
TRPV4 Stimulation Level Regulates Ca2+-Dependent Control of Human Corneal Endothelial Cell Viability and Survival

 , , , , ,
, , , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
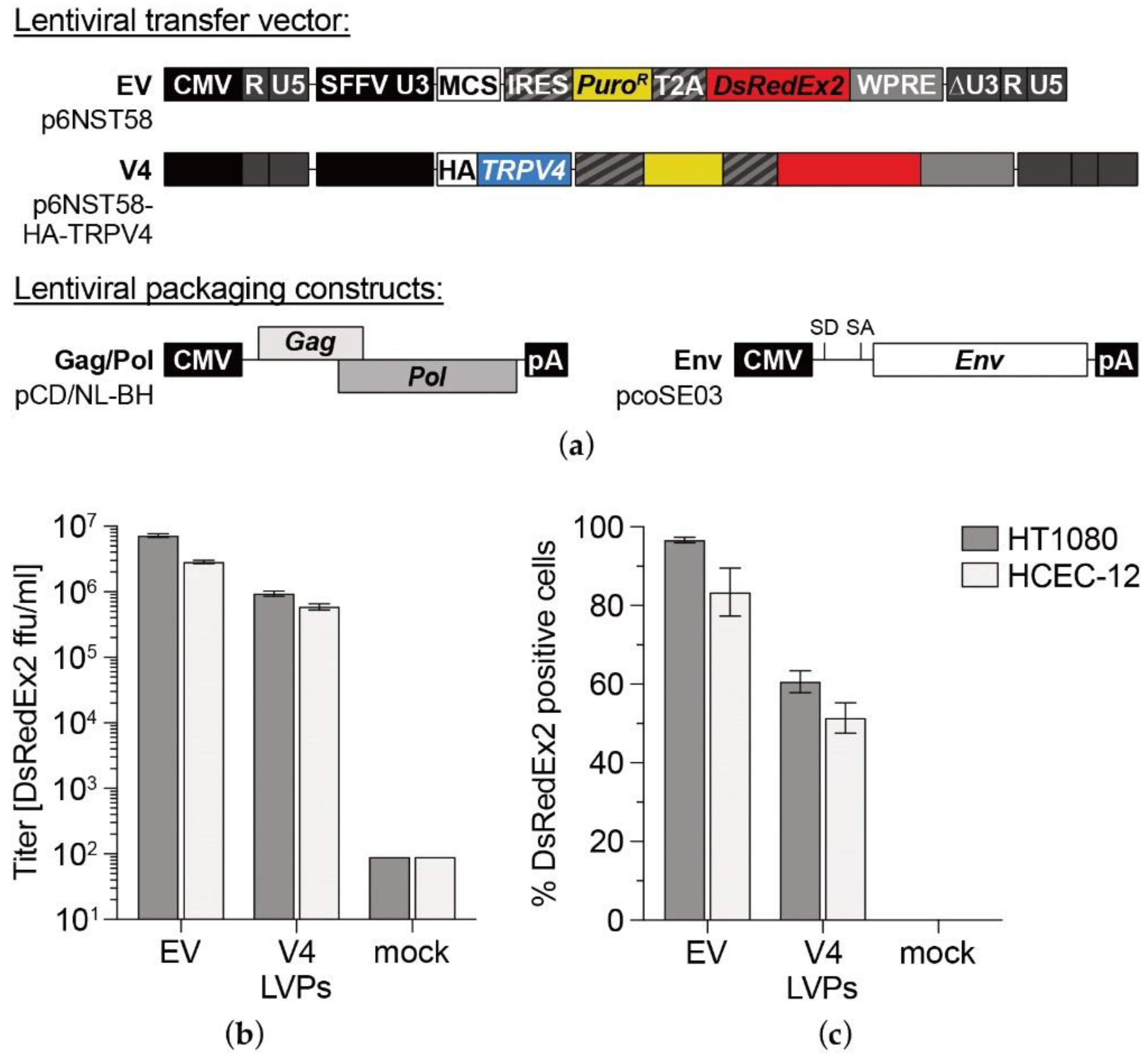
2.2. Cloning and Lentiviral Transduction
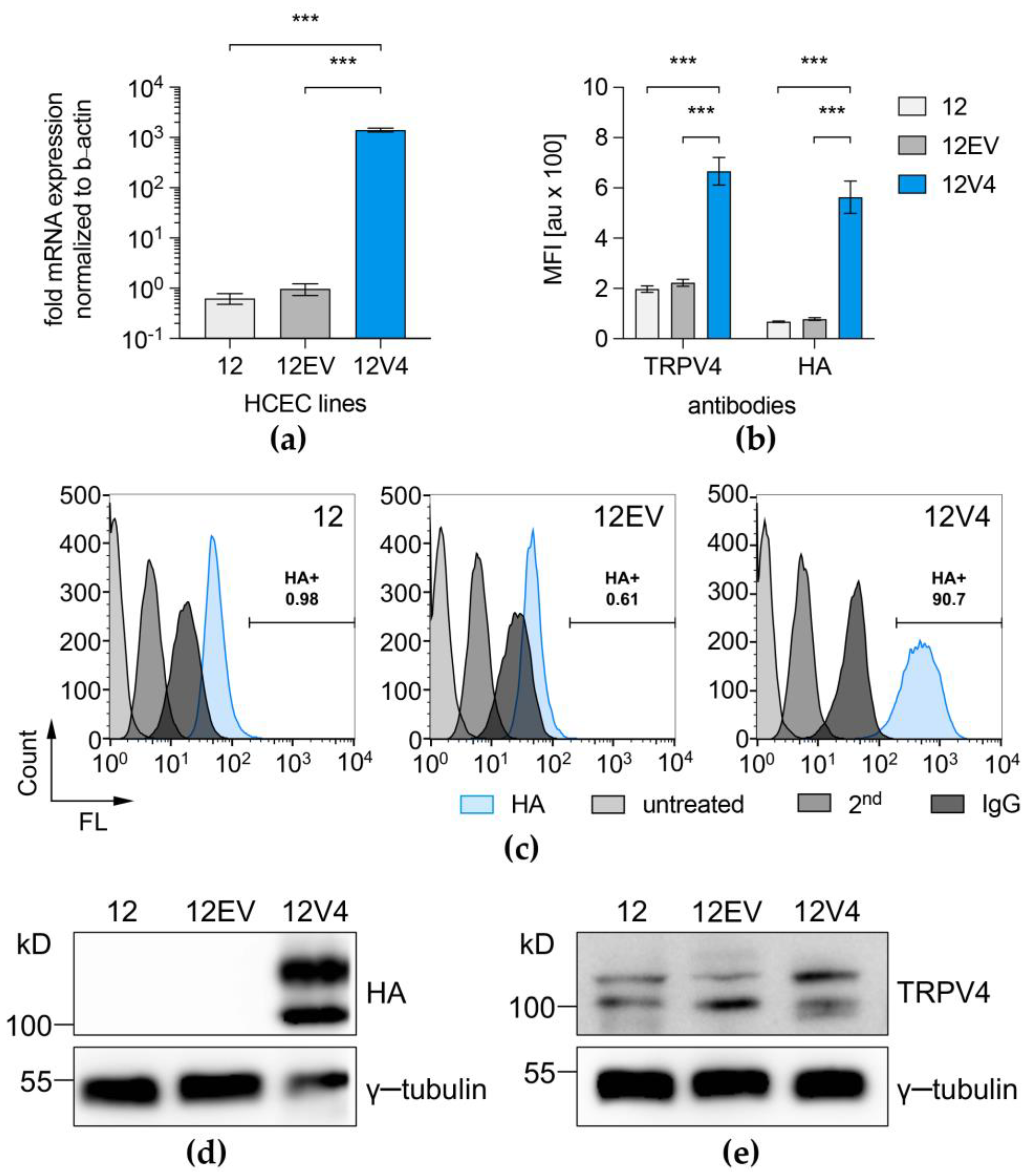
2.3. Quantitative Real-Time PCR
2.4. Western Blotting
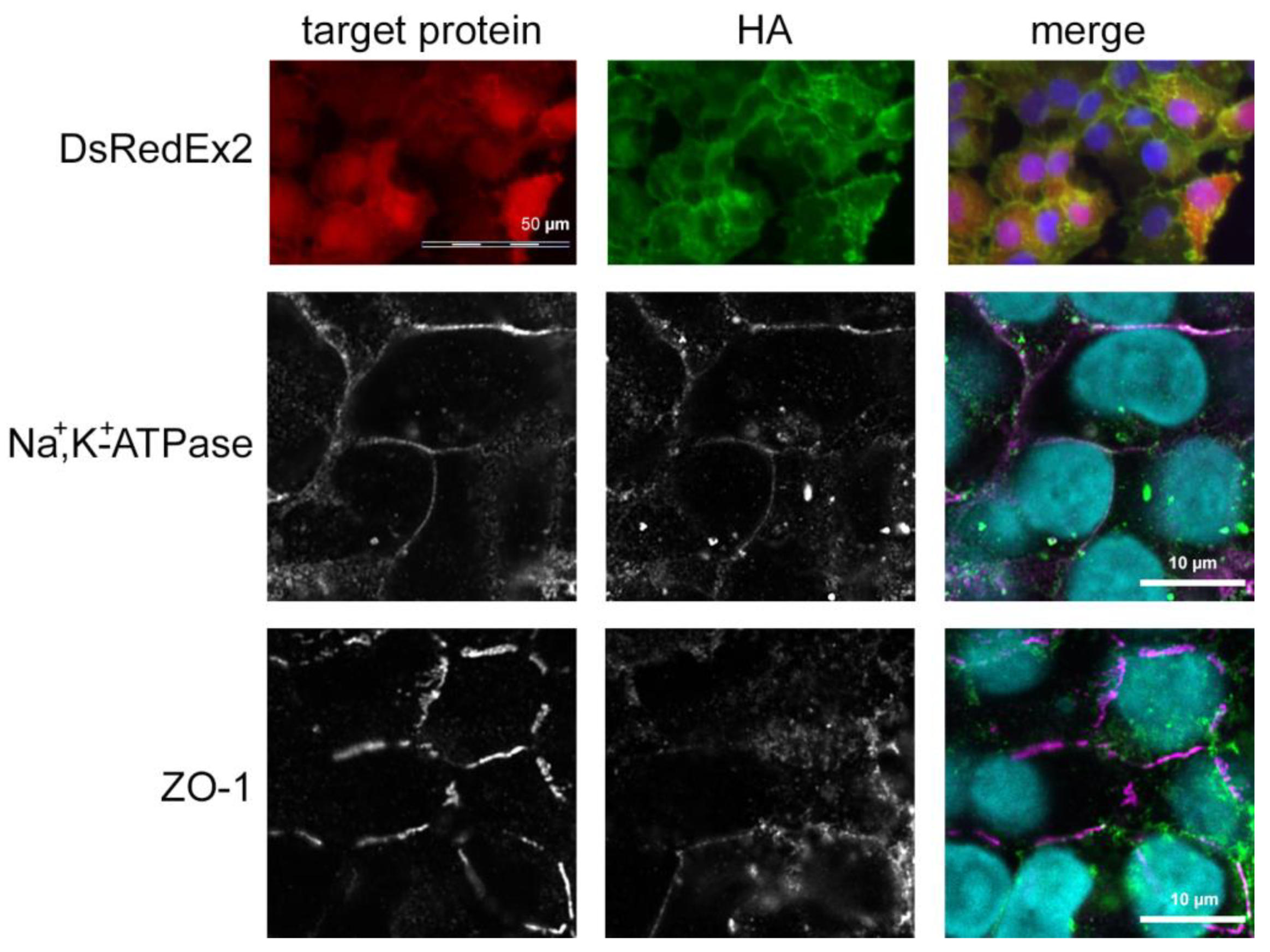
2.5. Immunofluorescence Staining
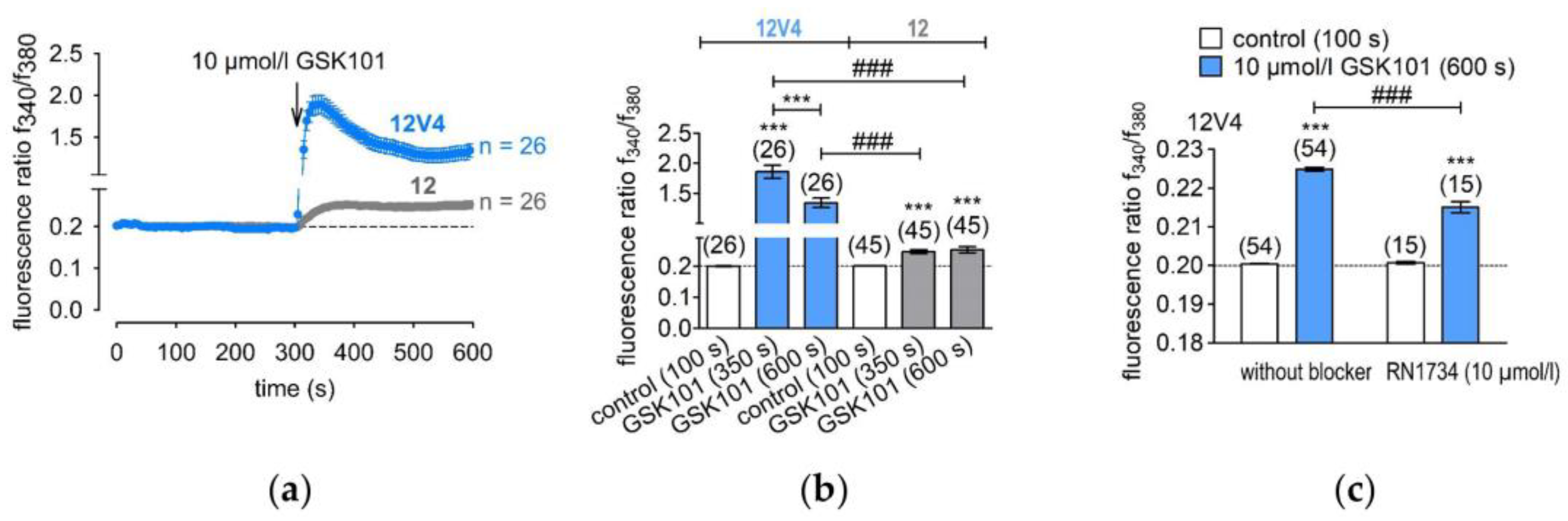
2.6. Fluorescence Calcium Imaging
2.7. Cell Viability Assays
2.8. Transepithelial Electrical Resistance (TEER) and Cellular Capacitance (Ccl) Measurement
2.9. Scanning Electron Microscopy
2.10. Statistical Analysis
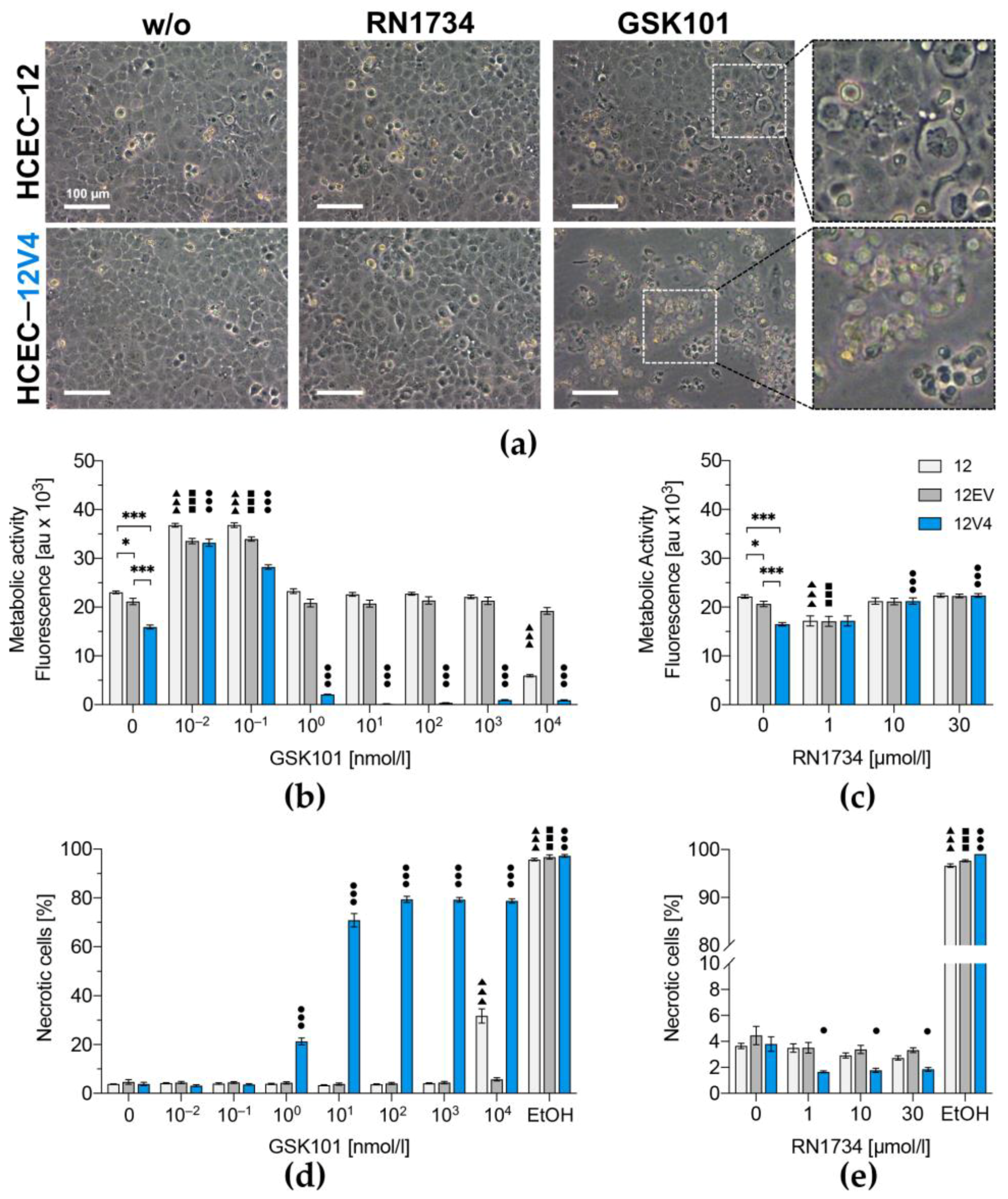
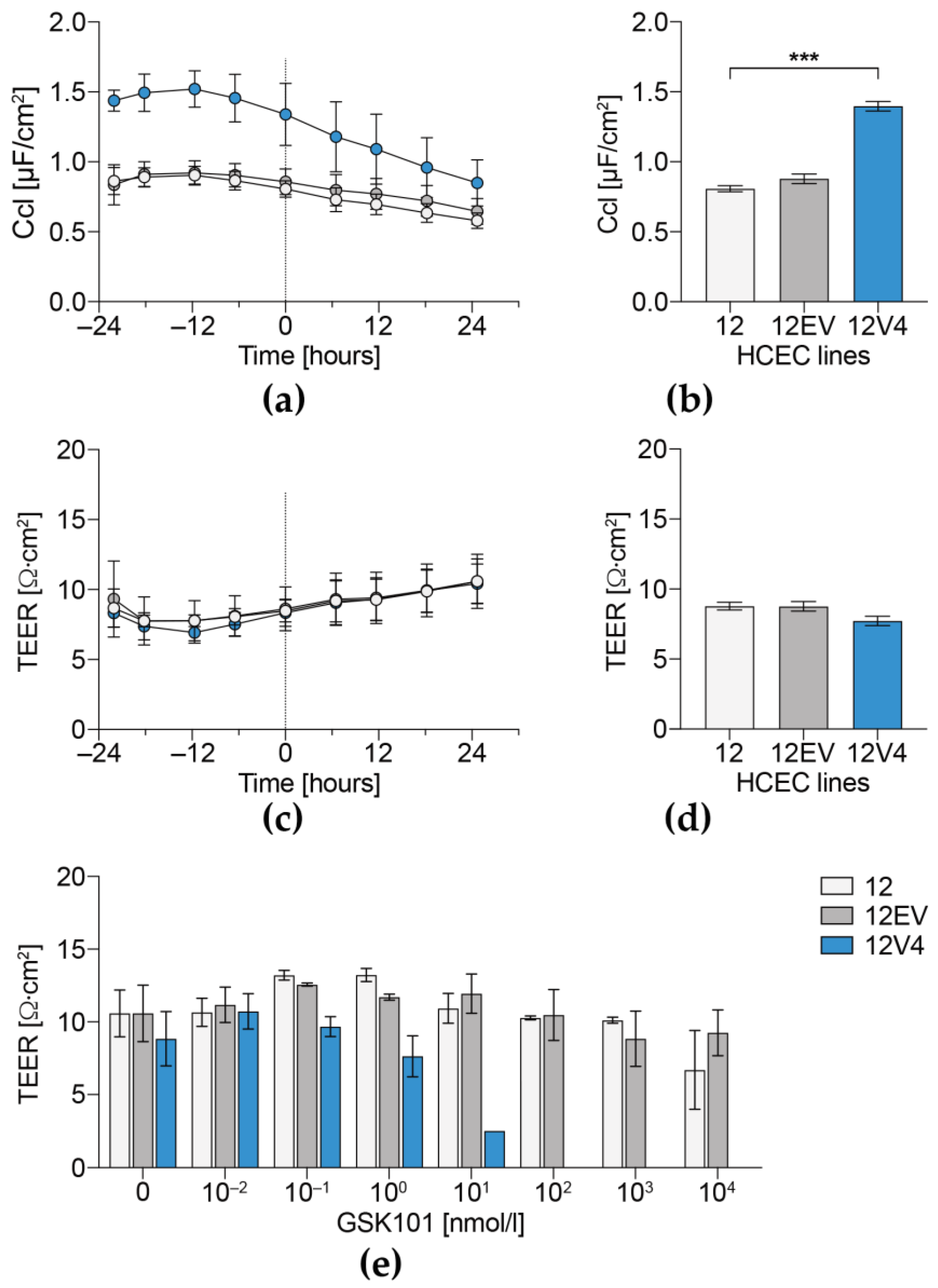
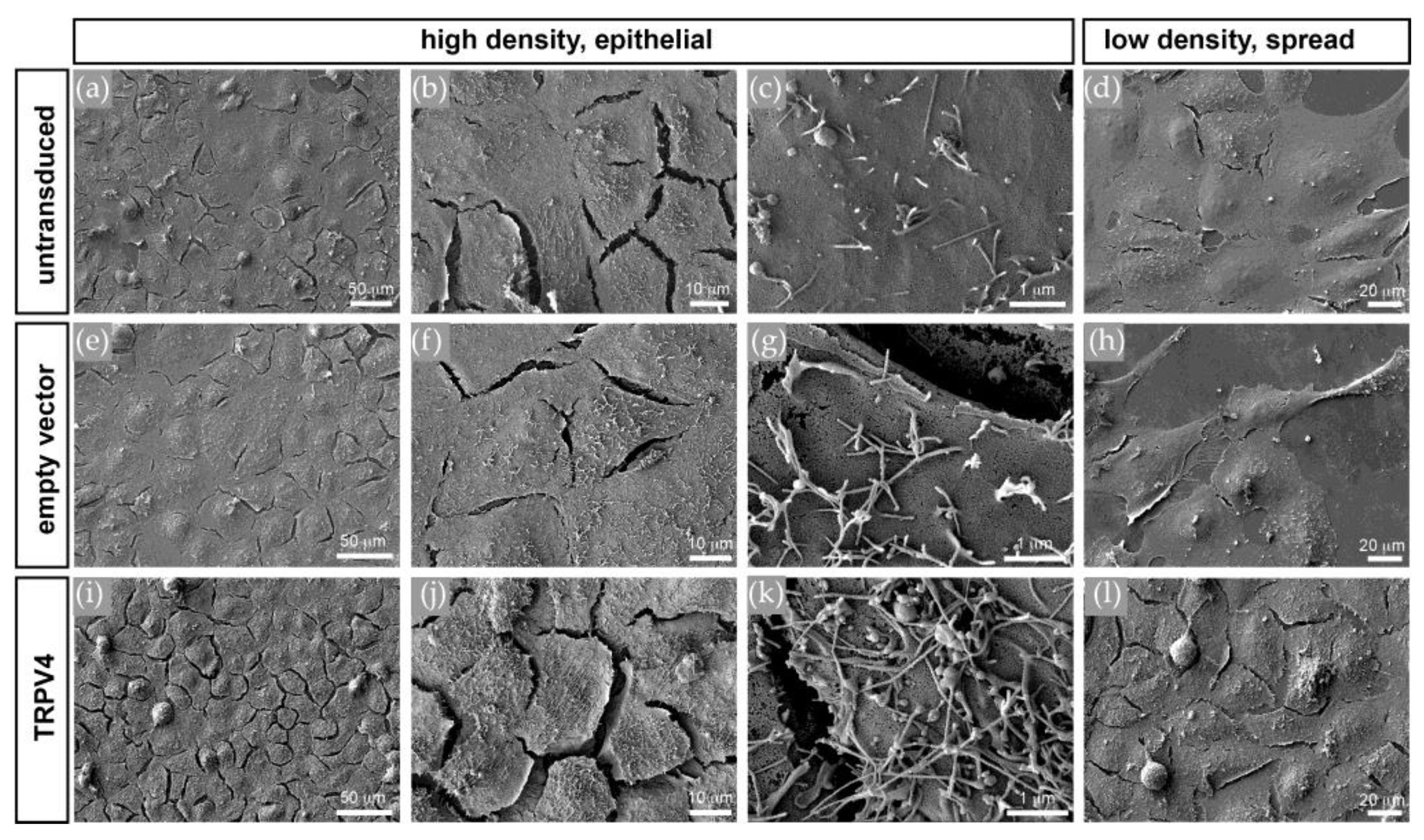
3. Results
3.1. TRPV4 Channel Overexpression in HCEC-12
3.2. Functional Overexpression of TRPV4
3.3. TRPV4 Overexpression Diminishes Cell Survival
3.4. Cellular Capacitance and Transepithelial Electrical Resistance
3.5. TRPV4 Overexpression Alters Cell Surface Ultrastructure but Not Cell Volume
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Joyce, N.C. Proliferative capacity of the corneal endothelium. Prog. Retin. Eye Res. 2003, 22, 359–389. [Google Scholar] [CrossRef]
- Swan, J.S.; Hodson, S.A. Rabbit corneal hydration and the bicarbonate pump. J. Membr. Biol. 2004, 201, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Senoo, T.; Obara, Y.; Joyce, N.C. EDTA: A Promoter of Proliferation in Human Corneal Endothelium. Invest. Ophthalmol. Vis. Sci. 2000, 41, 2930–2935. [Google Scholar]
- Green, K.; Cheeks, L.; Hull, D.S. Effects of calcium channel blockers on rabbit corneal endothelial function. Curr. Eye Res. 1994, 13, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Mergler, S.; Dannowski, H.; Bednarz, J.; Engelmann, K.; Hartmann, C.; Pleyer, U. Calcium influx induced by activation of receptor tyrosine kinases in SV40-transfected human corneal endothelial cells. Exp. Eye Res. 2003, 77, 485–495. [Google Scholar] [CrossRef]
