
Characterization and Roles of Membrane Lipids in Fatty Liver Disease


 and
and 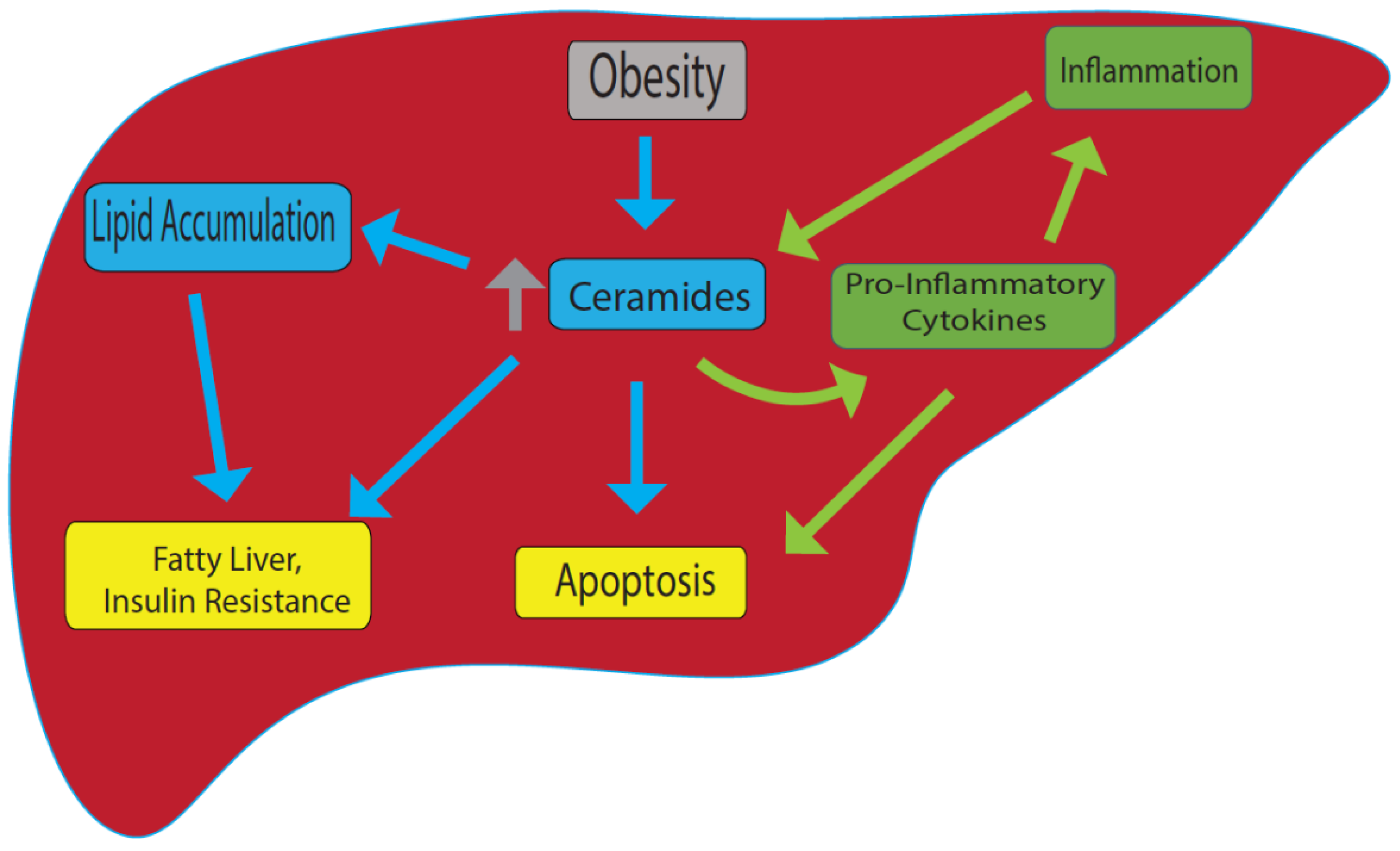{kind=link}
{kind=link}
Abstract
1. Introduction
2. Role of Sphingomyelin-Mediated Ceramide in Fatty Liver Disease
2.1. Hepatotoxicity of Ceramides in the Liver
2.2. Ceramides and Hepatic Inflammation
2.3. Ceramides and Hepatocyte Cell Death
2.4. Ceramides and Hepatic Fibrosis
3. Role of Glycolipids in NAFLD
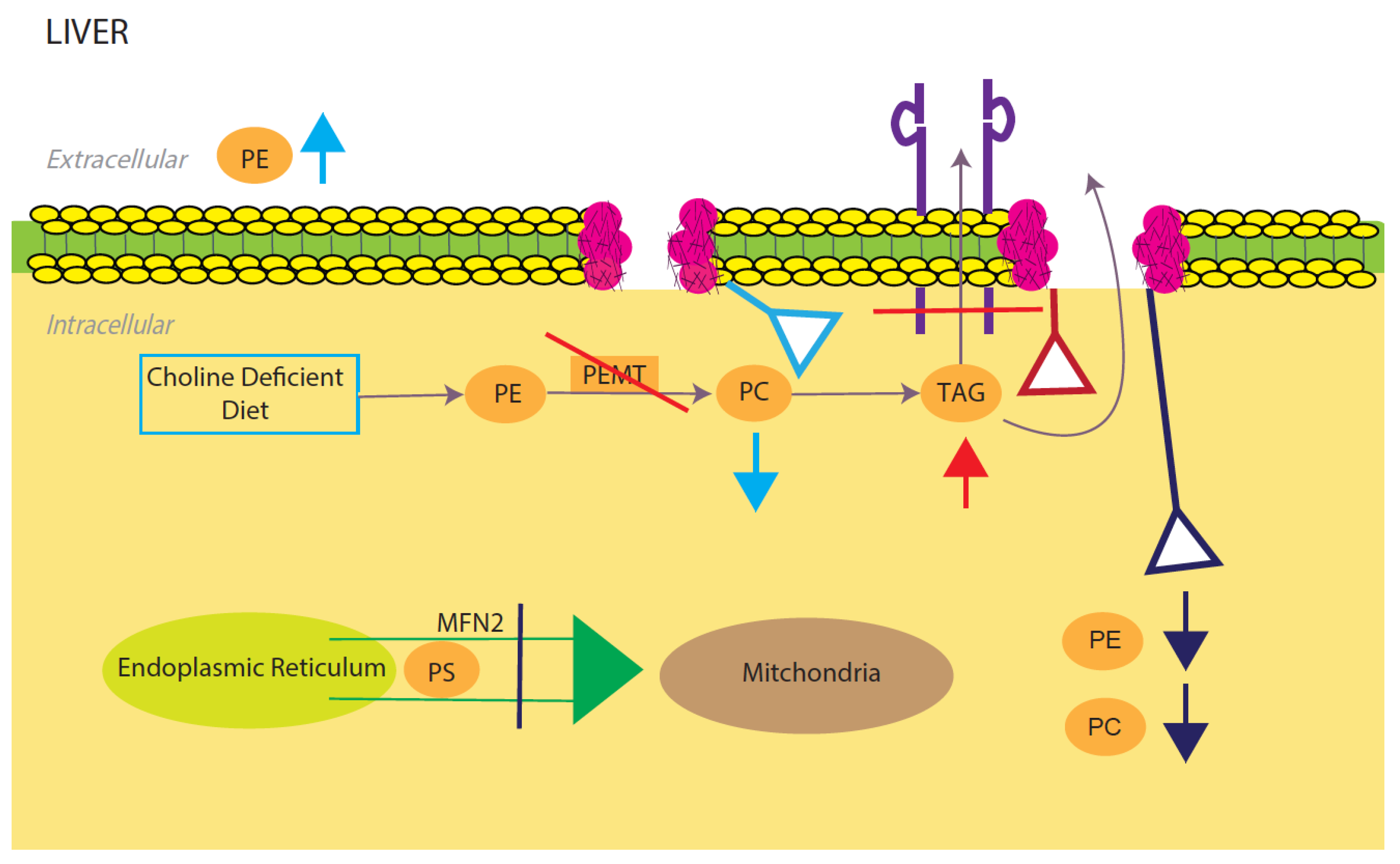
4. Phosphatidylcholine and Phosphatidylethanolamine-Mediated NAFLD
4.1. Decreasing PE/PC Ratio Results in NAFLD
4.2. Increasing PE/PC Ratio Results in NAFLD
5. Oxidized Phospholipids Contribution to NAFLD/NASH
6. Role of Cholesterol in NAFLD/NASH
Free Cholesterol Contribution to NAFLD/NASH
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rocchini, A.P. Childhood obesity and a diabetes epidemic. N. Engl. J. Med. 2002, 346, 854–855. [Google Scholar] [CrossRef] [PubMed]
- Ard, J. Obesity in the US: What is the best role for primary care? BMJ 2015, 350, g7846. [Google Scholar] [CrossRef] [PubMed]
- Arroyo-Johnson, C.; Mincey, K.D. Obesity Epidemiology Worldwide. Gastroenterol. Clin. N. Am. 2016, 45, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Gastaldelli, A.; Rebelos, E.; Bugianesi, E.; Messa, P.; Miele, L.; Svegliati-Baroni, G.; Valenti, L.; Bonino, F. Pathophysiology of Non Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 2082. [Google Scholar] [CrossRef] [PubMed]
- Williams, V.F.; Taubman, S.B.; Stahlman, S. Non-alcoholic fatty liver disease (NAFLD), active component, U.S. Armed Forces, 2000–2017. MSMR 2019, 26, 2–11. [Google Scholar] [PubMed]
- Rada, P.; González-Rodríguez, Á.; García-Monzón, C.; Valverde, Á. Understanding lipotoxicity in NAFLD pathogenesis: Is CD36 a key driver? Cell Death Dis. 2020, 11, 802. [Google Scholar] [CrossRef] [PubMed]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Stepanova, M.; De Avila, L.; Afendy, M.; Younossi, I.; Pham, H.; Cable, R.; Younossi, Z.M. Direct and Indirect Economic Burden of Chronic Liver Disease in the United States. Clin. Gastroenterol. Hepatol. 2017, 15, 759–766.e755. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Blissett, D.; Blissett, R.; Henry, L.; Stepanova, M.; Younossi, Y.; Racila, A.; Hunt, S.; Beckerman, R. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 2016, 64, 1577–1586. [Google Scholar] [CrossRef]
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef]
- Gundermann, K.J.; Gundermann, S.; Drozdzik, M.; Mohan Prasad, V.G. Essential phospholipids in fatty liver: A scientific update. Clin. Exp. Gastroenterol. 2016, 9, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, M.; Heacock, P.N.; Dowhan, W. A polytopic membrane protein displays a reversible topology dependent on membrane lipid composition. EMBO J. 2002, 21, 2107–2116. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; Fernández-Checa, J.C. Sphingolipid signalling and liver diseases. Liver Int. 2007, 27, 440–450. [Google Scholar] [CrossRef]
- Insausti-Urkia, N.; Solsona-Vilarrasa, E.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Sphingomyelinases and Liver Diseases. Biomolecules 2020, 10, 1497. [Google Scholar] [CrossRef] [PubMed]
