
Advances in Mass Spectrometry on Membrane Proteins


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. HDX-MS
2.1. Method Development
2.2. Applications
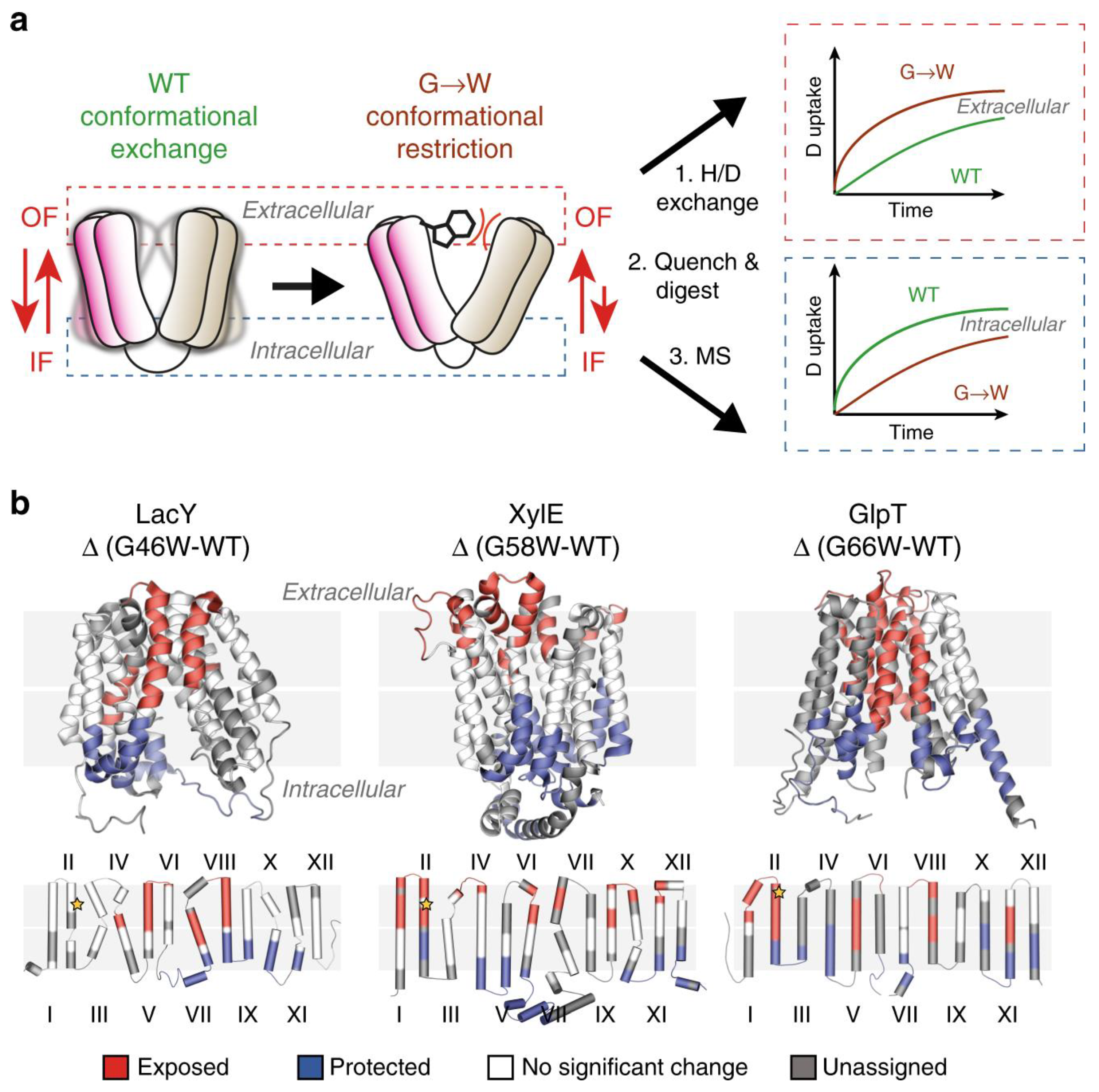
HDX Transmembrane Domains
2.3. Future Directions for HDX
3. Chemical Footprinting
3.1. Method Development
3.2. Applications
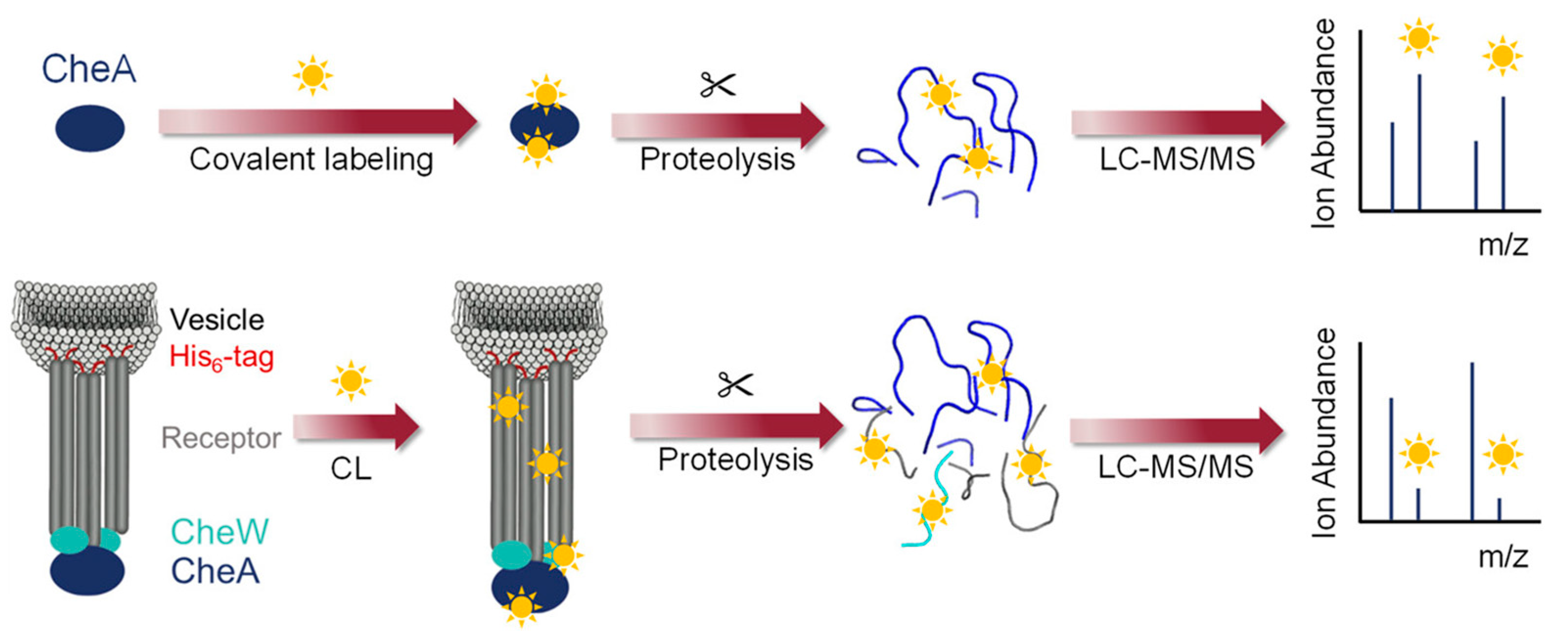
3.2.1. Footprinting Membrane-Associated Proteins
3.2.2. Footprinting Extramembrane Domains
3.2.3. Footprinting Transmembrane Domains
4. Summary and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Escribá, P.V.; González-Ros, J.M.; Goñi, F.M.; Kinnunen, P.K.J.; Vigh, L.; Sánchez-Magraner, L.; Fernández, A.M.; Busquets, X.; Horváth, I.; Barceló-Coblijn, G. Membranes: A Meeting Point for Lipids, Proteins and Therapies. J. Cell. Mol. Med. 2008, 12, 829–875. [Google Scholar] [CrossRef] [PubMed]
- Dessau, M.A.; Modis, Y. Protein Crystallization for X-Ray Crystallography. J. Vis. Exp. 2011, 47, 2285. [Google Scholar] [CrossRef]
- Hagn, F.; Nasr, M.L.; Wagner, G. Assembly of Phospholipid Nanodiscs of Controlled Size for Structural Studies of Membrane Proteins by NMR. Nat. Protoc. 2018, 13, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Christie, S.; Shi, X.; Smith, A.W. Resolving Membrane Protein-Protein Interactions in Live Cells with Pulsed Interleaved Excitation Fluorescence Cross-Correlation Spectroscopy. Acc. Chem. Res. 2020, 53, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Thonghin, N.; Kargas, V.; Clews, J.; Ford, R.C. Cryo-Electron Microscopy of Membrane Proteins. Methods 2018, 147, 176–186. [Google Scholar] [CrossRef]
- Benesch, J.L.P.; Ruotolo, B.T.; Simmons, D.A.; Robinson, C.V. Protein Complexes in the Gas Phase: Technology for Structural Genomics and Proteomics. Chem. Rev. 2007, 107, 3544–3567. [Google Scholar] [CrossRef]
- Konermann, L.; Pan, J.; Liu, Y.-H. Hydrogen Exchange Mass Spectrometry for Studying Protein Structure and Dynamics. Chem. Soc. Rev. 2011, 40, 1224–1234. [Google Scholar] [CrossRef]