- Mergler, S.; Pleyer, U.; Reinach, P.; Bednarz, J.; Dannowski, H.; Engelmann, K.; Hartmann, C.; Yousif, T. EGF suppresses hydrogen peroxide induced Ca2+ influx by inhibiting L-type channel activity in cultured human corneal endothelial cells. Exp. Eye Res. 2005, 80, 285–293. [Google Scholar] [CrossRef]
- Mergler, S.; Mertens, C.; Valtink, M.; Reinach, P.S.; Szekely, V.C.; Slavi, N.; Garreis, F.; Abdelmessih, S.; Turker, E.; Fels, G.; et al. Functional significance of thermosensitive transient receptor potential melastatin channel 8 (TRPM8) expression in immortalized human corneal endothelial cells. Exp. Eye Res. 2013, 116, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Mergler, S.; Valtink, M.; Coulson-Thomas, V.J.; Lindemann, D.; Reinach, P.S.; Engelmann, K.; Pleyer, U. TRPV channels mediate temperature-sensing in human corneal endothelial cells. Exp. Eye Res. 2010, 90, 758–770. [Google Scholar] [CrossRef]
- Mergler, S.; Valtink, M.; Taetz, K.; Sahlmuller, M.; Fels, G.; Reinach, P.S.; Engelmann, K.; Pleyer, U. Characterization of transient receptor potential vanilloid channel 4 (TRPV4) in human corneal endothelial cells. Exp. Eye Res. 2011, 93, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Krick, S.; Platoshyn, O.; Sweeney, M.; Kim, H.; Yuan, J.X. Activation of K+ channels induces apoptosis in vascular smooth muscle cells. Am. J. Physiol. Cell. Physiol. 2001, 280, C970–C979. [Google Scholar] [CrossRef]
- Satoh, S.; Tanaka, H.; Ueda, Y.; Oyama, J.; Sugano, M.; Sumimoto, H.; Mori, Y.; Makino, N. Transient receptor potential (TRP) protein 7 acts as a G protein-activated Ca2+ channel mediating angiotensin II-induced myocardial apoptosis. Mol. Cell. Biochem. 2007, 294, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Guarino, B.D.; Paruchuri, S.; Thodeti, C.K. The role of TRPV4 channels in ocular function and pathologies. Exp. Eye Res. 2020, 201, 108257. [Google Scholar] [CrossRef] [PubMed]