- Lawler, J.F.; Yin, M.; Diehl, A.M.; Roberts, E.; Chatterjee, S. Tumor necrosis factor-alpha stimulates the maturation of sterol regulatory element binding protein-1 in human hepatocytes through the action of neutral sphingomyelinase. J. Biol. Chem. 1998, 273, 5053–5059. [Google Scholar] [CrossRef] [PubMed]
- Nikolova-Karakashian, M.N.; Rozenova, K.A. Ceramide in stress response. Sphingolipids Signal. Regul. Mol. 2010, 86–108. [Google Scholar]
- Aslan, M.; Özcan, F.; Tuzcu, H.; Kıraç, E.; Elpek, G.O. Inhibition of neutral sphingomyelinase decreases arachidonic acid mediated inflammation in liver ischemia-reperfusion injury. Int. J. Clin. Exp. Pathol. 2014, 7, 7814. [Google Scholar]
- Schwabe, R.F.; Brenner, D.A. Mechanisms of liver injury. I. TNF-α-induced liver injury: Role of IKK, JNK, and ROS pathways. Am. J. Physiol.-Gastrointest. Liver Physiol. 2006, 290, G583–G589. [Google Scholar] [CrossRef]
- Claus, R.A.; Dorer, M.J.; Bunck, A.C.; Deigner, H.P. Inhibition of sphingomyelin hydrolysis: Targeting the lipid mediator ceramide as a key regulator of cellular fate. Curr. Med. Chem. 2009, 16, 1978–2000. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Miller, R.A.; Wang, Z.V.; Sun, K.; Barth, B.M.; Bui, H.H.; Davis, K.E.; Bikman, B.T.; Halberg, N.; Rutkowski, J.M. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med. 2011, 17, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Luberto, C. Ceramide in the eukaryotic stress response. Trends Cell Biol. 2000, 10, 73–80. [Google Scholar] [CrossRef]
- Malaguarnera, M.; Di Rosa, M.; Nicoletti, F.; Malaguarnera, L. Molecular mechanisms involved in NAFLD progression. J. Mol. Med. 2009, 87, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Kannan, R.; Jin, M.; Gamulescu, M.-A.; Hinton, D. Ceramide-induced apoptosis: Role of catalase and hepatocyte growth factor. Free Radic. Biol. Med. 2004, 37, 166–175. [Google Scholar] [CrossRef]
- Ding, W.X.; Yin, X.M. Dissection of the multiple mechanisms of TNF-alpha-induced apoptosis in liver injury. J. Cell. Mol. Med. 2004, 8, 445–454. [Google Scholar] [CrossRef]
- Alkhouri, N.; Dixon, L.J.; Feldstein, A.E. Lipotoxicity in nonalcoholic fatty liver disease: Not all lipids are created equal. Expert Rev. Gastroenterol. Hepatol. 2009, 3, 445–451. [Google Scholar] [CrossRef]
- Kim, M.H.; Ahn, H.K.; Lee, E.J.; Kim, S.J.; Kim, Y.R.; Park, J.W.; Park, W.J. Hepatic inflammatory cytokine production can be regulated by modulating sphingomyelinase and ceramide synthase 6. Int. J. Mol. Med. 2017, 39, 453–462. [Google Scholar] [CrossRef]
- Ichi, I.; Nakahara, K.; Fujii, K.; Iida, C.; Miyashita, Y.; Kojo, S. Increase of ceramide in the liver and plasma after carbon tetrachloride intoxication in the rat. J. Nutr. Sci. Vitaminol. 2007, 53, 53–56. [Google Scholar] [CrossRef]
- Jiang, M.; Li, C.; Liu, Q.; Wang, A.; Lei, M. Inhibiting Ceramide Synthesis Attenuates Hepatic Steatosis and Fibrosis in Rats with Non-alcoholic Fatty Liver Disease. Front. Endocrinol. 2019, 10, 665. [Google Scholar] [CrossRef]
