
Immunogenicity of Escherichia coli Outer Membrane Vesicles: Elucidation of Humoral Responses against OMV-Associated Antigens

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Studies
2.2. Bacterial Strains and Cultures
2.3. Cloning Strategies
2.4. Production and Purification of Recombinant Proteins
2.5. Western Blot and Enzyme-Linked Immunosorbent Assay (ELISA)
2.6. OMVs Purification and SDS-PAGE
2.7. Software and Data Analysis
3. Results
3.1. Theoretical Considerations

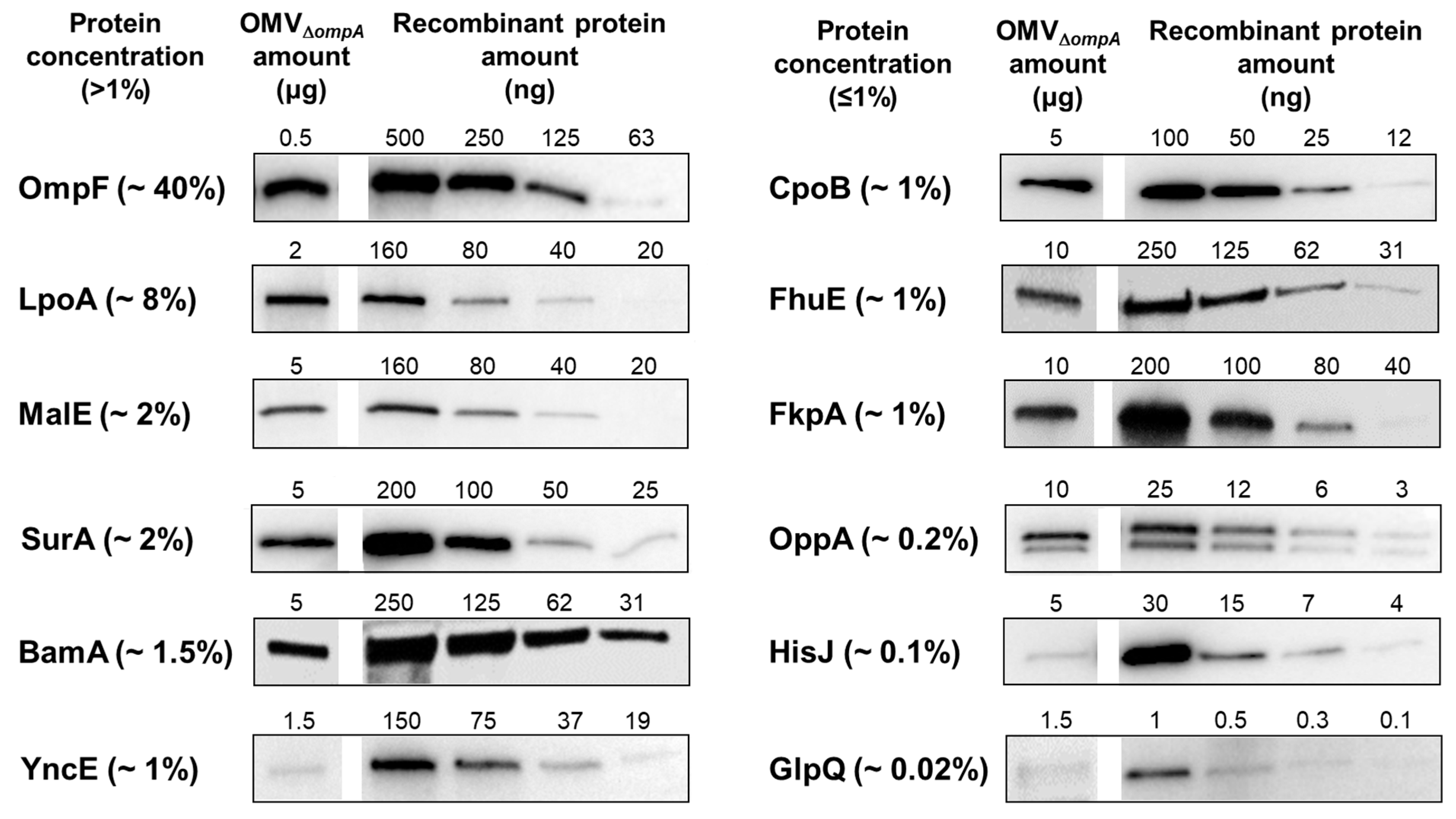
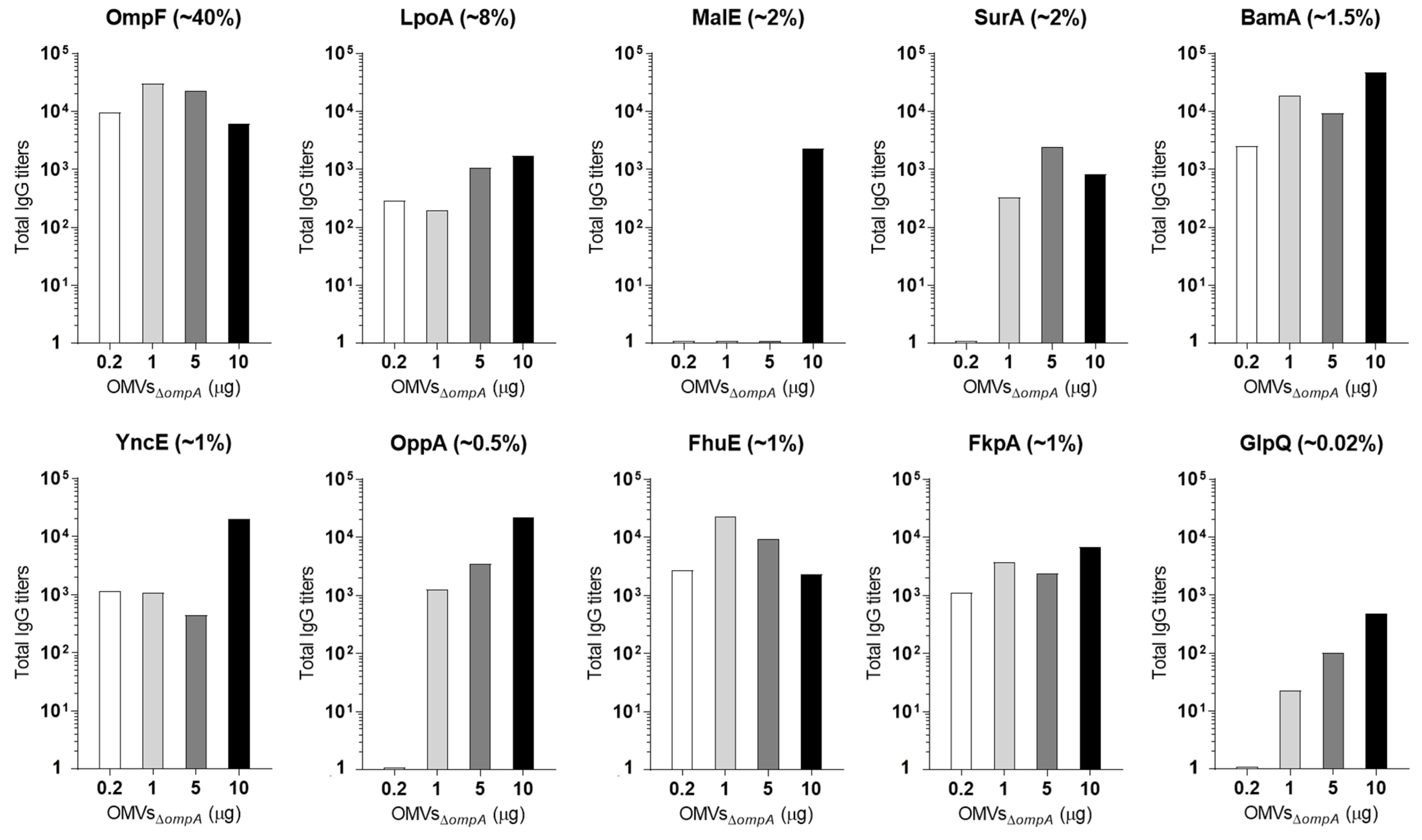
3.2. OMV Immunization Elicits Antibodies against OMV-Associated Proteins Expressed at Concentrations ≥1% of Total OMV Proteins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Annotation | Compartment 1 | Molecular Weight (kDa) |
|---|---|---|---|
| cpoB | Cell division coordinator | PP | 25.4 |
| lpoA | Penicillin-binding protein activator | LP | 70 |
| pal | Peptidoglycan-associated lipoprotein | LP | 16.6 |
| rlpA | Endolytic peptidoglycan transglycosylase | LP | 35.7 |
| surA | Chaperone | PP | 45 |
| yncE | Uncharacterized protein | PP | 35.3 |
| fkpA | FKBP-type peptidyl-prolyl cis-trans isomerase | PP | 26.2 |
| glpQ | Glycerophosphodiester phosphodiesterase | PP | 38.2 |
| malE | Maltose/maltodextrin-binding periplasmic protein | PP | 40.7 |
| ydcL | Uncharacterized lipoprotein | LP | 22.4 |
| hisJ | Histidine-binding periplasmic protein | PP | 26.2 |
| oppA | Periplasmic oligopeptide-binding protein | PP | 58.4 |
| ybiS | Probable L,D-transpeptidase | PP | 30.8 |
| bamA | Outer membrane protein assembly factor | OM | 88.4 |
| ompF | Outer membrane porine F | OM | 37 |
| fhuE | FhuE receptor | OM | 77.4 |
3.3. A Total of 5 μg of OMVs Are Generally Sufficient to Elicit Plateau Levels of Antibodies against OMV Proteins Expressed at Concentrations ≥2%
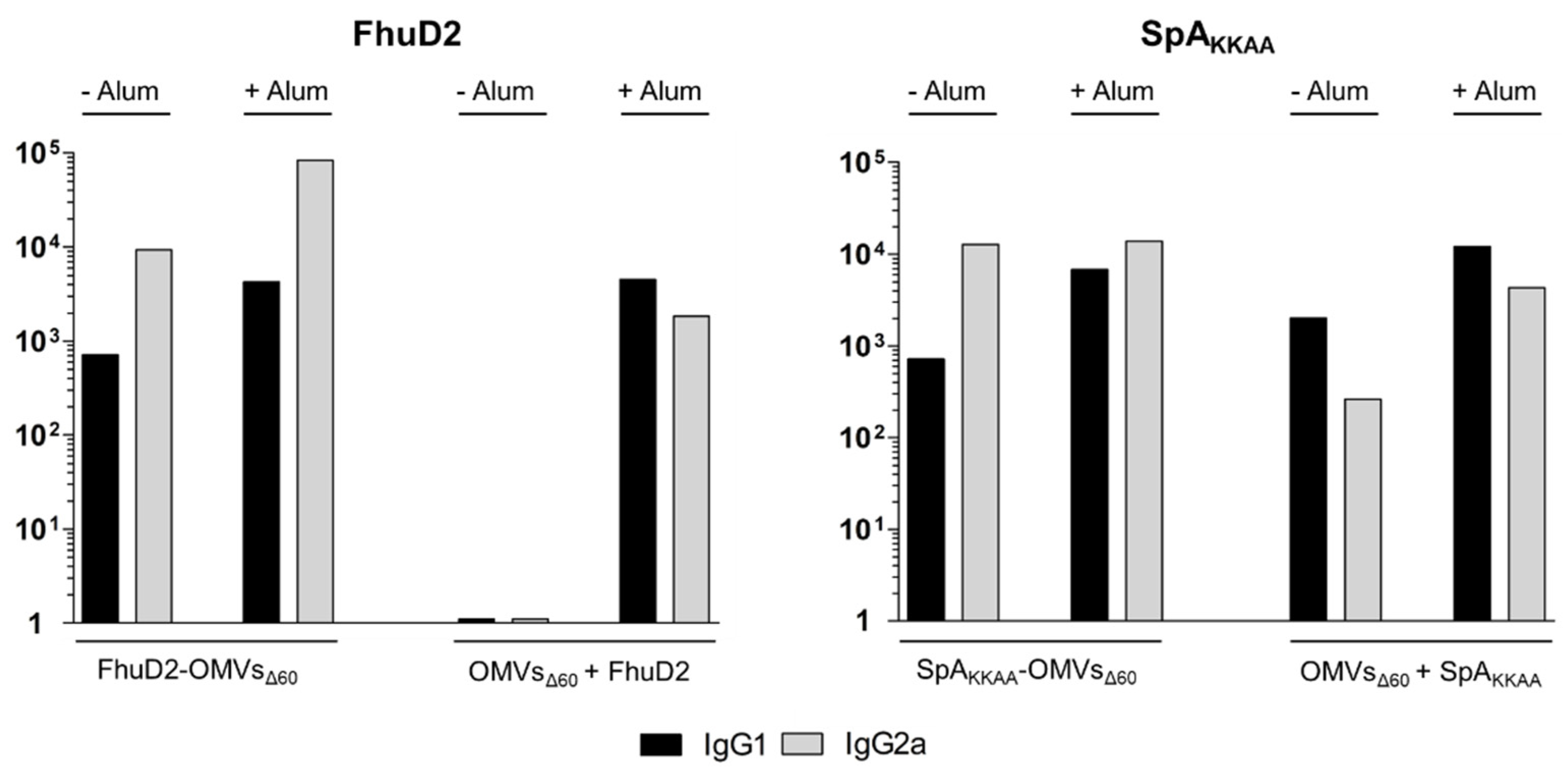
3.4. Comparison between Antigen-Specific Antibodies Elicited by Engineered OMVs and OMVs + Antigen Mixtures
3.5. The OMV–Alum Combination Guarantees High Immunogenicity of Any OMV–Antigen Mixture
