

The Infarct-Reducing Effect of the δ2 Opioid Receptor Agonist Deltorphin II: The Molecular Mechanism

 , ,
, ,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Coronary Artery Occlusion and Reperfusion
2.3. Detection of Infarct Size and the Area at Risk
2.4. Pharmacological Agents
2.5. Experimental Protocol
2.6. Detection of Cyclic Nucleotides
2.7. Statistical Analysis
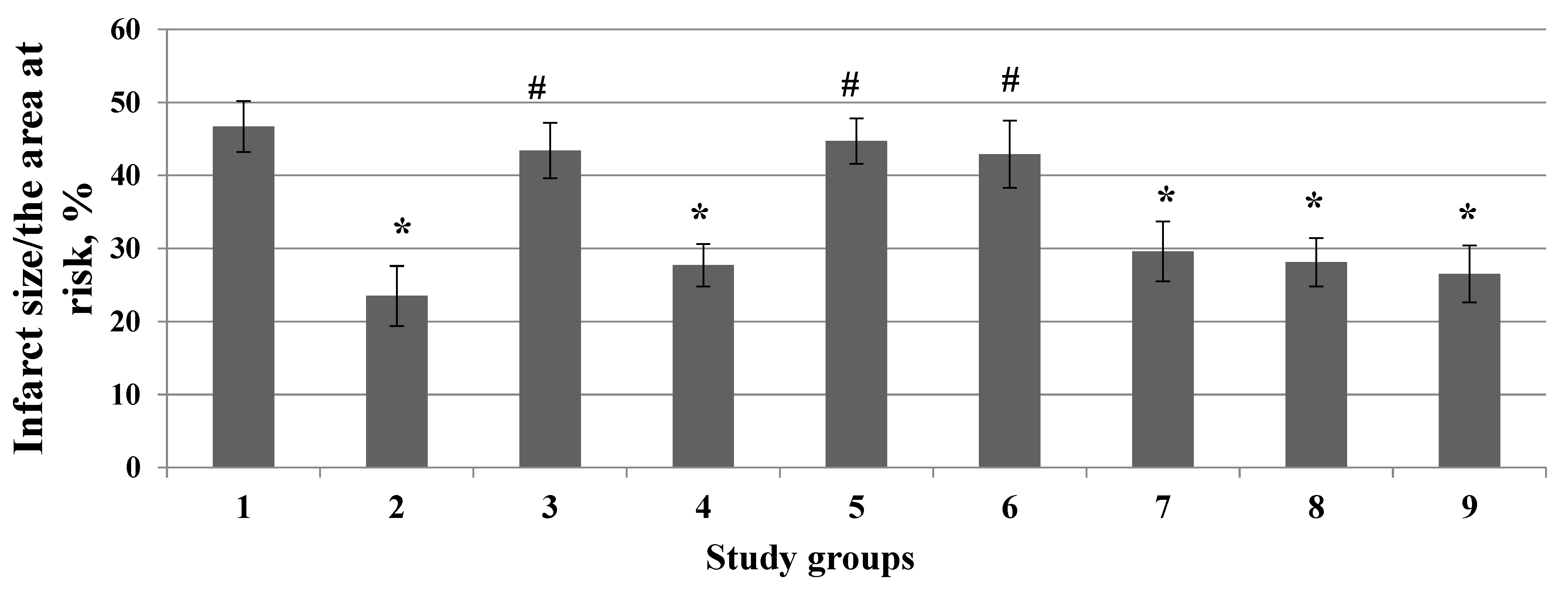
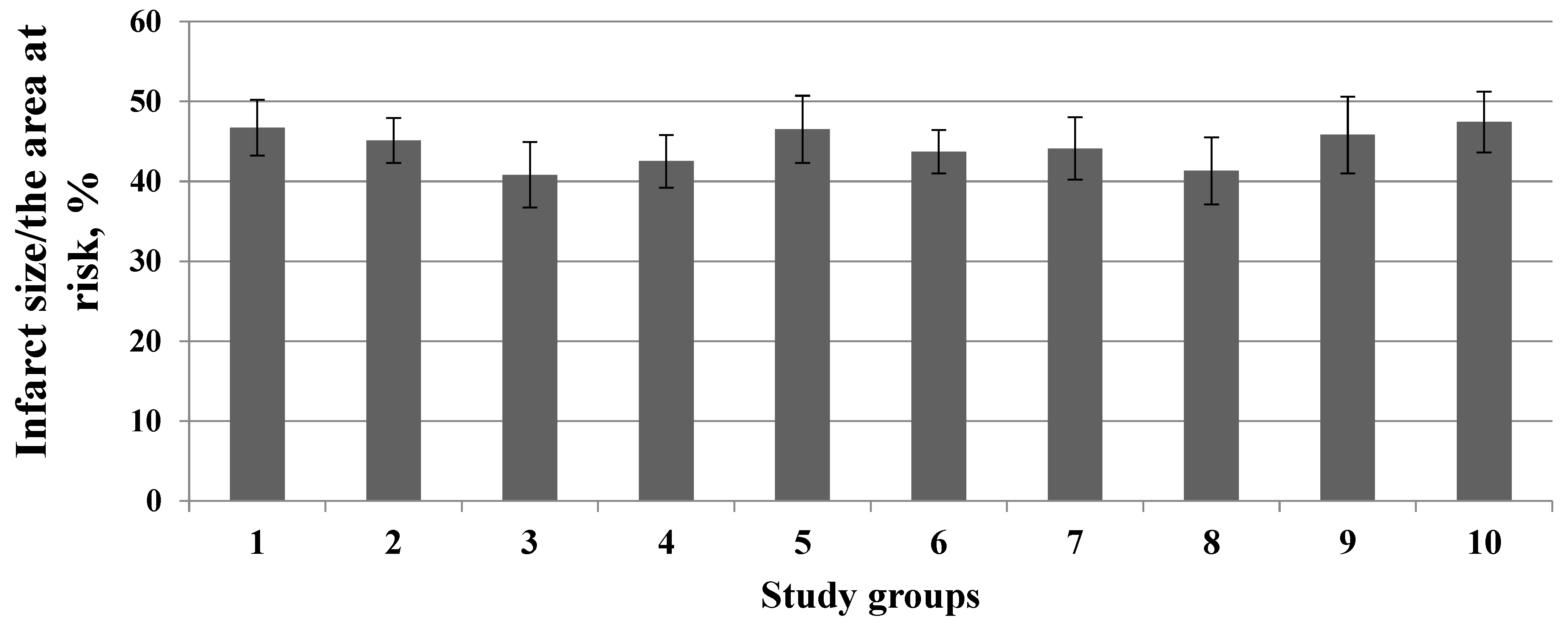
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olier, I.; Sirker, A.; Hildick-Smith, D.; Kinnaird, T.; Ludman, P.; de Belder, M.A.; Baumbach, A.; Byrne, J.; Rashid, M.; Curzen, N.; et al. British cardiovascular intervention society and the national institute for cardiovascular outcomes research. Association of different antiplatelet therapies with mortality after primary percutaneous coronary intervention. Heart 2018, 104, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- McCartney, P.J.; Berry, C. Redefining successful primary PCI. Eur. Heart J. Cardiovasc. Imaging 2019, 20, 133–135. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Samangouei, P.; Kalkhoran, S.B.; Hausenloy, D.J. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J. Mol. Cell Cardiol. 2015, 78, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Maslov, L.N.; Mukhomedzyanov, A.V.; Tsibulnikov, S.Y.; Suleiman, M.S.; Khaliulin, I.; Oeltgen, P.R. Activation of peripheral δ2-opioid receptor prevents reperfusion heart injury. Eur. J. Pharmacol. 2021, 907, 174302. [Google Scholar] [CrossRef] [PubMed]
- Maslov, L.N.; Lishmanov, Y.B.; Oeltgen, P.R.; Barzakh, E.I.; Krylatov, A.V.; Govindaswami, M.; Brown, S.A. Activation of peripheral δ2 opioid receptors increases cardiac tolerance to ischemia/reperfusion injury: Involvement of protein kinase C, NO-synthase, KATP channels and the autonomic nervous system. Life Sci. 2009, 84, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Maslov, L.N.; Naryzhnaia, N.V.; Tsibulnikov, S.Y.; Kolar, F.; Zhang, Y.; Wang, H.; Gusakova, A.M.; Lishmanov, Y.B. Role of endogenous opioid peptides in the infarct size-limiting effect of adaptation to chronic continuous hypoxia. Life Sci. 2013, 93, 373–379. [Google Scholar] [CrossRef]