- Lapajne, L.; Lakk, M.; Yarishkin, O.; Gubeljak, L.; Hawlina, M.; Krizaj, D. Polymodal Sensory Transduction in Mouse Corneal Epithelial Cells. Invest. Ophthalmol. Vis. Sci. 2020, 61, 2. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Rendon, J.; Sanchez-Guzman, E.; Rueda, A.; Gonzalez, J.; Gulias-Canizo, R.; Aquino-Jarquin, G.; Castro-Munozledo, F.; Garcia-Villegas, R. TRPV4 Regulates Tight Junctions and Affects Differentiation in a Cell Culture Model of the Corneal Epithelium. J. Cell Physiol. 2017, 232, 1794–1807. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Shirai, K.; Miyajima, M.; Reinach, P.S.; Yamanaka, O.; Sumioka, T.; Kokado, M.; Tomoyose, K.; Saika, S. Loss of TRPV4 Function Suppresses Inflammatory Fibrosis Induced by Alkali-Burning Mouse Corneas. PLoS ONE 2016, 11, e0167200. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Yang, H.; Mergler, S.; Liu, H.; Tachado, S.D.; Zhang, F.; Kao, W.W.; Koziel, H.; Pleyer, U.; Reinach, P.S. Dependence of regulatory volume decrease on transient receptor potential vanilloid 4 (TRPV4) expression in human corneal epithelial cells. Cell Calcium 2008, 44, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Elias, A.; Lorenzo, I.M.; Vicente, R.; Valverde, M.A. IP3 receptor binds to and sensitizes TRPV4 channel to osmotic stimuli via a calmodulin-binding site. J. Biol. Chem. 2008, 283, 31284–31288. [Google Scholar] [CrossRef]
- Kottgen, M.; Buchholz, B.; Garcia-Gonzalez, M.A.; Kotsis, F.; Fu, X.; Doerken, M.; Boehlke, C.; Steffl, D.; Tauber, R.; Wegierski, T.; et al. TRPP2 and TRPV4 form a polymodal sensory channel complex. J. Cell Biol. 2008, 182, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Donate-Macian, P.; Jungfleisch, J.; Perez-Vilaro, G.; Rubio-Moscardo, F.; Peralvarez-Marin, A.; Diez, J.; Valverde, M.A. The TRPV4 channel links calcium influx to DDX3X activity and viral infectivity. Nat. Commun. 2018, 9, 2307. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Shikano, M.; Kamiya, T.; Joh, T.; Ugawa, S. The TRPV4 channel is a novel regulator of intracellular Ca2+ in human esophageal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G138–G147. [Google Scholar] [CrossRef] [PubMed]