- Schmidt, C.; Robinson, C.V. A Comparative Cross-Linking Strategy to Probe Conformational Changes in Protein Complexes. Nat. Protoc. 2014, 9, 2224–2236. [Google Scholar] [CrossRef]
- Xu, G.; Chance, M.R. Hydroxyl Radical-Mediated Modification of Proteins as Probes for Structural Proteomics. Chem. Rev. 2007, 107, 3514–3543. [Google Scholar] [CrossRef]
- Hambly, D.; Gross, M. Laser Flash Photochemical Oxidation to Locate Heme Binding and Conformational Changes in Myoglobin. Int. J. Mass Spectrom. 2007, 259, 124–129. [Google Scholar] [CrossRef]
- Minkoff, B.B.; Blatz, J.M.; Choudhury, F.A.; Benjamin, D.; Shohet, J.L.; Sussman, M.R. Plasma-Generated OH Radical Production for Analyzing Three-Dimensional Structure in Protein Therapeutics. Sci. Rep. 2017, 7, 12946. [Google Scholar] [CrossRef] [PubMed]
- Maleknia, S.D.; Chance, M.R.; Downard, K.M. Electrospray-Assisted Modification of Proteins: A Radical Probe of Protein Structure. Rapid Commun. Mass Spectrom. 1999, 13, 2352–2358. [Google Scholar] [CrossRef]
- Downard, K.M.; Maleknia, S.D. Mass Spectrometry in Structural Proteomics: The Case for Radical Probe Protein Footprinting. TrAC Trends Anal. Chem. 2019, 110, 293–302. [Google Scholar] [CrossRef]
- Sharp, J.S.; Chea, E.E.; Misra, S.K.; Orlando, R.; Popov, M.; Egan, R.W.; Holman, D.; Weinberger, S.R. Flash Oxidation (FOX) System: A Novel Laser-Free Fast Photochemical Oxidation Protein Footprinting Platform. J. Am. Soc. Mass Spectrom. 2021, 32, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Martens, C.; Shekhar, M.; Lau, A.M.; Tajkhorshid, E.; Politis, A. Integrating Hydrogen–Deuterium Exchange Mass Spectrometry with Molecular Dynamics Simulations to Probe Lipid-Modulated Conformational Changes in Membrane Proteins. Nat. Protoc. 2019, 14, 3183–3204. [Google Scholar] [CrossRef]
- Yen, H.-Y.; Jazayeri, A.; Robinson, C.V. G Protein-Coupled Receptor Pharmacology—Insights from Mass Spectrometry. Pharmacol. Rev. 2023, 75, 397–415. [Google Scholar] [CrossRef]
- Keener, J.E.; Zhang, G.; Marty, M.T. Native Mass Spectrometry of Membrane Proteins. Anal. Chem. 2021, 93, 583–597. [Google Scholar] [CrossRef]