- Neckár, J.; Sźárszoi, O.; Herget, J.; Ostádal, B.; Kolár, F. Cardioprotective effect of chronic hypoxia is blunted by concomitant hypercapnia. Physiol. Res. 2003, 52, 171–175. [Google Scholar]
- Ebrahim, Z.; Yellon, D.M.; Baxter, G.F. Bradykinin elicits “second window” myocardial protection in rat heart through an NO-dependent mechanism. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, 1458–1464. [Google Scholar] [CrossRef]
- Zatta, A.J.; Kin, H.; Lee, G.; Wang, N.; Jiang, R.; Lust, R.; Reeves, J.G.; Mykytenko, J.; Guyton, R.A.; Zhao, Z.Q.; et al. Infarct-sparing effect of myocardial postconditioning is dependent on protein kinase C signaling. Cardiovasc. Res. 2006, 70, 315–334. [Google Scholar] [CrossRef]
- Lasley, R.D.; Keith, B.J.; Kristo, G.; Yoshimura, Y.; Mentzer, R.M., Jr. Delayed adenosine A1 receptor preconditioning in rat myocardium is MAPK dependent but iNOS independent. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, 785–791. [Google Scholar] [CrossRef][Green Version]
- Gross, E.R.; Hsu, A.K.; Gross, G.J. The JAK/STAT pathway is essential for opioid-induced cardioprotection: JAK2 as a mediator of STAT3, Akt, and GSK-3β. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Fettiplace, M.R.; Kowal, K.; Ripper, R.; Young, A.; Lis, K.; Rubinstein, I.; Bonini, M.; Minshall, R.; Weinberg, G. Insulin signaling in bupivacaine-induced cardiac toxicity: Sensitization during recovery and potentiation by lipid emulsion. Anesthesiology 2016, 124, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Sun, Y.; An, J.Z.; Wang, C.; Qiao, S.G. Sevoflurane preconditioning confers delayed cardioprotection by upregulating AMP-activated protein kinase levels to restore autophagic flux in ischemia-reperfusion rat hearts. Med. Sci. Monit. 2020, 26, e922176. [Google Scholar] [CrossRef] [PubMed]
- Schindler, U.; Strobel, H.; Schönafinger, K.; Linz, W.; Löhn, M.; Martorana, P.A.; Rütten, H.; Schindler, P.W.; Busch, A.E.; Sohn, M.; et al. Biochemistry and pharmacology of novel anthranilic acid derivatives activating heme-oxidized soluble guanylyl cyclase. Mol. Pharmacol. 2006, 69, 1260–1268. [Google Scholar] [CrossRef]