- Bednarz, J.; Teifel, M.; Friedl, P.; Engelmann, K. Immortalization of human corneal endothelial cells using electroporation protocol optimized for human corneal endothelial and human retinal pigment epithelial cells. Acta Ophthalmol. Scand. 2000, 78, 130–136. [Google Scholar] [CrossRef]
- DuBridge, R.B.; Tang, P.; Hsia, H.C.; Leong, P.M.; Miller, J.H.; Calos, M.P. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell. Biol. 1987, 7, 379–387. [Google Scholar] [PubMed]
- Rasheed, S.; Nelson-Rees, W.A.; Toth, E.M.; Arnstein, P.; Gardner, M.B. Characterization of a newly derived human sarcoma cell line (HT-1080). Cancer 1974, 33, 1027–1033. [Google Scholar] [CrossRef]
- Amer, D.A.; Kretzschmar, G.; Muller, N.; Stanke, N.; Lindemann, D.; Vollmer, G. Activation of transgenic estrogen receptor-beta by selected phytoestrogens in a stably transduced rat serotonergic cell line. J. Steroid Biochem. Mol. Biol. 2010, 120, 208–217. [Google Scholar] [CrossRef]
- Mochizuki, H.I.; Schwartz, J.P.; Tanaka, K.; Brady, R.O.; Reiser, J. High-Titer Human Immunodeficiency Virus Type 1-Based Vector Systems for Gene Delivery into Nondividing Cells. J. Virol. 1998, 72, 8873–8883. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, D.; Stirnnagel, K.; Luüftenegger, D. Foamy Viral Envelope Genes. European Patent 2,138,584B1A61K48/00, 14 August 2013. [Google Scholar]
- Voets, T.; Droogmans, G.; Wissenbach, U.; Janssens, A.; Flockerzi, V.; Nilius, B. The principle of temperature-dependent gating in cold- and heat-sensitive TRP channels. Nature 2004, 430, 748–754. [Google Scholar] [CrossRef]
- Wegener, J.; Abrams, D.; Willenbrink, W.; Galla, H.J.; Janshoff, A. Automated multi-well device to measure transepithelial electrical resistances under physiological conditions. Biotechniques 2004, 37, 590–597. [Google Scholar] [CrossRef]
- Thorneloe, K.S.; Cheung, M.; Holt, D.A.; Willette, R.N. Properties of theTRPV4 agonist GSK1016790A and the TRPV4 antagonist GSK2193874. Physiol. Rev. 2017, 97, 1231–1232. [Google Scholar] [CrossRef][Green Version]