- van Dyck, J.F.; Konijnenberg, A.; Sobott, F. Native Mass Spectrometry for the Characterization of Structure and Interactions of Membrane Proteins. In Membrane Protein Structure and Function Characterization: Methods and Protocols; Lacapere, J.-J., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; pp. 205–232. ISBN 978-1-4939-7151-0. [Google Scholar]
- Gault, J.; Liko, I.; Landreh, M.; Shutin, D.; Bolla, J.R.; Jefferies, D.; Agasid, M.; Yen, H.-Y.; Ladds, M.J.G.W.; Lane, D.P.; et al. Combining Native and ‘Omics’ Mass Spectrometry to Identify Endogenous Ligands Bound to Membrane Proteins. Nat. Methods 2020, 17, 505–508. [Google Scholar] [CrossRef]
- Chen, S.; Getter, T.; Salom, D.; Wu, D.; Quetschlich, D.; Chorev, D.S.; Palczewski, K.; Robinson, C.V. Capturing a Rhodopsin Receptor Signalling Cascade across a Native Membrane. Nature 2022, 604, 384–390. [Google Scholar] [CrossRef]
- Javed, W.; Griffiths, D.; Politis, A. Hydrogen/Deuterium Exchange-Mass Spectrometry of Integral Membrane Proteins in Native-like Environments: Current Scenario and the Way Forward. Essays Biochem. 2023, 67, 187–200. [Google Scholar] [CrossRef]
- Jia, R.; Martens, C.; Shekhar, M.; Pant, S.; Pellowe, G.A.; Lau, A.M.; Findlay, H.E.; Harris, N.J.; Tajkhorshid, E.; Booth, P.J.; et al. Hydrogen-Deuterium Exchange Mass Spectrometry Captures Distinct Dynamics upon Substrate and Inhibitor Binding to a Transporter. Nat. Commun. 2020, 11, 6162. [Google Scholar] [CrossRef] [PubMed]
- Reading, E.; Hall, Z.; Martens, C.; Haghighi, T.; Findlay, H.; Ahdash, Z.; Politis, A.; Booth, P.J. Interrogating Membrane Protein Conformational Dynamics within Native Lipid Compositions. Angew. Chem. Int. Ed. 2017, 56, 15654–15657. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, A.N.; Radford, S.E. Mass Spectrometry-Enabled Structural Biology of Membrane Proteins. Methods 2018, 147, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chien, E.Y.T.; Chalmers, M.J.; Pascal, B.D.; Gatchalian, J.; Stevens, R.C.; Griffin, P.R. Dynamics of the β 2 -Adrenergic G-Protein Coupled Receptor Revealed by Hydrogen−Deuterium Exchange. Anal. Chem. 2010, 82, 1100–1108. [Google Scholar] [CrossRef]
