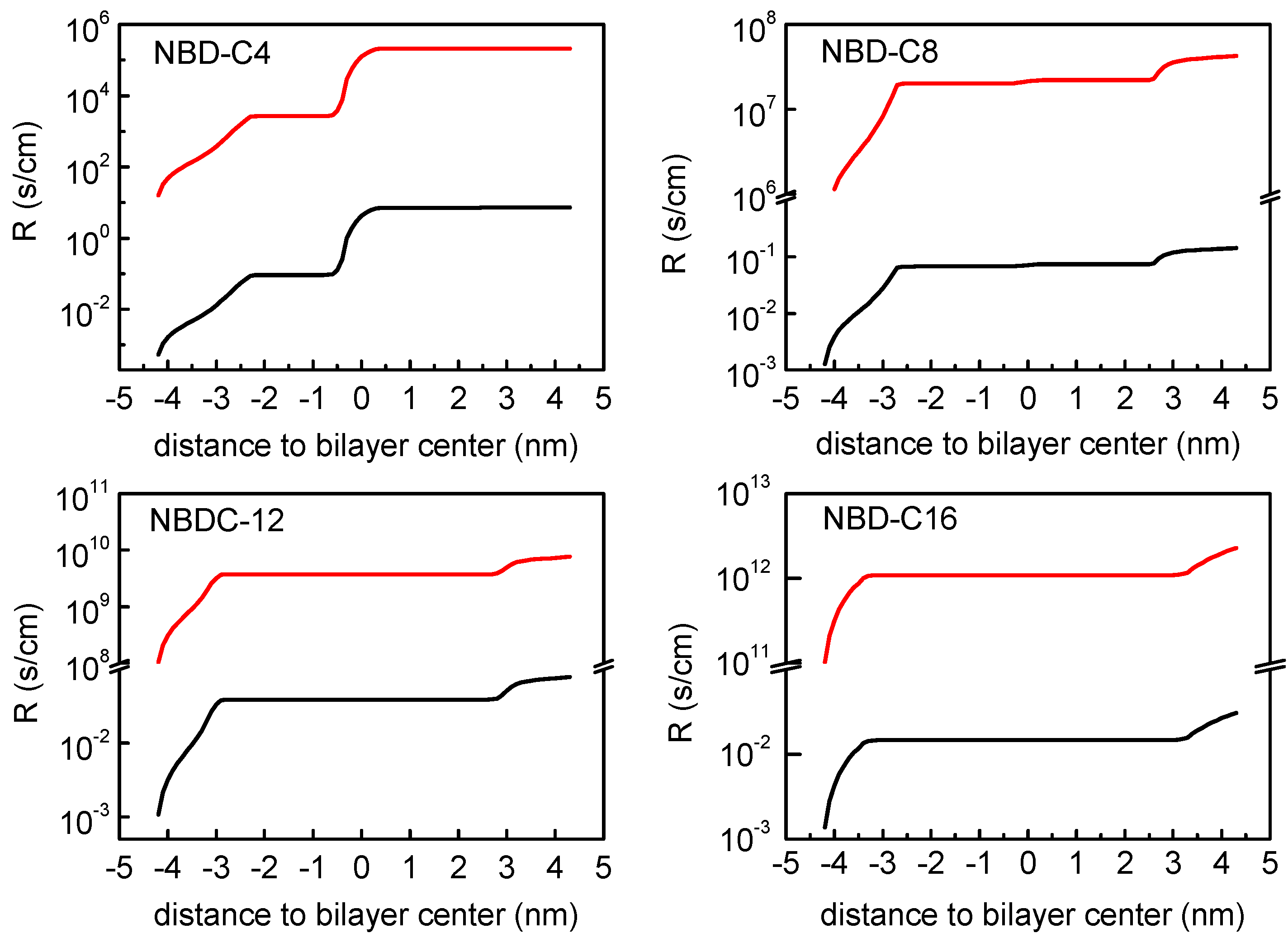
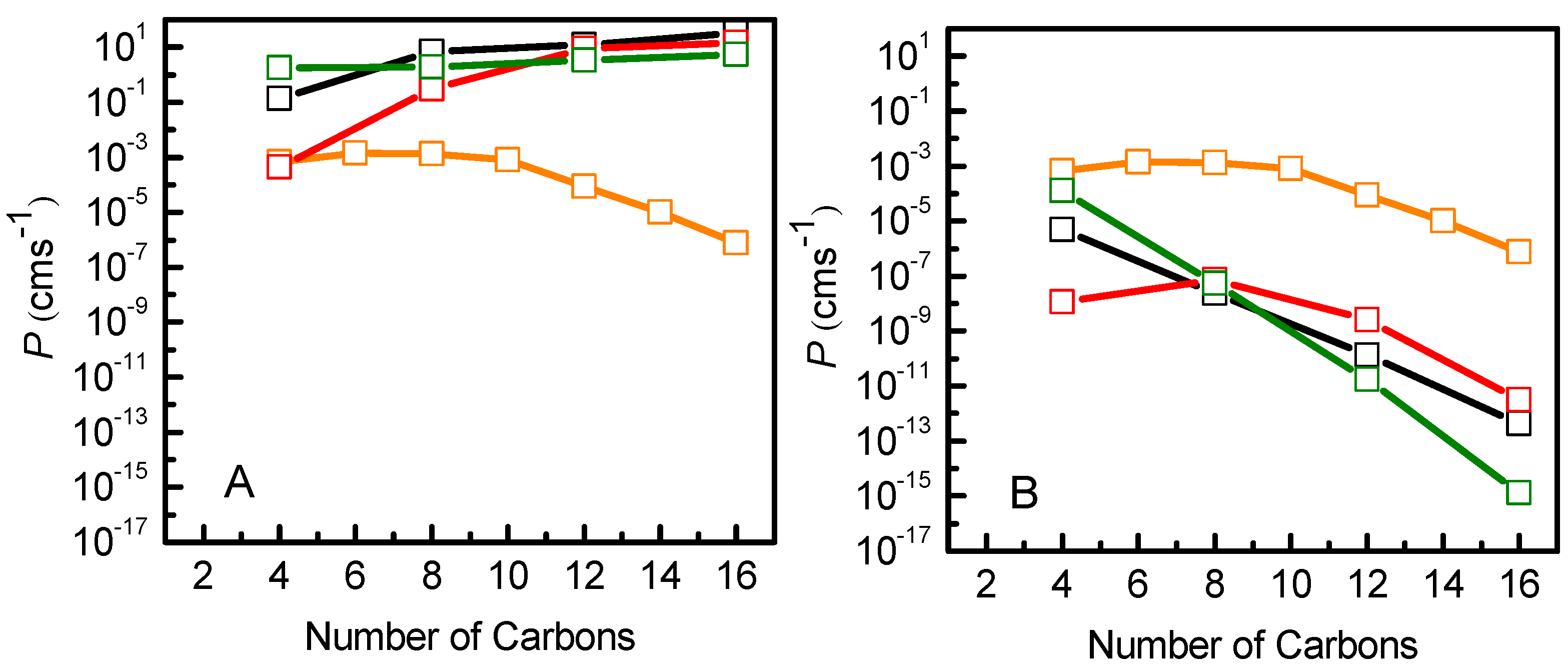
3.2. Quantitative Evaluation of Permeation of the Amphiphiles through the Lipid Bilayers—Application of the Inhomogeneous Solubility-Diffusion Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kell, D.B.; Dobson, P.D.; Oliver, S.G. Pharmaceutical drug transport: The issues and the implications that it is essentially carrier-mediated only. Drug. Discov. Today 2011, 16, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Sugano, K.; Kansy, M.; Artursson, P.; Avdeef, A.; Bendels, S.; Di, L.; Ecker, G.F.; Faller, B.; Fischer, H.; Gerebtzoff, G.; et al. Coexistence of passive and carrier-mediated processes in drug transport. Nat. Rev. Drug. Discov. 2010, 9, 597–614. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.; Artursson, P.; Avdeef, A.; Di, L.; Ecker, G.F.; Faller, B.; Houston, J.B.; Kansy, M.; Kerns, E.H.; Krämer, S.D.; et al. Passive Lipoidal Diffusion and Carrier-Mediated Cell Uptake Are Both Important Mechanisms of Membrane Permeation in Drug Disposition. Mol. Pharm. 2014, 11, 1727–1738. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, J.L.; Moreno, M.J.; Vaz, W.L.C. Kinetics and thermodynamics of association of a fluorescent lysophospholipid derivative with lipid bilayers in liquid-ordered and liquid-disordered phases. Biophys. J. 2005, 88, 4064–4071. [Google Scholar] [CrossRef]
- Coreta-Gomes, F.M.; Vaz, W.L.C.; Moreno, M.J. Effect of Acyl Chain Length on the Rate of Phospholipid Flip-Flop and Intermembrane Transfer. J. Membr. Biol. 2018, 251, 431–442. [Google Scholar] [CrossRef]
- Estronca, L.; Moreno, M.J.; Vaz, W.L.C. Kinetics and thermodynamics of the association of dehydroergosterol with lipid bilayer membranes. Biophys. J. 2007, 93, 4244–4253. [Google Scholar] [CrossRef]
- Cardoso, R.M.S.; Martins, P.A.T.; Gomes, F.; Doktorovova, S.; Vaz, W.L.C.; Moreno, M.J. Chain-Length Dependence of Insertion, Desorption, and Translocation of a Homologous Series of 7-Nitrobenz-2-oxa-1,3-diazol-4-yl-Labeled Aliphatic Amines in Membranes. J. Phys. Chem. B 2011, 115, 10098–10108. [Google Scholar] [CrossRef]
- Martins, P.T.; Velazquez-Campoy, A.; Vaz, W.L.C.; Cardoso, R.M.S.; Valério, J.; Moreno, M.J. Kinetics and Thermodynamics of Chlorpromazine Interaction with Lipid Bilayers: Effect of Charge and Cholesterol. J. Am. Chem. Soc. 2012, 134, 4184–4195. [Google Scholar] [CrossRef]
- Moreno, M.J.; Estronca, L.; Vaz, W.L.C. Translocation of phospholipids and dithionite permeability in liquid-ordered and liquid-disordered membranes. Biophys. J. 2006, 91, 873–881. [Google Scholar] [CrossRef]
- Abreu, M.S.C.; Moreno, M.J.; Vaz, W.L.C. Kinetics and thermodynamics of association of a phospholipid derivative with lipid bilayers in liquid-disordered and liquid-ordered phases. Biophys. J. 2004, 87, 353–365. [Google Scholar] [CrossRef]