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kulp, A.; Kuehn, M.J. Biological Functions and Biogenesis of Secreted Bacterial Outer Membrane Vesicles. Annu. Rev. Microbiol. 2010, 64, 163–184. [Google Scholar] [CrossRef] [PubMed]
- Ellis, T.N.; Kuehn, M.J. Virulence and Immunomodulatory Roles of Bacterial Outer Membrane Vesicles. Microbiol. Mol. Biol. Rev. 2010, 74, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Moshiri, A.; Dashtbani-Roozbehani, A.; Peerayeh, S.N.; Siadat, S.D. Outer membrane vesicle: A macromolecule with multifunctional activity. Hum. Vaccin. Immunother. 2012, 8, 953–955. [Google Scholar] [CrossRef] [PubMed]
- Ellis, T.N.; Leiman, S.A.; Kuehn, M.J. Naturally produced outer membrane vesicles from Pseudomonas aeruginosa elicit a potent innate immune response via combined sensing of both lipopolysaccharide and protein components. Infect. Immun. 2010, 78, 3822–3831. [Google Scholar] [CrossRef]
- Kaparakis, M.; Turnbull, L.; Carneiro, L.; Firth, S.; Coleman, H.A.; Parkington, H.C.; Le Bourhis, L.; Karrar, A.; Viala, J.; Mak, J.; et al. Bacterial membrane vesicles deliver peptidoglycan to NOD1 in epithelial cells. Cell Microbiol. 2010, 12, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.J.; Osterrieder, N.; Metzger, S.M.; Buckles, E.; Doody, A.M.; DeLisa, M.P.; Putnam, D. Delivery of foreign antigens by engineered outer membrane vesicle vaccines. Proc. Natl. Acad. Sci. USA 2010, 107, 3099–3104. [Google Scholar] [CrossRef]
- Kim, O.Y.; Hong, B.S.; Park, K.-S.; Yoon, Y.J.; Choi, S.J.; Lee, W.H.; Roh, T.-Y.; Lötvall, J.; Kim, Y.-K.; Gho, Y.S. Immunization with Escherichia coli Outer Membrane Vesicles Protects Bacteria-Induced Lethality via Th1 and Th17 Cell Responses. J. Immunol. 2013, 190, 4092–4102. [Google Scholar] [CrossRef]
- Kesty, N.C.; Kuehn, M.J. Incorporation of Heterologous Outer Membrane and Periplasmic Proteins into Escherichia coli Outer Membrane Vesicles. J. Biol. Chem. 2004, 279, 2069–2076. [Google Scholar] [CrossRef] [PubMed]
- Fantappiè, L.; de Santis, M.; Chiarot, E.; Carboni, F.; Bensi, G.; Jousson, O.; Margarit, I.; Grandi, G. Antibody-mediated immunity induced by engineered Escherichia coli OMVs carrying heterologous antigens in their lumen. J. Extracell. Vesicles 2014, 3, 24015. [Google Scholar] [CrossRef]
