

γ-Core Guided Antibiotic Design Based on Human Enteric Defensin 5

 , , , , and
, , , , and
Abstract
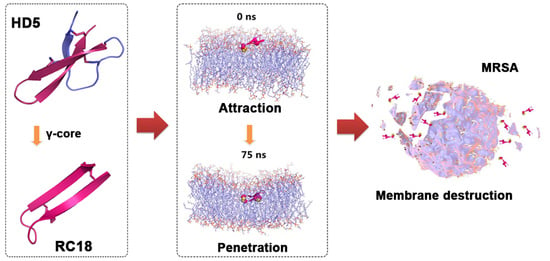
1. Introduction
2. Materials and Methods
2.1. Peptide Synthesis
2.2. Virtual Colony Count Antibacterial Assay
2.3. Circular Dichroism (CD) Spectroscopy
2.4. Broth Microdilution Assay
2.5. Biolayer Interferometry (BLI)
2.6. Zeta Potential Determination
2.7. NPN Uptake Assay
2.8. Inner Membrane Permeabilization Assay
2.9. Scanning Electron Microscopy (SEM)
2.10. Molecular Dynamics Simulation (MDS)
2.11. Toxicological Evaluation
2.12. Mouse Peritoneal Infection Model
2.13. Statistical Analysis
3. Results and Discussion
3.1. AMP Design and Antimicrobial Assessment
3.2. RC18 Attraction to the Bacterial Membrane
3.3. Bacterial Membrane Disruption Induced by RC18
3.4. MDS Modelling of the Membrane Disruption by RC18
3.5. Toxicological Evaluation of RC18
3.6. In Vivo Antibacterial Evaluation of RC18
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mancuso, G.; Midiri, A.; Gerace, E.; Biondo, C. Bacterial antibiotic resistance: The most critical pathogens. Pathogens 2021, 10, 1310. [Google Scholar] [CrossRef] [PubMed]
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Piddock, L.J.V. Reflecting on the final report of the O’Neill review on antimicrobial resistance. Lancet Infect. Dis. 2016, 16, 767–768. [Google Scholar] [CrossRef] [PubMed]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Ding, J.; Liao, C.; Xu, J.; Liu, X.; Lu, W. Defensins: The natural peptide antibiotic. Adv. Drug Deliv. Rev. 2021, 179, 114008. [Google Scholar] [CrossRef]
- Lehrer, R.I.; Lu, W. α-defensins in human innate immunity. Immunol. Rev. 2012, 245, 84–112. [Google Scholar] [CrossRef]
- Ganz, T.; Selsted, M.E.; Szklarek, D.; Harwig, S.S.; Daher, K.; Bainton, D.F.; Lehrer, R.I. Defensins. Natural peptide antibiotics of human neutrophils. J. Clin. Investig. 1985, 76, 1427–1435. [Google Scholar] [CrossRef]
- Wilde, C.G.; Griffith, J.E.; Marra, M.N.; Snable, J.L.; Scott, R.W. Purification and characterization of human neutrophil peptide 4, a novel member of the defensin family. J. Biol. Chem. 1989, 264, 11200–11203. [Google Scholar] [CrossRef]
- Jones, D.E.; Bevins, C.L. Paneth cells of the human small intestine express an antimicrobial peptide gene. J. Biol. Chem. 1992, 267, 23216–23225. [Google Scholar] [CrossRef]
- Jones, D.E.; Bevins, C.L. Defensin-6 mRNA in human paneth cells: Implications for antimicrobial peptides in host defense of the human bowel. FEBS Lett. 1993, 315, 187–192. [Google Scholar] [CrossRef]