- Filipe, H.A.L.; Salvador, A.; Silvestre, J.M.; Vaz, W.L.C.; Moreno, M.J. Beyond Overton’s Rule: Quantitative Modeling of Passive Permeation through Tight Cell Monolayers. Mol. Pharm. 2014, 11, 3696–3706. [Google Scholar] [CrossRef] [PubMed]
- Deamer, D.W.; Kleinzeller, A.; Fambrough, D.M. Membrane permeability: 100 years since Ernest Overton. In Current Topics in Membranes; Academic Press: Cambridge, MA, USA, 1999; Volume 48. [Google Scholar]
- Paloncýová, M.; DeVane, R.; Murch, B.; Berka, K.; Otyepka, M. Amphiphilic Drug-Like Molecules Accumulate in a Membrane below the Head Group Region. J. Phys. Chem. B 2014, 118, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Berka, K.; Hendrychová, T.; Anzenbacher, P.; Otyepka, M. Membrane Position of Ibuprofen Agrees with Suggested Access Path Entrance to Cytochrome P450 2C9 Active Site. J. Phys. Chem. A 2011, 115, 11248–11255. [Google Scholar] [CrossRef] [PubMed]
- Shahoei, R.; Tajkhorshid, E. Menthol Binding to the Human α4β2 Nicotinic Acetylcholine Receptor Facilitated by Its Strong Partitioning in the Membrane. J. Phys. Chem. B 2020, 124, 1866–1880. [Google Scholar] [CrossRef]
- Lolicato, F.; Juhola, H.; Zak, A.; Postila, P.A.; Saukko, A.; Rissanen, S.; Enkavi, G.; Vattulainen, I.; Kepczynski, M.; Róg, T. Membrane-Dependent Binding and Entry Mechanism of Dopamine into Its Receptor. ACS Chem. Neurosci. 2020, 11, 1914–1924. [Google Scholar] [CrossRef]
- Moreno, M.J.; Filipe, H.A.L.; Cunha, S.V.P.; Ramos, C.V.; Martins, P.A.T.; Abel, B.; Loura, L.M.S.; Ambudkar, S.V. Interaction of a Homologous Series of Amphiphiles with P-glycoprotein in a Membrane Environment—Contributions of Polar and Non-Polar Interactions. Pharmaceutics 2023, 15, 174. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.J.; Salvador, A. Ligand’s Partition to the Lipid Bilayer Should Be Accounted for When Estimating Their Affinity to Proteins. Molecules 2023, 28, 3136. [Google Scholar] [CrossRef]
- Košinová, P.; Berka, K.; Wykes, M.; Otyepka, M.; Trouillas, P. Positioning of Antioxidant Quercetin and Its Metabolites in Lipid Bilayer Membranes: Implication for Their Lipid-Peroxidation Inhibition. J. Phys. Chem. B 2011, 116, 1309–1318. [Google Scholar] [CrossRef]
- Filipe, H.A.L.; Sousa, C.; Marquês, J.T.; Vila-Viçosa, D.; de Granada-Flor, A.; Viana, A.S.; Santos, M.S.C.S.; Machuqueiro, M.; de Almeida, R.F.M. Differential targeting of membrane lipid domains by caffeic acid and its ester derivatives. Free Radic. Biol. Med. 2018, 115, 232–245. [Google Scholar] [CrossRef]