- Schroeder, J.; Aebischer, T. Recombinant outer membrane vesicles to augment antigen-specific live vaccine responses. Vaccine 2009, 27, 6748–6754. [Google Scholar] [CrossRef]
- Fantappiè, L.; Irene, C.; De Santis, M.; Armini, A.; Gagliardi, A.; Tomasi, M.; Parri, M.; Cafardi, V.; Bonomi, S.; Ganfini, L.; et al. Some gram-negative lipoproteins keep their surface topology when transplanted from one species to another and deliver foreign polypeptides to the bacterial surface. Mol. Cell. Proteom. 2017, 16, 1348–1364. [Google Scholar] [CrossRef]
- Granoff, D.M. Review of meningococcal group B vaccines. Clin. Infect. Dis. 2010, 50, S54–S65. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, G.; Garaguso, I.; Adu-Bobie, J.; Doro, F.; Taddei, A.R.; Biolchi, A.; Brunelli, B.; Giuliani, M.M.; Pizza, M.; Norais, N.; et al. Outer membrane vesicles from group B Neisseria meningitidis Δgna33 mutant: Proteomic and immunological comparison with detergent-derived outer membrane vesicles. Proteomics 2006, 6, 1856–1866. [Google Scholar] [CrossRef] [PubMed]
- Bernadac, A.; Gavioli, M.; Lazzaroni, J.C.; Raina, S.; Lloubès, R. Escherichia coli tol-pal Mutants Form Outer Membrane Vesicles. J. Bacteriol. 1998, 180, 4872–4878. [Google Scholar] [CrossRef]
- Deatherage, B.L.; Lara, J.C.; Bergsbaken, T.; Barrett, S.L.R.; Lara, S.; Cookson, B.T. Biogenesis of bacterial membrane vesicles. Mol. Microbiol. 2009, 72, 1395–1407. [Google Scholar] [CrossRef]
- McBroom, A.J.; Kuehn, M.J. Release of outer membrane vesicles by Gram-negative bacteria is a novel envelope stress response. Mol. Microbiol. 2007, 63, 545–558. [Google Scholar] [CrossRef]
- Scorza, F.B.; Colucci, A.M.; Maggiore, L.; Sanzone, S.; Rossi, O.; Ferlenghi, I.; Pesce, I.; Caboni, M.; Norais, N.; Di Cioccio, V.; et al. High yield production process for Shigella outer membrane particles. PLoS ONE 2012, 7, e35616. [Google Scholar] [CrossRef]
- Ladhani, S.N.; Ramsay, M.; Borrow, R.; Riordan, A.; Watson, J.M.; Pollard, A.J. Enter B and W: Two new meningococcal vaccine programmes launched. Arch. Dis. Child. 2016, 101, 91–95. [Google Scholar] [CrossRef]