- Ericksen, B.; Wu, Z.; Lu, W.; Lehrer, R.I. Antibacterial activity and specificity of the six human {alpha}-defensins. Antimicrob. Agents Chemother. 2005, 49, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.L.; Ouellette, A.J.; Satchell, D.P.; Ayabe, T.; Lopez-Boado, Y.S.; Stratman, J.L.; Hultgren, S.J.; Matrisian, L.M.; Parks, W.C. Regulation of intestinal alpha-defensin activation by the metalloproteinase matrilysin in innate host defense. Science 1999, 286, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Salzman, N.H.; Ghosh, D.; Huttner, K.M.; Paterson, Y.; Bevins, C.L. Protection against enteric salmonellosis in transgenic mice expressing a human intestinal defensin. Nature 2003, 422, 522–526. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, E.; Burks, S.R.; Li, X.; Kao, J.P.; Lu, W. Structure-dependent functional properties of human defensin 5. FEBS Lett. 2007, 581, 515–520. [Google Scholar] [CrossRef]
- Wang, C.; Zhao, G.; Wang, S.; Chen, Y.; Gong, Y.; Chen, S.; Xu, Y.; Hu, M.; Wang, X.; Zeng, H.; et al. A simplified derivative of human defensin 5 with potent and efficient activity against multidrug-resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 2018, 62, e01504–e15017. [Google Scholar] [CrossRef]
- Chen, F.; Tang, Y.; Zheng, H.; Xu, Y.; Wang, J.; Wang, C. Roles of the conserved amino acid residues in reduced human defensin 5: Cysteine and arginine are indispensable for its antibacterial action and LPS neutralization. ChemMedChem 2019, 14, 1457–1465. [Google Scholar] [CrossRef]
- Yount, N.Y.; Yeaman, M.R. Multidimensional signatures in antimicrobial peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 7363–7368. [Google Scholar] [CrossRef]
- Colicchio, R.; Nigro, E.; Colavita, I.; Pagliuca, C.; Di Maro, S.; Tomassi, S.; Scaglione, E.; Carbone, F.; Carriero, M.V.; Matarese, G.; et al. A novel smaller β-defensin-derived peptide is active against multidrug-resistant bacterial strains. FASEB J. 2021, 35, e22026. [Google Scholar] [CrossRef]
- Sonderegger, C.; Váradi, G.; Galgóczy, L.; Kocsubé, S.; Posch, W.; Borics, A.; Dubrac, S.; Tóth, G.K.; Wilflingseder, D.; Marx, F. The evolutionary conserved γ-core motif influences the anti-candida activity of the Penicillium chrysogenum antifungal protein PAF. Front. Microbiol. 2018, 9, 1655. [Google Scholar] [CrossRef]
- Gao, B.; Zhu, S. A fungal defensin targets the SARS-CoV-2 spike receptor-binding domain. J. Fungi 2021, 7, 553. [Google Scholar] [CrossRef]
- Wang, C.; Wang, S.; Li, D.; Wei, D.Q.; Zhao, J.; Wang, J. Human intestinal defensin 5 inhibits SARS-CoV-2 invasion by cloaking ACE2. Gastroenterology 2020, 159, 1145–1147. [Google Scholar] [CrossRef]
- Wang, C.; Shen, M.; Gohain, N.; Tolbert, W.D.; Chen, F.; Zhang, N.; Yang, K.; Wang, A.; Su, Y.; Cheng, T. Design of a potent antibiotic peptide based on the active region of human defensin 5. J. Med. Chem. 2015, 58, 3083–3093. [Google Scholar] [CrossRef]
- Wu, M.; Hancock, R.E. Interaction of the cyclic antimicrobial cationic peptide bactenecin with the outer and cytoplasmic membrane. J. Biol. Chem. 1999, 274, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Strahl, H.; Errington, J. Bacterial membranes: Structure, domains, and function. Annu. Rev. Microbiol. 2017, 71, 519–538. [Google Scholar] [CrossRef] [PubMed]
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef]