- de Granada-Flor, A.; Sousa, C.; Filipe, H.A.L.; Santos, M.S.C.S.; de Almeida, R.F.M. Quercetin dual interaction at the membrane level. Chem. Commun. 2019, 55, 1750–1753. [Google Scholar] [CrossRef]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.J.; Teles Martins, P.A.; Bernardino, E.F.; Abel, B.; Ambudkar, S.V. Characterization of the Lipidome and Biophysical Properties of Membranes from High Five Insect Cells Expressing Mouse P-Glycoprotein. Biomolecules 2021, 11, 426. [Google Scholar] [CrossRef] [PubMed]
- Op den Kamp, J.A.F. Lipid Asymmetry in Membranes. Annu. Rev. Biochem. 1979, 48, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Quinn, P. Plasma Membrane Phospholipid Asymmetry. In Phospholipid Metabolism in Apoptosis; Quinn, P., Kagan, V., Eds.; Subcellular Biochemistry; Springer: Berlin/Heidelberg, Germany, 2002; Volume 36, pp. 39–60. [Google Scholar]
- Jo, S.; Rui, H.; Lim, J.B.; Klauda, J.B.; Im, W. Cholesterol Flip-Flop: Insights from Free Energy Simulation Studies. J. Phys. Chem. B 2010, 114, 13342–13348. [Google Scholar] [CrossRef] [PubMed]
- Bennett, W.F.D.; Tieleman, D.P. Molecular simulation of rapid translocation of cholesterol, diacylglycerol, and ceramide in model raft and nonraft membranes. J. Lipid Res. 2012, 53, 421–429. [Google Scholar] [CrossRef]
- Steck, T.L.; Lange, Y. How Slow Is the Transbilayer Diffusion (Flip-Flop) of Cholesterol? Biophys. J. 2012, 102, 945–946. [Google Scholar] [CrossRef]
- Filipe, H.A.L.; Javanainen, M.; Salvador, A.; Galvão, A.M.; Vattulainen, I.; Loura, L.M.S.; Moreno, M.J. Quantitative Assessment of Methods Used to Obtain Rate Constants from Molecular Dynamics Simulations—Translocation of Cholesterol across Lipid Bilayers. J. Chem. Theory Comput. 2018, 14, 3840–3848. [Google Scholar] [CrossRef]
- Shaikh, S.R.; Cherezov, V.; Caffrey, M.; Stillwell, W.; Wassall, S.R. Interaction of Cholesterol with a Docosahexaenoic Acid-Containing Phosphatidylethanolamine: Trigger for Microdomain/Raft Formation? Biochemistry 2003, 42, 12028–12037. [Google Scholar] [CrossRef]
- Lange, Y.; Tabei, S.M.A.; Ye, J.; Steck, T.L. Stability and Stoichiometry of Bilayer Phospholipid–Cholesterol Complexes: Relationship to Cellular Sterol Distribution and Homeostasis. Biochemistry 2013, 52, 6950–6959. [Google Scholar] [CrossRef]
- Dodge, J.T.; Phillips, G.B. Composition of phospholipids and of phospholipid fatty acids and aldehydes in human red cells. J. Lipid Res. 1967, 8, 667–675. [Google Scholar] [CrossRef]
- Rothman, J.E.; Lenard, J. Membrane asymmetry. Science 1977, 195, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Devaux, P.F. Protein involvement in transmembrane lipid asymmetry. Annu. Rev. Biophys. Biomol. Struct. 1992, 21, 417–439. [Google Scholar] [CrossRef] [PubMed]