- Möller, I.R.; Slivacka, M.; Hausner, J.; Nielsen, A.K.; Pospíšilová, E.; Merkle, P.S.; Lišková, R.; Polák, M.; Loland, C.J.; Kádek, A.; et al. Improving the Sequence Coverage of Integral Membrane Proteins during Hydrogen/Deuterium Exchange Mass Spectrometry Experiments. Anal. Chem. 2019, 91, 10970–10978. [Google Scholar] [CrossRef]
- Calvaresi, V.; Redsted, A.; Norais, N.; Rand, K.D. Hydrogen–Deuterium Exchange Mass Spectrometry with Integrated Size-Exclusion Chromatography for Analysis of Complex Protein Samples. Anal. Chem. 2021, 93, 11406–11414. [Google Scholar] [CrossRef]
- Giladi, M.; Khananshvili, D. Hydrogen-Deuterium Exchange Mass-Spectrometry of Secondary Active Transporters: From Structural Dynamics to Molecular Mechanisms. Front. Pharmacol. 2020, 11, 70. [Google Scholar] [CrossRef]
- Martens, C.; Shekhar, M.; Borysik, A.J.; Lau, A.M.; Reading, E.; Tajkhorshid, E.; Booth, P.J.; Politis, A. Direct Protein-Lipid Interactions Shape the Conformational Landscape of Secondary Transporters. Nat. Commun. 2018, 9, 4151. [Google Scholar] [CrossRef]
- Jia, R.; Bradshaw, R.T.; Calvaresi, V.; Politis, A. Integrating Hydrogen Deuterium Exchange–Mass Spectrometry with Molecular Simulations Enables Quantification of the Conformational Populations of the Sugar Transporter XylE. J. Am. Chem. Soc. 2023, 145, 7768–7779. [Google Scholar] [CrossRef]
- Anderson, K.W.; Gallagher, E.S.; Hudgens, J.W. Automated Removal of Phospholipids from Membrane Proteins for H/D Exchange Mass Spectrometry Workflows. Anal. Chem. 2018, 90, 6409–6412. [Google Scholar] [CrossRef]
- Hammerschmid, D.; Calvaresi, V.; Bailey, C.; Russell Lewis, B.; Politis, A.; Morris, M.; Denbigh, L.; Anderson, M.; Reading, E. Chromatographic Phospholipid Trapping for Automated H/D Exchange Mass Spectrometry of Membrane Protein–Lipid Assemblies. Anal. Chem. 2023, 95, 3002–3011. [Google Scholar] [CrossRef] [PubMed]