- Kotronen, A.; Seppänen-Laakso, T.; Westerbacka, J.; Kiviluoto, T.; Arola, J.; Ruskeepää, A.L.; Yki-Järvinen, H.; Oresic, M. Comparison of lipid and fatty acid composition of the liver, subcutaneous and intra-abdominal adipose tissue, and serum. Obesity 2010, 18, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Promrat, K.; Longato, L.; Wands, J.R.; de la Monte, S.M. Weight loss amelioration of non-alcoholic steatohepatitis linked to shifts in hepatic ceramide expression and serum ceramide levels. Hepatol. Res. 2011, 41, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.; Ottenhoff, R.; Powlson, A.S.; Grefhorst, A.; van Eijk, M.; Dubbelhuis, P.F.; Aten, J.; Kuipers, F.; Serlie, M.J.; Wennekes, T.; et al. Pharmacological inhibition of glucosylceramide synthase enhances insulin sensitivity. Diabetes 2007, 56, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Tagami, S.; Inokuchi Ji, J.; Kabayama, K.; Yoshimura, H.; Kitamura, F.; Uemura, S.; Ogawa, C.; Ishii, A.; Saito, M.; Ohtsuka, Y.; et al. Ganglioside GM3 participates in the pathological conditions of insulin resistance. J. Biol. Chem. 2002, 277, 3085–3092. [Google Scholar] [CrossRef] [PubMed]
- Kabayama, K.; Sato, T.; Kitamura, F.; Uemura, S.; Kang, B.W.; Igarashi, Y.; Inokuchi, J. TNFalpha-induced insulin resistance in adipocytes as a membrane microdomain disorder: Involvement of ganglioside GM3. Glycobiology 2005, 15, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Mitsutake, S.; Date, T.; Yokota, H.; Sugiura, M.; Kohama, T.; Igarashi, Y. Ceramide kinase deficiency improves diet-induced obesity and insulin resistance. FEBS Lett. 2012, 586, 1300–1305. [Google Scholar] [CrossRef]
- Hanamatsu, H.; Ohnishi, S.; Sakai, S.; Yuyama, K.; Mitsutake, S.; Takeda, H.; Hashino, S.; Igarashi, Y. Altered levels of serum sphingomyelin and ceramide containing distinct acyl chains in young obese adults. Nutr. Diabetes 2014, 4, e141. [Google Scholar] [CrossRef]
- Kim, C.W.; Addy, C.; Kusunoki, J.; Anderson, N.N.; Deja, S.; Fu, X.; Burgess, S.C.; Li, C.; Ruddy, M.; Chakravarthy, M.; et al. Acetyl CoA Carboxylase Inhibition Reduces Hepatic Steatosis but Elevates Plasma Triglycerides in Mice and Humans: A Bedside to Bench Investigation. Cell Metab. 2017, 26, 576. [Google Scholar] [CrossRef]
- Chung, H.Y.; Witt, C.J.; Jbeily, N.; Hurtado-Oliveros, J.; Giszas, B.; Lupp, A.; Gräler, M.H.; Bruns, T.; Stallmach, A.; Gonnert, F.A.; et al. Acid Sphingomyelinase Inhibition Prevents Development of Sepsis Sequelae in the Murine Liver. Sci. Rep. 2017, 7, 12348. [Google Scholar] [CrossRef]
- Kolesnick, R.N.; Krönke, M. Regulation of ceramide production and apoptosis. Annu. Rev. Physiol. 1998, 60, 643–665. [Google Scholar] [CrossRef]
- Andrieu-Abadie, N.; Gouazé, V.; Salvayre, R.; Levade, T. Ceramide in apoptosis signaling: Relationship with oxidative stress. Free Radic. Biol. Med. 2001, 31, 717–728. [Google Scholar] [CrossRef]
- Jin, J.; Hou, Q.; Mullen, T.D.; Zeidan, Y.H.; Bielawski, J.; Kraveka, J.M.; Bielawska, A.; Obeid, L.M.; Hannun, Y.A.; Hsu, Y.-T. Ceramide generated by sphingomyelin hydrolysis and the salvage pathway is involved in hypoxia/reoxygenation-induced Bax redistribution to mitochondria in NT-2 cells. J. Biol. Chem. 2008, 283, 26509–26517. [Google Scholar] [CrossRef] [PubMed]
- García-Ruiz, C.; Colell, A.; Marí, M.; Morales, A.; Calvo, M.; Enrich, C.; Fernández-Checa, J.C. Defective TNF-alpha-mediated hepatocellular apoptosis and liver damage in acidic sphingomyelinase knockout mice. J. Clin. Investig. 2003, 111, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Djerir, N.E.H.; Danckaert, A.; Fernandes, J.; Roux, P.; Charrueau, C.; Lachagès, A.-M.; Charlotte, F.; Brocheriou, I.; Clément, K. Hepatic stellate cell hypertrophy is associated with metabolic liver fibrosis. Sci. Rep. 2020, 10, 3850. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Yetukuri, L.; Katajamaa, M.; Medina-Gomez, G.; Seppänen-Laakso, T.; Vidal-Puig, A.; Oresic, M. Bioinformatics strategies for lipidomics analysis: Characterization of obesity related hepatic steatosis. BMC Syst. Biol. 2007, 1, 12. [Google Scholar] [CrossRef] [PubMed]