- Owen, J.S.; Bruckdorfer, K.R.; Day, R.C.; McIntyre, N. Decreased Erythrocyte membrane fluidity and altered lipid composition in human liver disease. J. Lipid Res. 1982, 23, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Leidl, K.; Liebisch, G.; Richter, D.; Schmitz, G. Mass spectrometric analysis of lipid species of human circulating blood cells. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2008, 1781, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Barceló, F.; Perona, J.S.; Prades, J.; Funari, S.S.; Gomez-Gracia, E.; Conde, M.; Estruch, R.; Ruiz-Gutiérrez, V. Mediterranean-Style Diet Effect on the Structural Properties of the Erythrocyte Cell Membrane of Hypertensive Patients: The Prevencion con Dieta Mediterranea Study. Hypertension 2009, 54, 1143–1150. [Google Scholar] [CrossRef]
- Fisher, K.A. Analysis of membrane halves: Cholesterol. Proc. Natl. Acad. Sci. USA 1976, 73, 173–177. [Google Scholar] [CrossRef]
- Yeagle, P.L. Cholesterol and the cell membrane. Biochim. Biophys. Acta (BBA)—Rev. Biomembr. 1985, 822, 267–287. [Google Scholar] [CrossRef]
- Doktorova, M.; Heberle, F.A.; Eicher, B.; Standaert, R.F.; Katsaras, J.; London, E.; Pabst, G.; Marquardt, D. Preparation of asymmetric phospholipid vesicles for use as cell membrane models. Nat. Protoc. 2018, 13, 2086–2101. [Google Scholar] [CrossRef]
- Markones, M.; Fippel, A.; Kaiser, M.; Drechsler, C.; Hunte, C.; Heerklotz, H. Stairway to Asymmetry: Five Steps to Lipid-Asymmetric Proteoliposomes. Biophys. J. 2020, 118, 294–302. [Google Scholar] [CrossRef]
- Bhide, S.Y.; Zhang, Z.; Berkowitz, M.L. Molecular Dynamics Simulations of SOPS and Sphingomyelin Bilayers Containing Cholesterol. Biophys. J. 2007, 92, 1284–1295. [Google Scholar] [CrossRef]
- Gurtovenko, A.A.; Vattulainen, I. Lipid Transmembrane Asymmetry and Intrinsic Membrane Potential: Two Sides of the Same Coin. J. Am. Chem. Soc. 2007, 129, 5358–5359. [Google Scholar] [CrossRef] [PubMed]
- Gurtovenko, A.A.; Vattulainen, I. Membrane Potential and Electrostatics of Phospholipid Bilayers with Asymmetric Transmembrane Distribution of Anionic Lipids. J. Phys. Chem. B 2008, 112, 4629–4634. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, D.; Geier, B.; Pabst, G. Asymmetric Lipid Membranes: Towards More Realistic Model Systems. Membranes 2015, 5, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Ingólfsson, H.I.; Neale, C.; Carpenter, T.S.; Shrestha, R.; López, C.A.; Tran, T.H.; Oppelstrup, T.; Bhatia, H.; Stanton, L.G.; Zhang, X.; et al. Machine learning-driven multiscale modeling reveals lipid-dependent dynamics of RAS signaling proteins. Proc. Natl. Acad. Sci. USA 2022, 119, e2113297119. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Kästner, J. Umbrella sampling. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 932–942. [Google Scholar] [CrossRef]