- Thorneloe, K.S.; Sulpizio, A.C.; Lin, Z.; Figueroa, D.J.; Clouse, A.K.; McCafferty, G.P.; Chendrimada, T.P.; Lashinger, E.S.; Gordon, E.; Evans, L.; et al. N-((1S)-1-{[4-((2S)-2-{[(2,4-dichlorophenyl)sulfonyl]amino}-3-hydroxypropa noyl)-1-piperazinyl]carbonyl}-3-methylbutyl)-1-benzothiophene-2-carboxamid e (GSK1016790A), a novel and potent transient receptor potential vanilloid 4 channel agonist induces urinary bladder contraction and hyperactivity: Part I. J. Pharmacol. Exp. Ther. 2008, 326, 432–442. [Google Scholar]
- Krizaj, D. What is glaucoma? In Webvision: The Organization of the Retina and Visual System; Kolb, H., Fernandez, E., Nelson, R., Eds.; University of Utah Health Sciences Center: Salt Lake City, UT, USA, 1995. [Google Scholar]
- Delamere, N.A.; Mandal, A.; Shahidullah, M. The Significance of TRPV4 Channels and Hemichannels in the Lens and Ciliary Epithelium. J. Ocul. Pharmacol. Ther. 2016, 32, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Krizaj, D.; Ryskamp, D.A.; Tian, N.; Tezel, G.; Mitchell, C.H.; Slepak, V.Z.; Shestopalov, V.I. From mechanosensitivity to inflammatory responses: New players in the pathology of glaucoma. Curr. Eye Res. 2014, 39, 105–119. [Google Scholar] [CrossRef]
- Liedtke, W. TRPV4 plays an evolutionary conserved role in the transduction of osmotic and mechanical stimuli in live animals. J. Physiol. 2005, 567, 53–58. [Google Scholar] [CrossRef]
- White, J.P.; Cibelli, M.; Urban, L.; Nilius, B.; McGeown, J.G.; Nagy, I. TRPV4: Molecular Conductor of a Diverse Orchestra. Physiol. Rev. 2016, 96, 911–973. [Google Scholar] [CrossRef] [PubMed]
- Alptekin, M.; Eroglu, S.; Tutar, E.; Sencan, S.; Geyik, M.A.; Ulasli, M.; Demiryurek, A.T.; Camci, C. Gene expressions of TRP channels in glioblastoma multiforme and relation with survival. Tumour Biol. 2015, 36, 9209–9213. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.J.; Cormier, R.T.; Scott, P.M. Role of ion channels in gastrointestinal cancer. World J. Gastroenterol. 2019, 25, 5732–5772. [Google Scholar] [CrossRef] [PubMed]