- Donnarumma, D.; Maestri, C.; Giammarinaro, P.I.; Capriotti, L.; Bartolini, E.; Veggi, D.; Petracca, R.; Scarselli, M.; Norais, N. Native State Organization of Outer Membrane Porins Unraveled by HDx-MS. J. Proteome Res. 2018, 17, 1794–1800. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Anderson, K.W.; Turko, I.V. Assessment of Extracellular Vesicles Purity Using Proteomic Standards. Anal. Chem. 2017, 89, 11070–11075. [Google Scholar] [CrossRef] [PubMed]
- Manzi, L.; Barrow, A.S.; Hopper, J.T.S.; Kaminska, R.; Kleanthous, C.; Robinson, C.V.; Moses, J.E.; Oldham, N.J. Carbene Footprinting Reveals Binding Interfaces of a Multimeric Membrane-Spanning Protein. Angew. Chem. 2017, 129, 15069–15073. [Google Scholar] [CrossRef]
- Cheng, Z.; Mobley, C.; Misra, S.K.; Gadepalli, R.S.; Hammond, R.I.; Brown, L.S.; Rimoldi, J.M.; Sharp, J.S. Self-Organized Amphiphiles Are Poor Hydroxyl Radical Scavengers in Fast Photochemical Oxidation of Proteins Experiments. J. Am. Soc. Mass Spectrom. 2021, 32, 1155–1161. [Google Scholar] [CrossRef]
- Liu, X.R.; Zhang, M.M.; Gross, M.L. Mass Spectrometry-Based Protein Footprinting for Higher-Order Structure Analysis: Fundamentals and Applications. Chem. Rev. 2020, 120, 4355–4454. [Google Scholar] [CrossRef]
- Gau, B.C.; Sharp, J.S.; Rempel, D.L.; Gross, M.L. Fast Photochemical Oxidation of Protein Footprints Faster than Protein Unfolding. Anal. Chem. 2009, 81, 6563–6571. [Google Scholar] [CrossRef]
- Pan, X.; Vachet, R.W. Membrane Protein Structures And Interactions From Covalent Labeling Coupled With Mass Spectrometry. Mass Spec. Rev. 2022, 41, 51–69. [Google Scholar] [CrossRef]
- Sun, J.; Li, W.; Gross, M.L. Advances in Mass Spectrometry-based Footprinting of Membrane Proteins. Proteomics 2022, 22, 2100222. [Google Scholar] [CrossRef]
- Boes, D.M.; Godoy-Hernandez, A.; McMillan, D.G.G. Peripheral Membrane Proteins: Promising Therapeutic Targets across Domains of Life. Membranes 2021, 11, 346. [Google Scholar] [CrossRef]
- Elucidating the Structure of Membrane Proteins. Available online: https://www.future-science.com/doi/epub/10.2144/btn-2019-0030 (accessed on 26 February 2023).