- Shmarakov, I.O.; Jiang, H.; Liu, J.; Fernandez, E.J.; Blaner, W.S. Hepatic stellate cell activation: A source for bioactive lipids. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2019, 1864, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Moles, A.; Tarrats, N.; Morales, A.; Domínguez, M.; Bataller, R.; Caballería, J.; García-Ruiz, C.; Fernández-Checa, J.C.; Marí, M. Acidic sphingomyelinase controls hepatic stellate cell activation and in vivo liver fibrogenesis. Am. J. Pathol. 2010, 177, 1214–1224. [Google Scholar] [CrossRef]
- Hernández-Muñoz, I.; de la Torre, P.; Sánchez-Alcázar, J.A.; García, I.; Santiago, E.; Muñoz-Yagüe, M.T.; Solís-Herruzo, J.A. Tumor necrosis factor alpha inhibits collagen alpha 1(I) gene expression in rat hepatic stellate cells through a G protein. Gastroenterology 1997, 113, 625–640. [Google Scholar] [CrossRef]
- Li, Z.; Chiang, Y.-p.; He, M.; Worgall, T.S.; Zhou, H.; Jiang, X.-C. Liver sphingomyelin synthase 1 deficiency causes steatosis, steatohepatitis, fibrosis, and tumorigenesis: An effect of glucosylceramide accumulation. Iscience 2021, 24, 103449. [Google Scholar] [CrossRef]
- Coskun, Ü.; Grzybek, M.; Drechsel, D.; Simons, K. Regulation of human EGF receptor by lipids. Proc. Natl. Acad. Sci. USA 2011, 108, 9044–9048. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. The Ceramide-centric universe of lipid-mediated cell regulation: Stress encounters of the lipid kind. J. Biol. Chem. 2002, 277, 25847–25850. [Google Scholar] [CrossRef] [PubMed]
- Kirschbaum, C.; Greis, K.; Mucha, E.; Kain, L.; Deng, S.; Zappe, A.; Gewinner, S.; Schöllkopf, W.; von Helden, G.; Meijer, G.; et al. Unravelling the structural complexity of glycolipids with cryogenic infrared spectroscopy. Nat. Commun. 2021, 12, 1201. [Google Scholar] [CrossRef] [PubMed]
- Saddoughi, S.A.; Song, P.; Ogretmen, B. Roles of bioactive sphingolipids in cancer biology and therapeutics. Subcell Biochem. 2008, 49, 413–440. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Wiest, M.M.; Cheung, O.; Mirshahi, F.; Sargeant, C.; Min, H.K.; Contos, M.J.; Sterling, R.K.; Fuchs, M.; Zhou, H.; et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 2009, 50, 1827–1838. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulou, M.; Gordillo, R.; Koliaki, C.; Gancheva, S.; Jelenik, T.; De Filippo, E.; Herder, C.; Markgraf, D.; Jankowiak, F.; Esposito, I.; et al. Specific Hepatic Sphingolipids Relate to Insulin Resistance, Oxidative Stress, and Inflammation in Nonalcoholic Steatohepatitis. Diabetes Care 2018, 41, 1235–1243. [Google Scholar] [CrossRef]
- Holland, W.L.; Summers, S.A. Sphingolipids, insulin resistance, and metabolic disease: New insights from in vivo manipulation of sphingolipid metabolism. Endocr. Rev. 2008, 29, 381–402. [Google Scholar] [CrossRef]
- Chocian, G.; Chabowski, A.; Zendzian-Piotrowska, M.; Harasim, E.; Łukaszuk, B.; Górski, J. High fat diet induces ceramide and sphingomyelin formation in rat’s liver nuclei. Mol. Cell. Biochem. 2010, 340, 125–131. [Google Scholar] [CrossRef]