- Lanciotti, A.; Brignone, M.S.; Molinari, P.; Visentin, S.; De Nuccio, C.; Macchia, G.; Aiello, C.; Bertini, E.; Aloisi, F.; Petrucci, T.C.; et al. Megalencephalic leukoencephalopathy with subcortical cysts protein 1 functionally cooperates with the TRPV4 cation channel to activate the response of astrocytes to osmotic stress: Dysregulation by pathological mutations. Hum. Mol. Genet. 2012, 21, 2166–2180. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell. Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Ryskamp, D.A.; Witkovsky, P.; Barabas, P.; Huang, W.; Koehler, C.; Akimov, N.P.; Lee, S.H.; Chauhan, S.; Xing, W.; Renteria, R.C.; et al. The polymodal ion channel transient receptor potential vanilloid 4 modulates calcium flux, spiking rate, and apoptosis of mouse retinal ganglion cells. J. Neurosci. 2011, 31, 7089–7101. [Google Scholar] [CrossRef] [PubMed]
- Afshari, N.A.; Igo, R.P., Jr.; Morris, N.J.; Stambolian, D.; Sharma, S.; Pulagam, V.L.; Dunn, S.; Stamler, J.F.; Truitt, B.J.; Rimmler, J.; et al. Genome-wide association study identifies three novel loci in Fuchs endothelial corneal dystrophy. Nat. Commun. 2017, 8, 14898. [Google Scholar] [CrossRef]
- Jie, P.; Hong, Z.; Tian, Y.; Li, Y.; Lin, L.; Zhou, L.; Du, Y.; Chen, L.; Chen, L. Activation of transient receptor potential vanilloid 4 induces apoptosis in hippocampus through downregulating PI3K/Akt and upregulating p38 MAPK signaling pathways. Cell Death Dis. 2015, 6, e1775. [Google Scholar] [CrossRef] [PubMed]
- Mrkonjic, S.; Garcia-Elias, A.; Pardo-Pastor, C.; Bazellieres, E.; Trepat, X.; Vriens, J.; Ghosh, D.; Voets, T.; Vicente, R.; Valverde, M.A. TRPV4 participates in the establishment of trailing adhesions and directional persistence of migrating cells. Pflugers Arch. 2015, 467, 2107–2119. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Wu, P.F.; Ma, J.X.; Liao, M.J.; Xu, L.S.; Yi, L. TRPV4 activates the Cdc42/N-wasp pathway to promote glioblastoma invasion by altering cellular protrusions. Sci. Rep. 2020, 10, 14151. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wu, Y.T.; Zhu, Q.; Zhang, H.Y.; Huang, Z.; Zhang, D.; Qi, H.; Liang, G.L.; He, X.Q.; Wang, X.F.; et al. TRPV4 is involved in levonorgestrel-induced reduction in oviduct ciliary beating. J. Pathol. 2019, 248, 77–87. [Google Scholar] [CrossRef]