- Neale, C.; Madill, C.; Rauscher, S.; Pomès, R. Accelerating Convergence in Molecular Dynamics Simulations of Solutes in Lipid Membranes by Conducting a Random Walk along the Bilayer Normal. J. Chem. Theory Comput. 2013, 9, 3686–3703. [Google Scholar] [CrossRef]
- Dickson, C.J.; Hornak, V.; Pearlstein, R.A.; Duca, J.S. Structure–Kinetic Relationships of Passive Membrane Permeation from Multiscale Modeling. J. Am. Chem. Soc. 2017, 139, 442–452. [Google Scholar] [CrossRef]
- Juračka, J.; Šrejber, M.; Melíková, M.; Bazgier, V.; Berka, K. MolMeDB: Molecules on Membranes Database. Database 2019, 2019, baz078. [Google Scholar] [CrossRef]
- Benmameri, M.; Chantemargue, B.; Humeau, A.; Trouillas, P.; Fabre, G. MemCross: Accelerated Weight Histogram method to assess membrane permeability. Biochim. Biophys. Acta Biomembr. 2023, 1865, 184120. [Google Scholar] [CrossRef]
- MacCallum, J.L.; Tieleman, D.P. Chapter 8 Interactions between Small Molecules and Lipid Bilayers. In Current Topics in Membranes; Scott, E.F., Ed.; Academic Press: Cambridge, MA, USA, 2008; Volume 60, pp. 227–256. [Google Scholar]
- Bemporad, D.; Essex, J.W.; Luttmann, C. Permeation of small molecules through a lipid bilayer: A computer simulation study. J. Phys. Chem. B 2004, 108, 4875–4884. [Google Scholar] [CrossRef]
- Paloncýová, M.; Berka, K.; Otyepka, M. Convergence of Free Energy Profile of Coumarin in Lipid Bilayer. J. Chem. Theory Comput. 2012, 8, 1200–1211. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, R.M.S.; Filipe, H.A.L.; Gomes, F.; Moreira, N.D.; Vaz, W.L.C.; Moreno, M.J. Chain Length Effect on the Binding of Amphiphiles to Serum Albumin and to POPC Bilayers. J. Phys. Chem. B 2010, 114, 16337–16346. [Google Scholar] [CrossRef] [PubMed]
- Filipe, H.A.L.; Moreno, M.J.; Loura, L.M.S. Interaction of 7-Nitrobenz-2-oxa-1,3-diazol-4-yl-Labeled Fatty Amines with 1-Palmitoyl, 2-Oleoyl-sn-glycero-3-phosphocholine Bilayers: A Molecular Dynamics Study. J. Phys. Chem. B 2011, 115, 10109–10119. [Google Scholar] [CrossRef] [PubMed]
- Filipe, H.A.L.; Moreno, M.J.; Róg, T.; Vattulainen, I.; Loura, L.M.S. How To Tackle the Issues in Free Energy Simulations of Long Amphiphiles Interacting with Lipid Membranes: Convergence and Local Membrane Deformations. J. Phys. Chem. B 2014, 118, 3572–3581. [Google Scholar] [CrossRef] [PubMed]
- Filipe, H.A.L.; Bowman, D.; Palmeira, T.; Cardoso, R.M.S.; Loura, L.M.S.; Moreno, M.J. Interaction of NBD-labelled fatty amines with liquid-ordered membranes: A combined molecular dynamics simulation and fluorescence spectroscopy study. Phys. Chem. Chem. Phys. 2015, 17, 27534–27547. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Hoff, B.; Strandberg, E.; Ulrich, A.S.; Tieleman, D.P.; Posten, C. H-2-NMR study and molecular dynamics simulation of the location, alignment, and mobility of pyrene in POPC bilayers. Biophys. J. 2005, 88, 1818–1827. [Google Scholar] [CrossRef]