- Monetti, M.; Levin, M.C.; Watt, M.J.; Sajan, M.P.; Marmor, S.; Hubbard, B.K.; Stevens, R.D.; Bain, J.R.; Newgard, C.B.; Farese, R.V.; et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab. 2007, 6, 69–78. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Pacana, T. A Lipidomic Readout of Disease Progression in a Diet-Induced Mouse Model of Nonalcoholic Fatty Liver Disease. Trans. Am. Clin. Climatol. Assoc. 2015, 126, 271–288. [Google Scholar]
- Luukkonen, P.K.; Zhou, Y.; Sädevirta, S.; Leivonen, M.; Arola, J.; Orešič, M.; Hyötyläinen, T.; Yki-Järvinen, H. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Turpin, S.M.; Nicholls, H.T.; Willmes, D.M.; Mourier, A.; Brodesser, S.; Wunderlich, C.M.; Mauer, J.; Xu, E.; Hammerschmidt, P.; Brönneke, H.S.; et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 2014, 20, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Longato, L.; Tong, M.; Wands, J.R.; de la Monte, S.M. High fat diet induced hepatic steatosis and insulin resistance: Role of dysregulated ceramide metabolism. Hepatol. Res. 2012, 42, 412–427. [Google Scholar] [CrossRef] [PubMed]
- Zigmond, E.; Tayer-Shifman, O.; Lalazar, G.; Ben Ya’acov, A.; Weksler-Zangen, S.; Shasha, D.; Sklair-Levy, M.; Zolotarov, L.; Shalev, Z.; Kalman, R.; et al. β-glycosphingolipids ameliorated non-alcoholic steatohepatitis in the Psammomys obesus model. J. Inflamm. Res. 2014, 7, 151–158. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ren, Z.; Yang, Z.; Lu, Y.; Zhang, R.; Yang, H. Anti-glycolipid disorder effect of epigallocatechin-3-gallate on high-fat diet and STZ-induced T2DM in mice. Mol. Med. Rep. 2020, 21, 2475–2483. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Shi, Q.; Li, H.; Liu, J.; Wu, S.; Sun, H.; Zhou, B. Glycolipid Metabolism Disorder in the Liver of Obese Mice Is Improved by TUDCA via the Restoration of Defective Hepatic Autophagy. Int. J. Endocrinol. 2015, 2015, 687938. [Google Scholar] [CrossRef]
- van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef]
- Calzada, E.; Onguka, O.; Claypool, S.M. Phosphatidylethanolamine Metabolism in Health and Disease. Int. Rev. Cell Mol. Biol. 2016, 321, 29–88. [Google Scholar] [CrossRef]
- Traiffort, E.; O’Regan, S.; Ruat, M. The choline transporter-like family SLC44: Properties and roles in human diseases. Mol. Asp. Med. 2013, 34, 646–654. [Google Scholar] [CrossRef]
- Lykidis, A.; Baburina, I.; Jackowski, S. Distribution of CTP:phosphocholine cytidylyltransferase (CCT) isoforms. Identification of a new CCTbeta splice variant. J. Biol. Chem. 1999, 274, 26992–27001. [Google Scholar] [CrossRef]
- Karim, M.; Jackson, P.; Jackowski, S. Gene structure, expression and identification of a new CTP: Phosphocholine cytidylyltransferase beta isoform. Biochim. Biophys. Acta 2003, 1633, 1–12. [Google Scholar] [CrossRef]
- Jacobs, R.L.; Devlin, C.; Tabas, I.; Vance, D.E. Targeted deletion of hepatic CTP:phosphocholine cytidylyltransferase alpha in mice decreases plasma high density and very low density lipoproteins. J. Biol. Chem. 2004, 279, 47402–47410. [Google Scholar] [CrossRef] [PubMed]
- Noga, A.A.; Vance, D.E. Insights into the requirement of phosphatidylcholine synthesis for liver function in mice. J. Lipid Res. 2003, 44, 1998–2005. [Google Scholar] [CrossRef] [PubMed]