- Pogozheva, I.D.; Armstrong, G.A.; Kong, L.; Hartnagel, T.J.; Carpino, C.A.; Gee, S.E.; Picarello, D.M.; Rubin, A.S.; Lee, J.; Park, S.; et al. Comparative molecular dynamics simulation studies of realistic eukaryotic, prokaryotic, and archaeal membranes. J. Chem. Inf. Model. 2022, 62, 1036–1051. [Google Scholar] [CrossRef]
- Han, S.; Zhao, G.; Wei, Z.; Chen, Y.; Zhao, J.; He, Y.; He, Y.J.; Gao, J.; Chen, S.; Du, C.; et al. An angiotensin-converting enzyme-2-derived heptapeptide GK-7 for SARS-CoV-2 spike blockade. Peptides 2021, 145, 170638. [Google Scholar] [CrossRef]
- Wang, C.; Shen, M.; Zhang, N.; Wang, S.; Xu, Y.; Chen, S.; Chen, F.; Yang, K.; He, T.; Wang, A.; et al. Reduction impairs the antibacterial activity but benefits the LPS neutralization ability of human enteric defensin 5. Sci. Rep. 2016, 6, 22875. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wanniarachchi, Y.A.; Kaczmarek, P.; Wan, A.; Nolan, E.M. Human defensin 5 disulfide array mutants: Disulfide bond deletion attenuates antibacterial activity against Staphylococcus aureus. Biochemistry 2011, 50, 8005–8017. [Google Scholar] [CrossRef] [PubMed]
- Avitabile, C.; Capparelli, R.; Rigano, M.M.; Fulgione, A.; Barone, A.; Pedone, C.; Romanelli, A. Antimicrobial peptides from plants: Stabilization of the γ core of a tomato defensin by intramolecular disulfide bond. J. Pept. Sci. 2013, 19, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Chandrababu, K.B.; Ho, B.; Yang, D. Structure, dynamics, and activity of an all-cysteine mutated human beta defensin-3 peptide analogue. Biochemistry 2009, 48, 6052–6061. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Chen, Y.; He, Y.; Chen, F.; Gong, Y.; Chen, S.; Xu, Y.; Su, Y.; Wang, C.; Wang, J. Succinylated casein-coated peptide-mesoporous silica nanoparticles as an antibiotic against intestinal bacterial infection. Biomater. Sci. 2019, 7, 2440–2451. [Google Scholar] [CrossRef]
- Luo, G.; Zhang, J.; Wang, H.; Sun, Y.; Cheng, B.; Xu, Z.; Zhang, Y.; Li, H.; Lu, W.; Nemeth, E.; et al. Human defensin-inspired discovery of peptidomimetic antibiotics. Proc. Natl. Acad. Sci. USA 2022, 119, e2117283119. [Google Scholar] [CrossRef]
- Kimura, R.; Shibata, M.; Koeda, S.; Miyagawa, A.; Yamamura, H.; Mizuno, T. Development of new antimicrobial agents from cationic PG-surfactants containing oligo-Lys peptides. Bioconjug. Chem. 2018, 29, 4072–4082. [Google Scholar] [CrossRef]
- Machuqueiro, M.; Campos, S.R.; Soares, C.M.; Baptista, A.M. Membrane-induced conformational changes of kyotorphin revealed by molecular dynamics simulations. J. Phys. Chem. B 2010, 114, 11659–11667. [Google Scholar] [CrossRef]
- Hristova, K.; Wimley, W.C. A look at arginine in membranes. J. Membr. Biol. 2011, 239, 49–56. [Google Scholar] [CrossRef]
- Lee, E.; Shin, A.; Jeong, K.W.; Jin, B.; Jnawali, H.N.; Shin, S.; Shin, S.Y.; Kim, Y. Role of phenylalanine and valine10 residues in the antimicrobial activity and cytotoxicity of piscidin-1. PLoS ONE 2014, 9, e114453. [Google Scholar] [CrossRef]