- Dorsch, M.; Behmenburg, F.; Raible, M.; Blase, D.; Grievink, H.; Hollmann, M.W.; Heinen, A.; Huhn, R. Morphine-induced preconditioning: Involvement of protein kinase A and mitochondrial permeability transition pore. PLoS ONE 2016, 11, e0151025. [Google Scholar] [CrossRef]
- Nithipatikom, K.; Moore, J.M.; Isbell, M.A.; Falck, J.R.; Gross, G.J. Epoxyeicosatrienoic acids in cardioprotection: Ischemic versus reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, 537–542. [Google Scholar] [CrossRef]
- Fryer, R.M.; Eells, J.T.; Hsu, A.K.; Henry, M.M.; Gross, G.J. Ischemic preconditioning in rats: Role of mitochondrial KATP channel in preservation of mitochondrial function. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, 305–312. [Google Scholar] [CrossRef]
- Fryer, R.M.; Hsu, A.K.; Nagase, H.; Gross, G.J. Opioid-induced cardioprotection against myocardial infarction and arrhythmias: Mitochondrial versus sarcolemmal ATP-sensitive potassium channels. J. Pharmacol. Exp. Ther. 2000, 294, 451–457. [Google Scholar]
- Smith, B.L.; McLeay, L.M.; Embling, P.P. Effect of the mycotoxins penitrem, paxilline and lolitrem B on the electromyographic activity of skeletal and gastrointestinal smooth muscle of sheep. Res. Vet. Sci. 1996, 62, 111–116. [Google Scholar] [CrossRef]
- Tsutsumi, Y.M.; Yokoyama, T.; Horikawa, Y.; Roth, D.M.; Patel, H.H. Reactive oxygen species trigger ischemic and pharmacological postconditioning: In vivo and in vitro characterization. Life Sci. 2007, 81, 1223–1227. [Google Scholar] [CrossRef]
- Pınar, N.; Kaplan, M.; Özgür, T.; Özcan, O. Ameliorating effects of tempol on methotrexate-induced liver injury in rats. Biomed. Pharmacother. 2018, 102, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Maslov, L.N.; Lishmanov, Y.B. The anti-arrhythmic effect of D-Ala2, Leu5,Arg6-enkephalin and its possible mechanism. Int. J. Cardiol. 1993, 40, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Peart, J.N.; Gross, E.R.; Reichelt, M.E.; Hsu, A.; Headrick, J.P.; Gross, G.J. Activation of kappa-opioid receptors at reperfusion affords cardioprotection in both rat and mouse hearts. Basic. Res. Cardiol. 