- Noga, A.A.; Vance, D.E. A gender-specific role for phosphatidylethanolamine N-methyltransferase-derived phosphatidylcholine in the regulation of plasma high density and very low density lipoproteins in mice. J. Biol. Chem. 2003, 278, 21851–21859. [Google Scholar] [CrossRef]
- Li, Z.; Agellon, L.B.; Vance, D.E. Phosphatidylcholine homeostasis and liver failure. J. Biol. Chem. 2005, 280, 37798–37802. [Google Scholar] [CrossRef]
- Li, Z.; Agellon, L.B.; Allen, T.M.; Umeda, M.; Jewell, L.; Mason, A.; Vance, D.E. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 2006, 3, 321–331. [Google Scholar] [CrossRef]
- Voshol, P.J.; Minich, D.M.; Havinga, R.; Elferink, R.P.; Verkade, H.J.; Groen, A.K.; Kuipers, F. Postprandial chylomicron formation and fat absorption in multidrug resistance gene 2 P-glycoprotein-deficient mice. Gastroenterology 2000, 118, 173–182. [Google Scholar] [CrossRef]
- Noureddin, M.; Mato, J.M.; Lu, S.C. Nonalcoholic fatty liver disease: Update on pathogenesis, diagnosis, treatment and the role of S-adenosylmethionine. Exp. Biol. Med. 2015, 240, 809–820. [Google Scholar] [CrossRef]
- Leonardi, R.; Frank, M.W.; Jackson, P.D.; Rock, C.O.; Jackowski, S. Elimination of the CDP-ethanolamine pathway disrupts hepatic lipid homeostasis. J. Biol. Chem. 2009, 284, 27077–27089. [Google Scholar] [CrossRef]
- Hernández-Alvarez, M.I.; Sebastián, D.; Vives, S.; Ivanova, S.; Bartoccioni, P.; Kakimoto, P.; Plana, N.; Veiga, S.R.; Hernández, V.; Vasconcelos, N.; et al. Deficient Endoplasmic Reticulum-Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell 2019, 177, 881–895.e817. [Google Scholar] [CrossRef]
- Martínez-Uña, M.; Varela-Rey, M.; Cano, A.; Fernández-Ares, L.; Beraza, N.; Aurrekoetxea, I.; Martínez-Arranz, I.; García-Rodríguez, J.L.; Buqué, X.; Mestre, D.; et al. Excess S-adenosylmethionine reroutes phosphatidylethanolamine towards phosphatidylcholine and triglyceride synthesis. Hepatology 2013, 58, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Uña, M.; Varela-Rey, M.; Mestre, D.; Fernández-Ares, L.; Fresnedo, O.; Fernandez-Ramos, D.; Gutiérrez-de Juan, V.; Martin-Guerrero, I.; García-Orad, A.; Luka, Z.; et al. S-Adenosylmethionine increases circulating very-low density lipoprotein clearance in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Aldrovandi, M.; Hammond, V.J.; Podmore, H.; Hornshaw, M.; Clark, S.R.; Marnett, L.J.; Slatter, D.A.; Murphy, R.C.; Collins, P.W.; O’Donnell, V.B. Human platelets generate phospholipid-esterified prostaglandins via cyclooxygenase-1 that are inhibited by low dose aspirin supplementation. J. Lipid Res. 2013, 54, 3085–3097. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Guy, C.J.; Scurr, M.J.; Taylor, P.R.; Kift-Morgan, A.P.; Hammond, V.J.; Thomas, C.P.; Coles, B.; Roberts, G.W.; Eberl, M.; et al. Esterified eicosanoids are acutely generated by 5-lipoxygenase in primary human neutrophils and in human and murine infection. Blood 2011, 117, 2033–2043. [Google Scholar] [CrossRef] [PubMed]
- Bochkov, V.N.; Oskolkova, O.V.; Birukov, K.G.; Levonen, A.L.; Binder, C.J.; Stöckl, J. Generation and biological activities of oxidized phospholipids. Antioxid. Redox Signal. 2010, 12, 1009–1059. [Google Scholar] [CrossRef]
- Ikura, Y.; Ohsawa, M.; Suekane, T.; Fukushima, H.; Itabe, H.; Jomura, H.; Nishiguchi, S.; Inoue, T.; Naruko, T.; Ehara, S.; et al. Localization of oxidized phosphatidylcholine in nonalcoholic fatty liver disease: Impact on disease progression. Hepatology 2006, 43, 506–514. [Google Scholar] [CrossRef]