- Serruto, D.; Bottomley, M.J.; Ram, S.; Giuliani, M.M.; Rappuoli, R. The new multicomponent vaccine against meningococcal serogroup B, 4CMenB: Immunological, functional and structural characterization of the antigens. Vaccine 2012, 30, B87–B97. [Google Scholar] [CrossRef]
- Gerke, C.; Colucci, A.M.; Giannelli, C.; Sanzone, S.; Vitali, C.G.; Sollai, L.; Rossi, O.; Martin, L.B.; Auerbach, J.; Di Cioccio, V.; et al. Production of a Shigella sonnei vaccine based on generalized modules for membrane antigens (GMMA), 1790GAHB. PLoS ONE 2015, 10, e0134478. [Google Scholar] [CrossRef] [PubMed]
- Rossi, O.; Caboni, M.; Negrea, A.; Necchi, F.; Alfini, R.; Micoli, F.; Saul, A.; MacLennan, C.A.; Rondini, S.; Gerke, C. Toll-Like Receptor Activation by Generalized Modules for Membrane Antigens from Lipid A Mutants of Salmonella enterica Serovars Typhimurium and Enteritidis. Clin. Vaccine Immunol. 2016, 23, 304–314. [Google Scholar] [CrossRef]
- Orenstein, W.; Offit, P.A.; Edwards, K.M.; Plotkin, S.A. Plotkin’s Vaccines, 7th ed.; Elsevier Health Sciences: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Huang, W.; Wang, S.; Yao, Y.; Xia, Y.; Yang, X.; Li, K.; Sun, P.; Liu, C.; Sun, W.; Bai, H.; et al. Employing Escherichia coli-derived outer membrane vesicles as an antigen delivery platform elicits protective immunity against Acinetobacter baumannii infection. Sci. Rep. 2016, 6, 37242. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, K.; Daleke-Schermerhorn, M.H.; Jong, W.S.; Hagen-Jongman, C.M.T.; van Opzeeland, F.; Simonetti, E.; Luirink, J.; de Jonge, M.I. Salmonella outer membrane vesicles displaying high densities of pneumococcal antigen at the surface offer protection against colonization. Vaccine 2015, 33, 2022–2029. [Google Scholar] [CrossRef]
- Irene, C.; Fantappiè, L.; Caproni, E.; Zerbini, F.; Anesi, A.; Tomasi, M.; Zanella, I.; Stupia, S.; Prete, S.; Valensin, S.; et al. Bacterial outer membrane vesicles engineered with lipidated antigens as a platform for Staphylococcus aureus vaccine. Proc. Natl. Acad. Sci. USA 2019, 116, 21780–21788. [Google Scholar] [CrossRef] [PubMed]