2008, 103, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, H.; Li, T.; Zhang, B. Involvement of adenosine monophosphate-activated protein kinase in morphine-induced cardioprotection. J. Surg. Res. 2011, 169, 179–187. [Google Scholar] [CrossRef]
- Gross, G.J.; Hsu, A.; Nithipatikom, K.; Pfeiffer, A.W.; Bobrova, I.; Bissessar, E. Acute and chronic cardioprotection by the enkephalin analogue, Eribis peptide 94, is mediated via activation of nitric oxide synthase and adenosine triphosphate-regulated potassium channels. Pharmacology 2012, 90, 110–116. [Google Scholar] [CrossRef]
- Zhang, Y.; Irwin, M.G.; Wong, T.M.; Chen, M.; Cao, C.M. Remifentanil preconditioning confers cardioprotection via cardiac kappa- and delta-opioid receptors. Anesthesiology 2005, 102, 371–378. [Google Scholar] [CrossRef]
- Cao, C.M.; Chen, M.; Wong, T.M. The KCa channel as a trigger for the cardioprotection induced by kappa-opioid receptor stimulation—Its relationship with protein kinase C. Br. J. Pharmacol. 2009, 145, 984–991. [Google Scholar] [CrossRef]
- Krylatov, A.V.; Maslov, L.N.; Voronkov, N.S.; Boshchenko, A.A.; Popov, S.V.; Gomez, L.; Wang, H.; Jaggi, A.S.; Downey, J.M. Reactive oxygen species as intracellular signaling molecules in the cardiovascular system. Curr. Cardiol. Rev. 2018, 14, 290–300. [Google Scholar] [CrossRef]
- Krylatov, A.V.; Tsibulnikov, S.Y.; Mukhomedzyanov, A.V.; Boshchenko, A.A.; Goldberg, V.E.; Jaggi, A.S.; Erben, R.G.; Maslov, L.N. The role of natriuretic peptides in the regulation of cardiac tolerance to ischemia/reperfusion and postinfarction heart remodeling. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 131–148. [Google Scholar] [CrossRef]
- Yang, X.M.; Philipp, S.; Downey, J.M.; Cohen, M.V. Atrial natriuretic peptide administered just prior to reperfusion limits infarction in rabbit hearts. Basic Res. Cardiol. 2006, 10, 311–318. [Google Scholar] [CrossRef]