- Wojciechowska, M.; Macyszyn, J.; Miszkiewicz, J.; Grzela, R.; Trylska, J. Stapled anoplin as an antibacterial agent. Front. Microbiol. 2021, 12, 772038. [Google Scholar] [CrossRef] [PubMed]
- Paray, B.A.; Ahmad, A.; Khan, J.M.; Taufiq, F.; Pathan, A.; Malik, A.; Ahmed, M.Z. The role of the multifunctional antimicrobial peptide melittin in gene delivery. Drug Discov. Today 2021, 26, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Budagavi, D.P.; Chugh, A. Antibacterial properties of latarcin 1 derived cell-penetrating peptides. Eur. J. Pharm. Sci. 2018, 115, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, D.; Roy, N.; Kulkarni, O.; Nanajkar, N.; Datey, A.; Ravichandran, S.; Thakur, C.; T., S.; Aprameya, I.V.; Sarma, S.P.; et al. Ω76: A designed antimicrobial peptide to combat carbapenem- and tigecycline-resistant Acinetobacter baumannii. Sci. Adv. 2019, 5, eaax1946. [Google Scholar] [CrossRef]
- Roy, R.N.; Lomakin, I.B.; Gagnon, M.G.; Steitz, T.A. The mechanism of inhibition of protein synthesis by the proline-rich peptide oncocin. Nat. Struct. Mol. Biol. 2015, 22, 466–469. [Google Scholar] [CrossRef]
- Panteleev, P.V.; Safronova, V.N.; Kruglikov, R.N.; Bolosov, I.A.; Bogdanov, I.V.; Ovchinnikova, T.V. A novel proline-rich cathelicidin from the alpaca vicugna pacos with potency to combat antibiotic-resistant bacteria: Mechanism of action and the functional role of the c-terminal region. Membranes 2022, 12, 515. [Google Scholar] [CrossRef]
- Shah, P.; Chen, C.S. Systematical screening of intracellular protein targets of polyphemusin-I using Escherichia coli proteome microarrays. Int. J. Mol. Sci. 2021, 22, 9158. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimicrobial Agents | A. baumannii | S. aureus | ||
|---|---|---|---|---|
| MDRAB | ATCC19606 | MRSA(ATCC43300) | ATCC25923 | |
| HD5 | 320 (89.3 μM ) | 320 (89.3 μM ) | 160 (44.7 μM ) | 160 (44.7 μM ) |
| RC18 | 160 (71.6 μM) | 160 (71.6 μM) | 40 (17.9 μM) | 40 (17.9 μM) |
| CTX a | 640 | 10 | >640 | ≤2.5 |
| CIP b | 320 | ≤2.5 | 160 | ≤2.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, G.; Jia, C.; Zhu, C.; Fang, M.; Li, C.; Chen, Y.; He, Y.; Han, S.; He, Y.; Gao, J.; et al. γ-Core Guided Antibiotic Design Based on Human Enteric Defensin 5. Membranes 2023, 13, 51. https://doi.org/10.3390/membranes13010051
Zhao G, Jia C, Zhu C, Fang M, Li C, Chen Y, He Y, Han S, He Y, Gao J, et al. γ-Core Guided Antibiotic Design Based on Human Enteric Defensin 5. Membranes. 2023; 13(1):51. https://doi.org/10.3390/membranes13010051
Chicago/Turabian StyleZhao, Gaomei, Changsheng Jia, Cheng Zhu, Minchao Fang, Chenwenya Li, Yin Chen, Yingjuan He, Songling Han, Yongwu He, Jining Gao, and et al. 2023. "γ-Core Guided Antibiotic Design Based on Human Enteric Defensin 5" Membranes 13, no. 1: 51. https://doi.org/10.3390/membranes13010051
APA StyleZhao, G., Jia, C., Zhu, C., Fang, M., Li, C., Chen, Y., He, Y., Han, S., He, Y., Gao, J., Wang, T., Wang, C., & Wang, J. (2023). γ-Core Guided Antibiotic Design Based on Human Enteric Defensin 5. Membranes, 13(1), 51. https://doi.org/10.3390/membranes13010051