- Berger, O.; Edholm, O.; Jahnig, F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 1997, 72, 2002–2013. [Google Scholar] [CrossRef]
- Martinez-Seara, H.; Róg, T.; Karttunen, M.; Reigada, R.; Vattulainen, I. Influence of cis double-bond parametrization on lipid membrane properties: How seemingly insignificant details in force-field change even qualitative trends. J. Chem. Phys. 2008, 129, 105103. [Google Scholar] [CrossRef] [PubMed]
- Niemelä, P.; Hyvönen, M.T.; Vattulainen, I. Structure and Dynamics of Sphingomyelin Bilayer: Insight Gained through Systematic Comparison to Phosphatidylcholine. Biophys. J. 2004, 87, 2976–2989. [Google Scholar] [CrossRef] [PubMed]
- Domański, J.; Stansfeld, P.; Sansom, M.P.; Beckstein, O. Lipidbook: A Public Repository for Force-Field Parameters Used in Membrane Simulations. J. Membr. Biol. 2010, 236, 255–258. [Google Scholar] [CrossRef]
- Höltje, M.; Förster, T.; Brandt, B.; Engels, T.; von Rybinski, W.; Höltje, H.-D. Molecular dynamics simulations of stratum corneum lipid models: Fatty acids and cholesterol. Biochim. Biophys. Acta Biomembr. 2001, 1511, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Paloncýová, M.; Fabre, G.; DeVane, R.H.; Trouillas, P.; Berka, K.; Otyepka, M. Benchmarking of Force Fields for Molecule–Membrane Interactions. J. Chem. Theory Comput. 2014, 10, 4143–4151. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Gunsteren, W.F.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration. In Intermolecular Forces; Pullman, B., Ed.; The Jerusalem Symposia on Quantum Chemistry and Biochemistry; Springer: Berlin/Heidelberg, Germany, 1981; Volume 14, pp. 331–342. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Hofsass, C.; Lindahl, E.; Edholm, O. Molecular dynamics simulations of phospholipid bilayers with cholesterol. Biophys. J. 2003, 84, 2192–2206. [Google Scholar] [CrossRef]
- Seelig, J. Deuterium magnetic resonance: Theory and application to lipid membranes. Q. Rev. Biophys. 1977, 10, 353–418. [Google Scholar] [CrossRef] [PubMed]
- Roux, B. The calculation of the potential of mean force using computer simulations. Comput. Phys. Commun. 1995, 91, 275–282. [Google Scholar] [CrossRef]
- Hub, J.S.; de Groot, B.L.; van der Spoel, D. g_wham—A Free Weighted Histogram Analysis Implementation Including Robust Error and Autocorrelation Estimates. J. Chem. Theory Comput. 2010, 6, 3713–3720. [Google Scholar] [CrossRef]
- Marrink, S.J.; Berendsen, H.J.C. Simulation of water transport through a lipid membrane. J. Phys. Chem. 1994, 98, 4155–4168. [Google Scholar] [CrossRef]