- Klock, H.E.; Lesley, S.A. The Polymerase Incomplete Primer Extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. Methods Mol. Biol. 2009, 498, 91–103. [Google Scholar] [PubMed]
- Zanella, I.; König, E.; Tomasi, M.; Gagliardi, A.; Frattini, L.; Fantappiè, L.; Irene, C.; Zerbini, F.; Caproni, E.; Isaac, S.J.; et al. Proteome-minimized outer membrane vesicles from Escherichia coli as a generalized vaccine platform. J. Extracell. Vesicles 2021, 10, e12066. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.Y.; Park, H.T.; Dinh, N.T.H.; Choi, S.J.; Lee, J.; Kim, J.H.; Lee, S.-W.; Gho, Y.S.; Thi, N. Bacterial outer membrane vesicles suppress tumor by interferon-γ-mediated antitumor response. Nat. Commun. 2017, 8, 626. [Google Scholar] [CrossRef]
- Thoma, J.; Manioglu, S.; Kalbermatter, D.; Bosshart, P.D.; Fotiadis, D.; Müller, D.J. Protein-enriched outer membrane vesicles as a native platform for outer membrane protein studies. Commun. Biol. 2018, 1, 23. [Google Scholar] [CrossRef] [PubMed]
- Bhakdi, S.; Tranum-Jensen, J. Alpha-Toxin of Staphylococcus aureus. Microbiol. Rev. 1991, 55, 733–751. [Google Scholar] [CrossRef]
- Mariotti, P.; Malito, E.; Biancucci, M.; Lo Surdo, P.; Mishra, R.P.N.; Nardi-Dei, V.; Savino, S.; Nissum, M.; Spraggon, G.; Grandi, G.; et al. Structural and functional characterization of the Staphylococcus aureus virulence factor and vaccine candidate FhuD2. Biochem. J. 2013, 449, 683–693. [Google Scholar] [CrossRef]
- Falugi, F.; Kim, H.K.; Missiakas, D.M.; Schneewind, O. Role of protein a in the evasion of host adaptive immune responses by Staphylococcus aureus. mBio 2013, 4, e00575-13. [Google Scholar] [CrossRef]
- Weller, U.; Muller, L.; Messner, M.; Palmer, M.; Valeva, A.; Tranum-Jensen, J.; Agrawal, P.; Biermann, C.; Dobereiner, A.; Kehoe, M.A.; et al. Expression of active streptolysin O in Escherichia coli as a maltose-binding-protein-streptolysin-O fusion protein. The N-terminal 70 amino acids are not required for hemolytic activity. Eur. J. Biochem. 1996, 236, 34–39. [Google Scholar] [CrossRef]