- Sun, X.; Seidman, J.S.; Zhao, P.; Troutman, T.D.; Spann, N.J.; Que, X.; Zhou, F.; Liao, Z.; Pasillas, M.; Yang, X.; et al. Neutralization of Oxidized Phospholipids Ameliorates Non-alcoholic Steatohepatitis. Cell Metab. 2020, 31, 189–206.e188. [Google Scholar] [CrossRef]
- Mendel, I.; Yacov, N.; Shoham, A.; Ishai, E.; Breitbart, E. Treatment with Oxidized Phospholipids Directly Inhibits Nonalcoholic Steatohepatitis and Liver Fibrosis without Affecting Steatosis. Dig. Dis. Sci. 2016, 61, 2545–2553. [Google Scholar] [CrossRef]
- Yimin; Furumaki, H.; Matsuoka, S.; Sakurai, T.; Kohanawa, M.; Zhao, S.; Kuge, Y.; Tamaki, N.; Chiba, H. A novel murine model for non-alcoholic steatohepatitis developed by combination of a high-fat diet and oxidized low-density lipoprotein. Lab. Investig. 2012, 92, 265–281. [Google Scholar] [CrossRef]
- Liangpunsakul, S.; Chalasani, N. Lipid mediators of liver injury in nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G75–G81. [Google Scholar] [CrossRef]
- Oemer, G.; Lackner, K.; Muigg, K.; Krumschnabel, G.; Watschinger, K.; Sailer, S.; Lindner, H.; Gnaiger, E.; Wortmann, S.B.; Werner, E.R.; et al. Molecular structural diversity of mitochondrial cardiolipins. Proc. Natl. Acad. Sci. USA 2018, 115, 4158–4163. [Google Scholar] [CrossRef] [PubMed]
- Maxfield, F.R.; Tabas, I. Role of cholesterol and lipid organization in disease. Nature 2005, 438, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Ikonen, E. Mechanisms for cellular cholesterol transport: Defects and human disease. Physiol. Rev. 2006, 86, 1237–1261. [Google Scholar] [CrossRef] [PubMed]
- Bloch, K. Sterol molecule: Structure, biosynthesis, and function. Steroids 1992, 57, 378–383. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef]
- Horton, J.D.; Shimomura, I.; Brown, M.S.; Hammer, R.E.; Goldstein, J.L.; Shimano, H. Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J. Clin. Investig. 1998, 101, 2331–2339. [Google Scholar] [CrossRef]
- Dietschy, J.M.; Turley, S.D.; Spady, D.K. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J. Lipid Res. 1993, 34, 1637–1659. [Google Scholar] [CrossRef]
- Malhotra, P.; Gill, R.K.; Saksena, S.; Alrefai, W.A. Disturbances in Cholesterol Homeostasis and Non-alcoholic Fatty Liver Diseases. Front. Med. 2020, 7, 467. [Google Scholar] [CrossRef]
- Arguello, G.; Balboa, E.; Arrese, M.; Zanlungo, S. Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. Biochim. Biophys. Acta 2015, 1852, 1765–1778. [Google Scholar] [CrossRef]
- Subramanian, S.; Goodspeed, L.; Wang, S.; Kim, J.; Zeng, L.; Ioannou, G.N.; Haigh, W.G.; Yeh, M.M.; Kowdley, K.V.; O’Brien, K.D.; et al. Dietary cholesterol exacerbates hepatic steatosis and inflammation in obese LDL receptor-deficient mice. J. Lipid Res. 2011, 52, 1626–1635. [Google Scholar] [CrossRef]
- Malhi, H.; Gores, G.J. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin. Liver Dis. 2008, 28, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Min, H.K.; Kapoor, A.; Fuchs, M.; Mirshahi, F.; Zhou, H.; Maher, J.; Kellum, J.; Warnick, R.; Contos, M.J.; Sanyal, A.J. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012, 15, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Wouters, K.; van Bilsen, M.; van Gorp, P.J.; Bieghs, V.; Lütjohann, D.; Kerksiek, A.; Staels, B.; Hofker, M.H.; Shiri-Sverdlov, R. Intrahepatic cholesterol influences progression, inhibition and reversal of non-alcoholic steatohepatitis in hyperlipidemic mice. FEBS Lett. 2010, 584, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Gores, G.J. Cellular and molecular mechanisms of liver injury. Gastroenterology 2008, 134, 1641–1654. [Google Scholar] [CrossRef] [PubMed]