- Maslov, L.N.; Khaliulin, I.; Oeltgen, P.R.; Naryzhnaya, N.V.; Pei, J.M.; Brown, S.A.; Lishmanov, Y.B.; Downey, J.M. Prospects for creation of cardioprotective and antiarrhythmic drugs based on opioid receptor agonists. Med. Res. Rev. 2016, 36, 871–923. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Before Ischemia | Before Reperfusion | After 30 min Reperfusion | After 2 h Reperfusion | |
|---|---|---|---|---|
| Heart rate (beats/min) | ||||
| Control | 365 ± 4 | 359 ± 4 | 352 ± 5 | 342 ± 9 |
| Deltorphin II | 367 ± 5 | 362 ± 5 | 354 ± 4 | 348 ± 8 |
| Naltrexone | 359 ± 8 | 353 ± 6 | 348 ± 5 | 340 ± 7 |
| Naloxone methiodide | 355 ± 6 | 349 ± 4 | 345 ± 5 | 336 ± 6 |
| TIPP [ψ] | 366 ± 3 | 358 ± 4 | 353 ± 4 | 344 ± 7 |
| Naltriben | 358 ± 5 | 352 ± 4 | 346 ± 3 | 337 ± 7 |
| L-NAME | 366 ± 5 | 335 ± 5 * | 318 ± 4 * | 304 ± 9 * |
| Chelerythrine | 361 ± 4 | 354 ± 3 | 347 ± 5 | 339 ± 6 |
| Rottlerin | 370 ± 3 | 363 ± 5 | 356 ± 4 | 345 ± 7 |
| PD098059 | 362 ± 4 | 355 ± 4 | 350 ± 5 | 341 ± 8 |
| AG490 | 367 ± 3 | 340 ± 4 * | 337 ± 5 * | 321 ± 7 * |
| Wortmannin | 360 ± 5 | 354 ± 3 | 349 ± 4 | 339 ± 9 |
| ODQ | 365 ± 4 | 359 ± 4 | 354 ± 5 | 345 ± 7 |
| H-89 | 367 ± 5 | 361 ± 5 | 356 ± 4 | 344 ± 8 |
| Compound C | 364 ± 4 | 360 ± 6 | 354 ± 5 | 346 ± 9 |
| Glibenclamide | 359 ± 4 | 354 ± 3 | 349 ± 5 | 338 ± 6 |
| 5-HD | 368 ± 3 | 363 ± 3 | 358 ± 4 | 347 ± 5 |
| HMR1098 | 362 ± 4 | 356 ± 5 | 350 ± 4 | 336 ± 7 |
| Paxilline | 357 ± 5 | 352 ± 4 | 345 ± 5 | 335 ± 8 |
| Atractyloside | 366 ± 4 | 360 ± 4 | 356 ± 3 | 345 ± 5 |
| MPG | 361 ± 3 | 355 ± 4 | 347 ± 4 | 337 ± 7 |
| Tempol | 356 ± 5 | 351 ± 4 | 344 ± 3 | 334 ± 8 |
| Mean systolic blood pressure (mmHg) | ||||
| Control | 126 ± 5 | 122 ± 3 | 119 ± 4 | 114 ± 6 |
| Deltorphin II | 122 ± 4 | 119 ± 4 | 115 ± 5 | 109 ± 7 |
| Naltrexone | 125 ± 3 | 120 ± 5 | 117 ± 3 | 111 ± 5 |
| Naloxone methiodide | 128 ± 4 | 124 ± 4 | 120 ± 5 | 113 ± 7 |
| TIPP[ψ] | 124 ± 3 | 119 ± 5 | 116 ± 3 | 111 ± 8 |
| Naltriben | 120 ± 3 | 117 ± 4 | 113 ± 3 | 106 ± 5 |
| L-NAME | 123 ± 4 | 145 ± 3 * | 155 ± 5 * | 161 ± 6 * |
| Chelerythrine | 126 ± 3 | 123 ± 4 | 118 ± 4 | 113 ± 5 |
| Rottlerin | 125 ± 4 | 121 ± 4 | 116 ± 5 | 111 ± 6 |
| PD098059 | 128 ± 3 | 124 ± 5 | 120 ± 3 | 112 ± 7 |