- Corrigan, M.A.; Johnson, G.P.; Stavenschi, E.; Riffault, M.; Labour, M.N.; Hoey, D.A. TRPV4-mediates oscillatory fluid shear mechanotransduction in mesenchymal stem cells in part via the primary cilium. Sci. Rep. 2018, 8, 3824. [Google Scholar] [CrossRef] [PubMed]
- Nabissi, M.; Marinelli, O.; Morelli, M.B.; Nicotra, G.; Iannarelli, R.; Amantini, C.; Santoni, G.; Maggi, F. Thyme extract increases mucociliary-beating frequency in primary cell lines from chronic obstructive pulmonary disease patients. Biomed. Pharmacother. 2018, 105, 1248–1253. [Google Scholar] [CrossRef]
- Gallagher, B.C. Primary cilia of the corneal endothelium. Am. J. Anat. 1980, 159, 475–484. [Google Scholar] [CrossRef]
- Blitzer, A.L.; Panagis, L.; Gusella, G.L.; Danias, J.; Mlodzik, M.; Iomini, C. Primary cilia dynamics instruct tissue patterning and repair of corneal endothelium. Proc. Natl. Acad. Sci. USA 2011, 108, 2819–2824. [Google Scholar] [CrossRef]
- Doughty, M.J. Changes in cell surface primary cilia and microvilli concurrent with measurements of fluid flow across the rabbit corneal endothelium ex vivo. Tissue Cell 1998, 30, 634–643. [Google Scholar] [CrossRef]
- Collin, S.P.; Barry Collin, H. Primary cilia in vertebrate corneal endothelial cells. Cell Biol. Int. 2004, 28, 125–130. [Google Scholar] [CrossRef]
- Luo, N.; Conwell, M.D.; Chen, X.; Kettenhofen, C.I.; Westlake, C.J.; Cantor, L.B.; Wells, C.D.; Weinreb, R.N.; Corson, T.W.; Spandau, D.F.; et al. Primary cilia signaling mediates intraocular pressure sensation. Proc. Natl. Acad. Sci. USA 2014, 111, 12871–12876. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen 1 | Host 1 | Clone 2 | Isotype | Supplier | Dilution, Method 3 |
|---|---|---|---|---|---|
| TRPV4 | rb | pc | IgG | Alomone Labs | WB 1:1000 ICC/IF 1:100 |
| HA | rb | pc | IgG | Bethyl Laboratories | WB 1:1000 ICC/IF 1:100 |
| HA | mu | HA.C5 | IgG3 | Abcam | WB 1:1000 |