- Zingaretti, C.; Falugi, F.; Nardi-Dei, V.; Pietrocola, G.; Mariani, M.; Liberatori1 Marilena Gallotta, S.; Tontini, M.; Tani, C.; Speziale, P.; Grandi, G.; et al. Streptococcus pyogenes 95 SpyCEP: A chemokine-inactivating protease with unique structural and biochemical features. FASEB J. 2010, 24, 2839–2848. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Moyle, P.M. Bioconjugation Approaches to Producing Subunit Vaccines Composed of Protein or Peptide Antigens and Covalently Attached Toll-Like Receptor Ligands. Bioconjugate Chem. 2017, 29, 572–586. [Google Scholar] [CrossRef]
- Cookson, B.T.; Alaniz, R.C.; Deatherage, B.L.; Lara, J.C. Protective Immunity In Vivo and T Cell Responses, and Stimulate Potently Activate Dendritic Cells, Prime B That Salmonella typhimurium Facsimiles of Membrane Vesicles Are Immunogenic. J. Immunol. Ref. 2022, 179, 7692–7701. [Google Scholar]
- Adriani, R.; Gargari, S.L.M.; Nazarian, S.; Sarvary, S.; Noroozi, N. Immunogenicity of Vibrio cholerae outer membrane vesicles secreted at various environmental conditions. Vaccine 2018, 36, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Qasim, M.; Wrage, M.; Nüse, B.; Mattner, J. Shigella Outer Membrane Vesicles as Promising Targets for Vaccination. Int. J. Mol. Sci. 2022, 23, 994. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, R.; Fernã¡Ndez, S.; Zayas, C.; Acosta, A.; Sarmiento, M.E.; Ferro, V.A.; Rosenqvist, E.; Campa, C.; Cardoso, D.; Garcia, L.; et al. Bacterial outer membrane vesicles and vaccine applications. Front. Immunol. 2014, 5, 121. [Google Scholar] [CrossRef]
- Bai, X.; Findlow, J.; Borrow, R. Recombinant protein meningococcal serogroup B vaccine combined with outer membrane vesicles. Expert Opin. Biol. Ther. 2011, 11, 969–985. [Google Scholar] [CrossRef]
- Cheng, K.; Zhao, R.; Li, Y.; Qi, Y.; Wang, Y.; Zhang, Y.; Qin, H.; Qin, Y.; Chen, L.; Li, C.; et al. Bioengineered bacteria-derived outer membrane vesicles as a versatile antigen display platform for tumor vaccination via Plug-and-Display technology. Nat. Commun. 2021, 12, 2041. [Google Scholar] [CrossRef]