| AG490 | 127 ± 4 | 140 ± 3 * | 138 ± 4 * | 134 ± 5 * |
| Wortmannin | 126 ± 5 | 121 ± 4 | 117 ± 3 | 113 ± 7 |
| ODQ | 122 ± 3 | 117 ± 3 | 114 ± 5 | 108 ± 5 |
| H-89 | 127 ± 4 | 123 ± 4 | 118 ± 5 | 113 ± 8 |
| Compound C | 124 ± 6 | 121 ± 4 | 119 ± 3 | 114 ± 5 |
| Glibenclamide | 128 ± 5 | 125 ± 5 | 121 ± 7 | 117 ± 6 |
| 5-HD | 121 ± 3 | 118 ± 4 | 114 ± 6 | 109 ± 5 |
| HMR1098 | 125 ± 3 | 120 ± 5 | 117 ± 3 | 112 ± 7 |
| Paxilline | 119 ± 6 | 115 ± 4 | 111 ± 5 | 105 ± 8 |
| Atractyloside | 128 ± 3 | 123 ± 4 | 118 ± 4 | 113 ± 6 |
| MPG | 124 ± 4 | 121 ± 3 | 117 ± 4 | 110 ± 7 |
| Tempol | 121 ± 5 | 116 ± 4 | 112 ± 3 | 107 ± 8 |
| Group | cAMP (nmol/g) | cGMP (nmol/g) |
|---|---|---|
| Control | 18.2 ± 1.1 | 9 ± 1.1 |
| I/R | 13.1 ± 3.9 * | 24 ± 4.4 * |
| Deltorphin II | 11.8 ± 2.8 # | 44 ± 4.7 *,# |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popov, S.V.; Mukhomedzyanov, A.V.; Maslov, L.N.; Naryzhnaya, N.V.; Kurbatov, B.K.; Prasad, N.R.; Singh, N.; Fu, F.; Azev, V.N. The Infarct-Reducing Effect of the δ2 Opioid Receptor Agonist Deltorphin II: The Molecular Mechanism. Membranes 2023, 13, 63. https://doi.org/10.3390/membranes13010063
Popov SV, Mukhomedzyanov AV, Maslov LN, Naryzhnaya NV, Kurbatov BK, Prasad NR, Singh N, Fu F, Azev VN. The Infarct-Reducing Effect of the δ2 Opioid Receptor Agonist Deltorphin II: The Molecular Mechanism. Membranes. 2023; 13(1):63. https://doi.org/10.3390/membranes13010063
Chicago/Turabian StylePopov, Sergey V., Alexandr V. Mukhomedzyanov, Leonid N. Maslov, Natalia V. Naryzhnaya, Boris K. Kurbatov, N. Rajendra Prasad, Nirmal Singh, Feng Fu, and Viacheslav N. Azev. 2023. "The Infarct-Reducing Effect of the δ2 Opioid Receptor Agonist Deltorphin II: The Molecular Mechanism" Membranes 13, no. 1: 63. https://doi.org/10.3390/membranes13010063
APA StylePopov, S. V., Mukhomedzyanov, A. V., Maslov, L. N., Naryzhnaya, N. V., Kurbatov, B. K., Prasad, N. R., Singh, N., Fu, F., & Azev, V. N. (2023). The Infarct-Reducing Effect of the δ2 Opioid Receptor Agonist Deltorphin II: The Molecular Mechanism. Membranes, 13(1), 63. https://doi.org/10.3390/membranes13010063