- Cardoso, R.M.S.; Martins, P.A.T.; Ramos, C.V.; Cordeiro, M.M.; Leote, R.J.B.; Razi Naqvi, K.; Vaz, W.L.C.; Moreno, M.J. Effect of dipole moment on amphiphile solubility and partition into liquid ordered and liquid disordered phases in lipid bilayers. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183157. [Google Scholar] [CrossRef] [PubMed]
- Bennett, W.F.D.; MacCallum, J.L.; Hinner, M.J.; Marrink, S.J.; Tieleman, D.P. Molecular View of Cholesterol Flip-Flop and Chemical Potential in Different Membrane Environments. J. Am. Chem. Soc. 2009, 131, 12714–12720. [Google Scholar] [CrossRef] [PubMed]
- Bennett, W.F.D.; MacCallum, J.L.; Tieleman, D.P. Thermodynamic Analysis of the Effect of Cholesterol on Dipalmitoylphosphatidylcholine Lipid Membranes. J. Am. Chem. Soc. 2009, 131, 1972–1978. [Google Scholar] [CrossRef]
- Harroun, T.A.; Katsaras, J.; Wassall, S.R. Cholesterol Hydroxyl Group Is Found To Reside in the Center of a Polyunsaturated Lipid Membrane. Biochemistry 2006, 45, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Harroun, T.A.; Katsaras, J.; Wassall, S.R. Cholesterol Is Found To Reside in the Center of a Polyunsaturated Lipid Membrane. Biochemistry 2008, 47, 7090–7096. [Google Scholar] [CrossRef] [PubMed]
- Homan, R.; Pownall, H.J. Transbilayer diffusion of phospholipids: Dependence on headgroup structure and acyl chain length. Biochim. Biophys. Acta Biomembr. 1988, 938, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Neves, M.C.; Filipe, H.A.L.; Reis, R.L.; Prates Ramalho, J.P.; Coreta-Gomes, F.; Moreno, M.J.; Loura, L.M.S. Interaction of Bile Salts With Lipid Bilayers: An Atomistic Molecular Dynamics Study. Front. Physiol. 2019, 10. [Google Scholar] [CrossRef]
- Coreta-Gomes, F.M.; Martins, P.A.T.; Velazquez-Campoy, A.; Vaz, W.L.C.; Geraldes, C.F.G.; Moreno, M.J. Interaction of Bile Salts with Model Membranes Mimicking the Gastrointestinal Epithelium: A Study by Isothermal Titration Calorimetry. Langmuir 2015, 31, 9097–9104. [Google Scholar] [CrossRef] [PubMed]
- Paloncýová, M.; DeVane, R.H.; Murch, B.P.; Berka, K.; Otyepka, M. Rationalization of Reduced Penetration of Drugs through Ceramide Gel Phase Membrane. Langmuir 2014, 30, 13942–13948. [Google Scholar] [CrossRef] [PubMed]
- Kramers, H.A. Brownian motion in a field of force and the diffusion model of chemical reactions. Physica 1940, 7, 284–304. [Google Scholar] [CrossRef]
- Aniansson, E.A.G. On the Rate of Many-Step Processes. In Chemical and Biological Applications of Relaxation Spectrometry; Springer: Dordrecht, The Netherlands, 1975; pp. 245–253. [Google Scholar]
- Cheng, H.-T.; Megha; London, E. Preparation and Properties of Asymmetric Vesicles That Mimic Cell Membranes*. J. Biol. Chem. 2009, 284, 6079–6092. [Google Scholar] [CrossRef]
- Wang, Q.; London, E. Lipid Structure and Composition Control Consequences of Interleaflet Coupling in Asymmetric Vesicles. Biophys. J. 2018, 115, 664–678. [Google Scholar] [CrossRef]