- Zeng, M.Y.; Cisalpino, D.; Varadarajan, S.; Hellman, J.; Warren, H.S.; Cascalho, M.; Inohara, N.; Núñez, G. Gut Microbiota-Induced Immunoglobulin G Controls Systemic Infection by Symbiotic Bacteria and Pathogens. Immunity 2016, 44, 647–658. [Google Scholar] [CrossRef] [PubMed]


| Protein Name | Immunogenicity | Concentration (%) | |
|---|---|---|---|
| WB | ELISA | ||
| OmpF | −/+ | + | 40 |
| LpoA | + | + | 8 |
| MalE | + | + | 2 |
| SurA | + | + | 2 |
| BamA | + | + | 1.5 |
| YncE | + | + | 1 |
| CpoB | −/+ | + | 1 |
| FhuE | + | + | 1 |
| FkpA | + | + | 1 |
| OppA | + | + | 0.2 |
| HisJ | − | − | 0.1 |
| GlpQ | − | + | 0.02 |
| RlpA | + | + | ND |
| YbiS | − | − | ND |
| YdcL | −/+ | − | ND |
| Pal | − | + | ND |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Croia, L.; Boscato Sopetto, G.; Zanella, I.; Caproni, E.; Gagliardi, A.; Tamburini, S.; König, E.; Benedet, M.; Di Lascio, G.; Corbellari, R.; et al. Immunogenicity of Escherichia coli Outer Membrane Vesicles: Elucidation of Humoral Responses against OMV-Associated Antigens. Membranes 2023, 13, 882. https://doi.org/10.3390/membranes13110882
Croia L, Boscato Sopetto G, Zanella I, Caproni E, Gagliardi A, Tamburini S, König E, Benedet M, Di Lascio G, Corbellari R, et al. Immunogenicity of Escherichia coli Outer Membrane Vesicles: Elucidation of Humoral Responses against OMV-Associated Antigens. Membranes. 2023; 13(11):882. https://doi.org/10.3390/membranes13110882
Chicago/Turabian StyleCroia, Lorenzo, Giulia Boscato Sopetto, Ilaria Zanella, Elena Caproni, Assunta Gagliardi, Silvia Tamburini, Enrico König, Mattia Benedet, Gabriele Di Lascio, Riccardo Corbellari, and et al. 2023. "Immunogenicity of Escherichia coli Outer Membrane Vesicles: Elucidation of Humoral Responses against OMV-Associated Antigens" Membranes 13, no. 11: 882. https://doi.org/10.3390/membranes13110882
APA StyleCroia, L., Boscato Sopetto, G., Zanella, I., Caproni, E., Gagliardi, A., Tamburini, S., König, E., Benedet, M., Di Lascio, G., Corbellari, R., Grandi, A., Tomasi, M., & Grandi, G. (2023). Immunogenicity of Escherichia coli Outer Membrane Vesicles: Elucidation of Humoral Responses against OMV-Associated Antigens. Membranes, 13(11), 882. https://doi.org/10.3390/membranes13110882