- Bieghs, V.; Hendrikx, T.; van Gorp, P.J.; Verheyen, F.; Guichot, Y.D.; Walenbergh, S.M.; Jeurissen, M.L.; Gijbels, M.; Rensen, S.S.; Bast, A.; et al. The cholesterol derivative 27-hydroxycholesterol reduces steatohepatitis in mice. Gastroenterology 2013, 144, 167–178.e161. [Google Scholar] [CrossRef] [PubMed]
- Van Rooyen, D.M.; Larter, C.Z.; Haigh, W.G.; Yeh, M.M.; Ioannou, G.; Kuver, R.; Lee, S.P.; Teoh, N.C.; Farrell, G.C. Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology 2011, 141, 1393–1403.e5. [Google Scholar] [CrossRef]
- DeBose-Boyd, R.A. Significance and regulation of lipid metabolism. Semin. Cell Dev. Biol. 2018, 81, 97. [Google Scholar] [CrossRef]
- DeBose-Boyd, R.A.; Ye, J. SREBPs in Lipid Metabolism, Insulin Signaling, and Beyond. Trends Biochem. Sci. 2018, 43, 358–368. [Google Scholar] [CrossRef]
- Li, H.; Yu, X.H.; Ou, X.; Ouyang, X.P.; Tang, C.K. Hepatic cholesterol transport and its role in non-alcoholic fatty liver disease and atherosclerosis. Prog. Lipid Res. 2021, 83, 101109. [Google Scholar] [CrossRef]
- Rinella, M.E.; Siddiqui, M.S.; Gardikiotes, K.; Gottstein, J.; Elias, M.; Green, R.M. Dysregulation of the unfolded protein response in db/db mice with diet-induced steatohepatitis. Hepatology 2011, 54, 1600–1609. [Google Scholar] [CrossRef]
- Lu, K.; Lee, M.H.; Hazard, S.; Brooks-Wilson, A.; Hidaka, H.; Kojima, H.; Ose, L.; Stalenhoef, A.F.; Mietinnen, T.; Bjorkhem, I.; et al. Two genes that map to the STSL locus cause sitosterolemia: Genomic structure and spectrum of mutations involving sterolin-1 and sterolin-2, encoded by ABCG5 and ABCG8, respectively. Am. J. Hum. Genet. 2001, 69, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Cortes, V.A.; Busso, D.; Maiz, A.; Arteaga, A.; Nervi, F.; Rigotti, A. Physiological and pathological implications of cholesterol. Front. Biosci. 2014, 19, 416–428. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Welch, M.; Secunda, C.; Ghimire, N.; Martinez, I.; Mathus, A.; Patel, U.; Bhogoju, S.; Al-Mutairi, M.; Min, K.; Lawan, A. Characterization and Roles of Membrane Lipids in Fatty Liver Disease. Membranes 2022, 12, 410. https://doi.org/10.3390/membranes12040410
Welch M, Secunda C, Ghimire N, Martinez I, Mathus A, Patel U, Bhogoju S, Al-Mutairi M, Min K, Lawan A. Characterization and Roles of Membrane Lipids in Fatty Liver Disease. Membranes. 2022; 12(4):410. https://doi.org/10.3390/membranes12040410
Chicago/Turabian StyleWelch, Morgan, Cassandra Secunda, Nabin Ghimire, Isabel Martinez, Amber Mathus, Urja Patel, Sarayu Bhogoju, Mashael Al-Mutairi, Kisuk Min, and Ahmed Lawan. 2022. "Characterization and Roles of Membrane Lipids in Fatty Liver Disease" Membranes 12, no. 4: 410. https://doi.org/10.3390/membranes12040410
APA StyleWelch, M., Secunda, C., Ghimire, N., Martinez, I., Mathus, A., Patel, U., Bhogoju, S., Al-Mutairi, M., Min, K., & Lawan, A. (2022). Characterization and Roles of Membrane Lipids in Fatty Liver Disease. Membranes, 12(4), 410. https://doi.org/10.3390/membranes12040410