- Van, Q.N.; López, C.A.; Tonelli, M.; Taylor, T.; Niu, B.; Stanley, C.B.; Bhowmik, D.; Tran, T.H.; Frank, P.H.; Messing, S.; et al. Uncovering a Membrane-Distal Conformation of KRAS Available to Recruit RAF to the Plasma Membrane. Proc. Natl. Acad. Sci. USA 2020, 117, 24258–24268. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Zhang, H.; Gross, M.L.; Blankenship, R.E. Membrane Orientation of the FMO Antenna Protein from Chlorobaculum Tepidum as Determined by Mass Spectrometry-Based Footprinting. Proc. Natl. Acad. Sci. USA 2009, 106, 6134–6139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wen, J.; Huang, R.Y.-C.; Blankenship, R.E.; Gross, M.L. Mass Spectrometry-Based Carboxyl Footprinting of Proteins: Method Evaluation. Int. J. Mass Spectrom. 2012, 312, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.M.; Rempel, D.L.; Gross, M.L. Modeling Data from Titration, Amide H/D Exchange, and Mass Spectrometry to Obtain Protein-Ligand Binding Constants. J. Am. Soc. Mass Spectrom. 2004, 15, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Hambly, D.M.; Gross, M.L. Laser Flash Photolysis of Hydrogen Peroxide to Oxidize Protein Solvent-Accessible Residues on the Microsecond Timescale. J. Am. Soc. Mass Spectrom. 2005, 16, 2057–2063. [Google Scholar] [CrossRef]
- Pan, X.; Tran, T.; Kirsch, Z.J.; Thompson, L.K.; Vachet, R.W. Diethylpyrocarbonate-Based Covalent Labeling Mass Spectrometry of Protein Interactions in a Membrane Complex System. J. Am. Soc. Mass Spectrom. 2023, 34, 82–91. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, H.; Niedzwiedzki, D.M.; Jiang, J.; Blankenship, R.E.; Gross, M.L. Fast Photochemical Oxidation of Proteins Maps the Topology of Intrinsic Membrane Proteins: Light-Harvesting Complex 2 in a Nanodisc. Anal. Chem. 2016, 88, 8827–8834. [Google Scholar] [CrossRef]
- Denisov, I.G.; Sligar, S.G. Nanodiscs in Membrane Biochemistry and Biophysics. Chem. Rev. 2017, 117, 4669–4713. [Google Scholar] [CrossRef]
- Zhou, F.; Yang, Y.; Chemuru, S.; Cui, W.; Liu, S.; Gross, M.; Li, W. Footprinting Mass Spectrometry of Membrane Proteins: Ferroportin Reconstituted in Saposin A Picodiscs. Anal. Chem. 2021, 93, 11370–11378. [Google Scholar] [CrossRef]
- Leney, A.C.; Rezaei Darestani, R.; Li, J.; Nikjah, S.; Kitova, E.N.; Zou, C.; Cairo, C.W.; Xiong, Z.J.; Privé, G.G.; Klassen, J.S. Picodiscs for Facile Protein-Glycolipid Interaction Analysis. Anal. Chem. 2015, 87, 4402–4408. [Google Scholar] [CrossRef]
- Li, J.; Richards, M.R.; Bagal, D.; Campuzano, I.D.G.; Kitova, E.N.; Xiong, Z.J.; Privé, G.G.; Klassen, J.S. Characterizing the Size and Composition of Saposin A Lipoprotein Picodiscs. Anal. Chem. 2016, 88, 9524–9531. [Google Scholar] [CrossRef]
- Shen, G.; Li, S.; Cui, W.; Liu, S.; Yang, Y.; Gross, M.; Li, W. Membrane Protein Structure in Live Cells: Methodology for Studying Drug Interaction by Mass Spectrometry-Based Footprinting. Biochemistry 2018, 57, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Zhang, B.; Cui, W.; Gross, M.L. Laser-Initiated Radical Trifluoromethylation of Peptides and Proteins: Application to Mass-Spectrometry-Based Protein Footprinting. Angew. Chem. Int. Ed. 2017, 56, 14007–14010. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Asuru, A.; Kiselar, J.; Mathai, G.; Chance, M.R.; Gross, M.L. Fast Protein Footprinting by X-Ray Mediated Radical Trifluoromethylation. J. Am. Soc. Mass Spectrom. 