- Markones, M.; Drechsler, C.; Kaiser, M.; Kalie, L.; Heerklotz, H.; Fiedler, S. Engineering Asymmetric Lipid Vesicles: Accurate and Convenient Control of the Outer Leaflet Lipid Composition. Langmuir 2018, 34, 1999–2005. [Google Scholar] [CrossRef]
- St. Clair, J.W.; London, E. Effect of sterol structure on ordered membrane domain (raft) stability in symmetric and asymmetric vesicles. Biochim. Biophys. Acta Biomembr. 2019, 1861, 1112–1122. [Google Scholar] [CrossRef]
- London, E. Membrane Structure–Function Insights from Asymmetric Lipid Vesicles. Acc. Chem. Res. 2019, 52, 2382–2391. [Google Scholar] [CrossRef]
- Estronca, L.; Moreno, M.J.; Abreu, M.S.C.; Melo, E.; Vaz, W.L.C. Solubility of amphiphiles in membranes: Influence of phase properties and amphiphile head group. Biochem. Biophys. Res. Commun. 2002, 296, 596–603. [Google Scholar] [CrossRef]
- Bedrov, D.; Smith, G.D.; Davande, H.; Li, L. Passive Transport of C60 Fullerenes through a Lipid Membrane: A Molecular Dynamics Simulation Study. J. Phys. Chem. B 2008, 112, 2078–2084. [Google Scholar] [CrossRef] [PubMed]
- Orsi, M.; Essex, J.W. Permeability of drugs and hormones through a lipid bilayer: Insights from dual-resolution molecular dynamics. Soft Matter 2010, 6, 3797–3808. [Google Scholar] [CrossRef]
- Sousa, C.F.; Kamal, M.A.M.; Richter, R.; Elamaldeniya, K.; Hartmann, R.W.; Empting, M.; Lehr, C.-M.; Kalinina, O.V. Modeling the Effect of Hydrophobicity on the Passive Permeation of Solutes across a Bacterial Model Membrane. J. Chem. Inf. Model. 2022, 62, 5023–5033. [Google Scholar] [CrossRef] [PubMed]
- Venable, R.M.; Krämer, A.; Pastor, R.W. Molecular Dynamics Simulations of Membrane Permeability. Chem. Rev. 2019, 119, 5954–5997. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filipe, H.A.L.; Loura, L.M.S.; Moreno, M.J. Permeation of a Homologous Series of NBD-Labeled Fatty Amines through Lipid Bilayers: A Molecular Dynamics Study. Membranes 2023, 13, 551. https://doi.org/10.3390/membranes13060551
Filipe HAL, Loura LMS, Moreno MJ. Permeation of a Homologous Series of NBD-Labeled Fatty Amines through Lipid Bilayers: A Molecular Dynamics Study. Membranes. 2023; 13(6):551. https://doi.org/10.3390/membranes13060551
Chicago/Turabian StyleFilipe, Hugo A. L., Luís M. S. Loura, and Maria João Moreno. 2023. "Permeation of a Homologous Series of NBD-Labeled Fatty Amines through Lipid Bilayers: A Molecular Dynamics Study" Membranes 13, no. 6: 551. https://doi.org/10.3390/membranes13060551
APA StyleFilipe, H. A. L., Loura, L. M. S., & Moreno, M. J. (2023). Permeation of a Homologous Series of NBD-Labeled Fatty Amines through Lipid Bilayers: A Molecular Dynamics Study. Membranes, 13(6), 551. https://doi.org/10.3390/membranes13060551