| ZO-1 | mu | ZO1-1A12 | IgG1 | Invitrogen | ICC 1:100 |
| y-tubulin | mu | GTU-88 | IgG | Sigma | WB 1:5000 |
| Na+/K+-ATPase | mu | 464.6 | IgG | Abcam | ICC 1:200 |
| Isotype control | rb | IgG | Invitrogen | ICC/IF 1:100 | |
| Isotype control | mu | IgG | Invitrogen | ICC/IF 1:500 | |
| HRP-anti mu | gt | pc | IgG | Dako | WB 1:1000 |
| HRP-anti rb | sw | pc | IgG | Dako | WB 1:1000 |
| Alexa Fluor 488 | gt | pc | IgG | Invitrogen | ICC/IF 1:800 |
| Anti rb-IgG | gt | pc | IgG | Invitrogen | ICC 1:800 |
| Alexa Fluor 568 | rb | pc | IgG | Alomone Labs | WB 1:1000 ICC/IF 1:100 |
| Anti mu-IgG | rb | pc | IgG | Bethyl Laboratories | WB 1:1000 ICC/IF 1:100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donau, J.; Luo, H.; Virta, I.; Skupin, A.; Pushina, M.; Loeffler, J.; Haertel, F.V.; Das, A.; Kurth, T.; Gerlach, M.; et al. TRPV4 Stimulation Level Regulates Ca2+-Dependent Control of Human Corneal Endothelial Cell Viability and Survival. Membranes 2022, 12, 281. https://doi.org/10.3390/membranes12030281
Donau J, Luo H, Virta I, Skupin A, Pushina M, Loeffler J, Haertel FV, Das A, Kurth T, Gerlach M, et al. TRPV4 Stimulation Level Regulates Ca2+-Dependent Control of Human Corneal Endothelial Cell Viability and Survival. Membranes. 2022; 12(3):281. https://doi.org/10.3390/membranes12030281
Chicago/Turabian StyleDonau, Jennifer, Huan Luo, Iiris Virta, Annett Skupin, Margarita Pushina, Jana Loeffler, Frauke V. Haertel, Anupam Das, Thomas Kurth, Michael Gerlach, and et al. 2022. "TRPV4 Stimulation Level Regulates Ca2+-Dependent Control of Human Corneal Endothelial Cell Viability and Survival" Membranes 12, no. 3: 281. https://doi.org/10.3390/membranes12030281
APA StyleDonau, J., Luo, H., Virta, I., Skupin, A., Pushina, M., Loeffler, J., Haertel, F. V., Das, A., Kurth, T., Gerlach, M., Lindemann, D., Reinach, P. S., Mergler, S., & Valtink, M. (2022). TRPV4 Stimulation Level Regulates Ca2+-Dependent Control of Human Corneal Endothelial Cell Viability and Survival. Membranes, 12(3), 281. https://doi.org/10.3390/membranes12030281