2020, 31, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Li, S.; Li, W.; Gross, M.L. Carbocation Footprinting of Soluble and Transmembrane Proteins. Anal. Chem. 2021, 93, 13101–13105. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, V.L.; Vachet, R.W. Probing Protein Structure by Amino Acid-Specific Covalent Labeling and Mass Spectrometry. Mass Spectrom. Rev. 2009, 28, 785–815. [Google Scholar] [CrossRef]
- Schmidt, C.; Macpherson, J.A.; Lau, A.M.; Tan, K.W.; Fraternali, F.; Politis, A. Surface Accessibility and Dynamics of Macromolecular Assemblies Probed by Covalent Labeling Mass Spectrometry and Integrative Modeling. Anal. Chem. 2017, 89, 1459–1468. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, Z.; Zhang, J.; Dou, T.; Chen, J.; Ge, G.; Zhu, S.; Wang, F. Prediction of Ligand Modulation Patterns on Membrane Receptors via Lysine Reactivity Profiling. Chem. Commun. 2019, 55, 4311–4314. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Membrane Proteins. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Carpenter, E.P.; Beis, K.; Cameron, A.D.; Iwata, S. Overcoming the Challenges of Membrane Protein Crystallography. Curr. Opin. Struct. Biol. 2008, 18, 581–586. [Google Scholar] [CrossRef]
- Frankel, L.K.; Sallans, L.; Bellamy, H.; Goettert, J.S.; Limbach, P.A.; Bricker, T.M. Radiolytic Mapping of Solvent-Contact Surfaces in Photosystem II of Higher Plants. J. Biol. Chem. 2013, 288, 23565–23572. [Google Scholar] [CrossRef]
- Zhang, B.; Rempel, D.L.; Gross, M.L. Protein Footprinting by Carbenes on a Fast Photochemical Oxidation of Proteins (FPOP) Platform. J. Am. Soc. Mass Spectrom. 2016, 27, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liu, X.R.; Li, S.; He, P.; Li, W.; Gross, M.L. Nanoparticles and Photochemistry for Native-like Transmembrane Protein Footprinting. Nat. Commun. 2021, 12, 7270. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Guo, C.; Li, W.; Gross, M.L. Free-Radical Membrane Protein Footprinting by Photolysis of Perfluoroisopropyl Iodide Partitioned to Detergent Micelle by Sonication. Angew. Chem. Int. Ed. 2021, 60, 8867–8873. [Google Scholar] [CrossRef]
- Guo, C.; Cheng, M.; Li, W.; Gross, M.L. Diethylpyrocarbonate Footprints a Membrane Protein in Micelles. J. Am. Soc. Mass Spectrom. 2021, 32, 2636–2643. [Google Scholar] [CrossRef] [PubMed]
- Espino, J.A.; Jones, L.M. Illuminating Biological Interactions with in Vivo Protein Footprinting. Anal. Chem. 2019, 91, 6577–6584. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.-C.; Li, W.; Sun, J.; Gross, M.L. Advances in Mass Spectrometry on Membrane Proteins. Membranes 2023, 13, 457. https://doi.org/10.3390/membranes13050457
Yang H-C, Li W, Sun J, Gross ML. Advances in Mass Spectrometry on Membrane Proteins. Membranes. 2023; 13(5):457. https://doi.org/10.3390/membranes13050457
Chicago/Turabian StyleYang, Hsin-Chieh, Weikai Li, Jie Sun, and Michael L. Gross. 2023. "Advances in Mass Spectrometry on Membrane Proteins" Membranes 13, no. 5: 457. https://doi.org/10.3390/membranes13050457
APA StyleYang, H.-C., Li, W., Sun, J., & Gross, M. L. (2023). Advances in Mass Spectrometry on Membrane Proteins. Membranes, 13(5), 457. https://doi.org/10.3390/membranes13050457