
P-Loop Channels: Experimental Structures, and Physics-Based and Neural Networks-Based Models


Abstract
1. Introduction
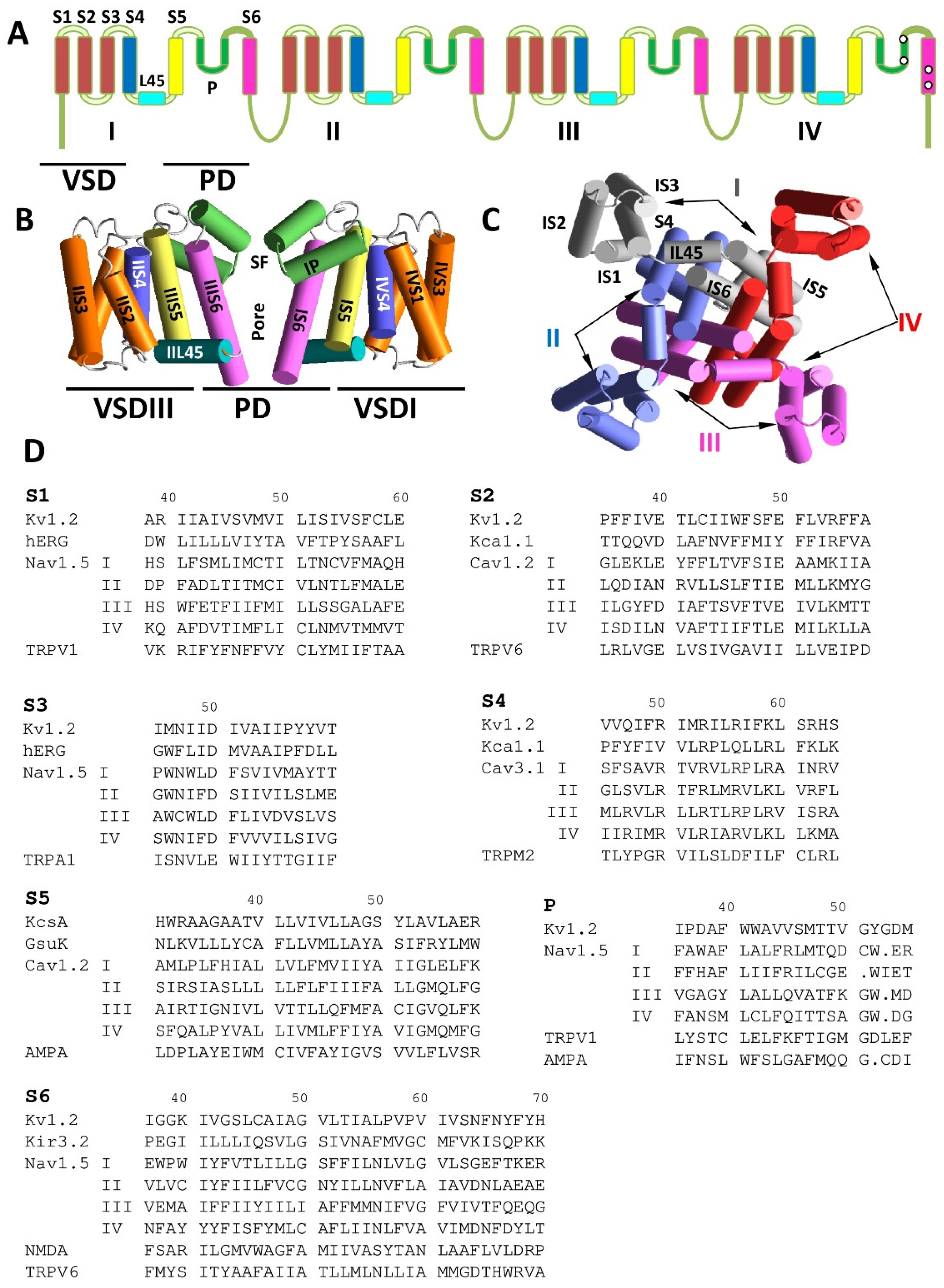
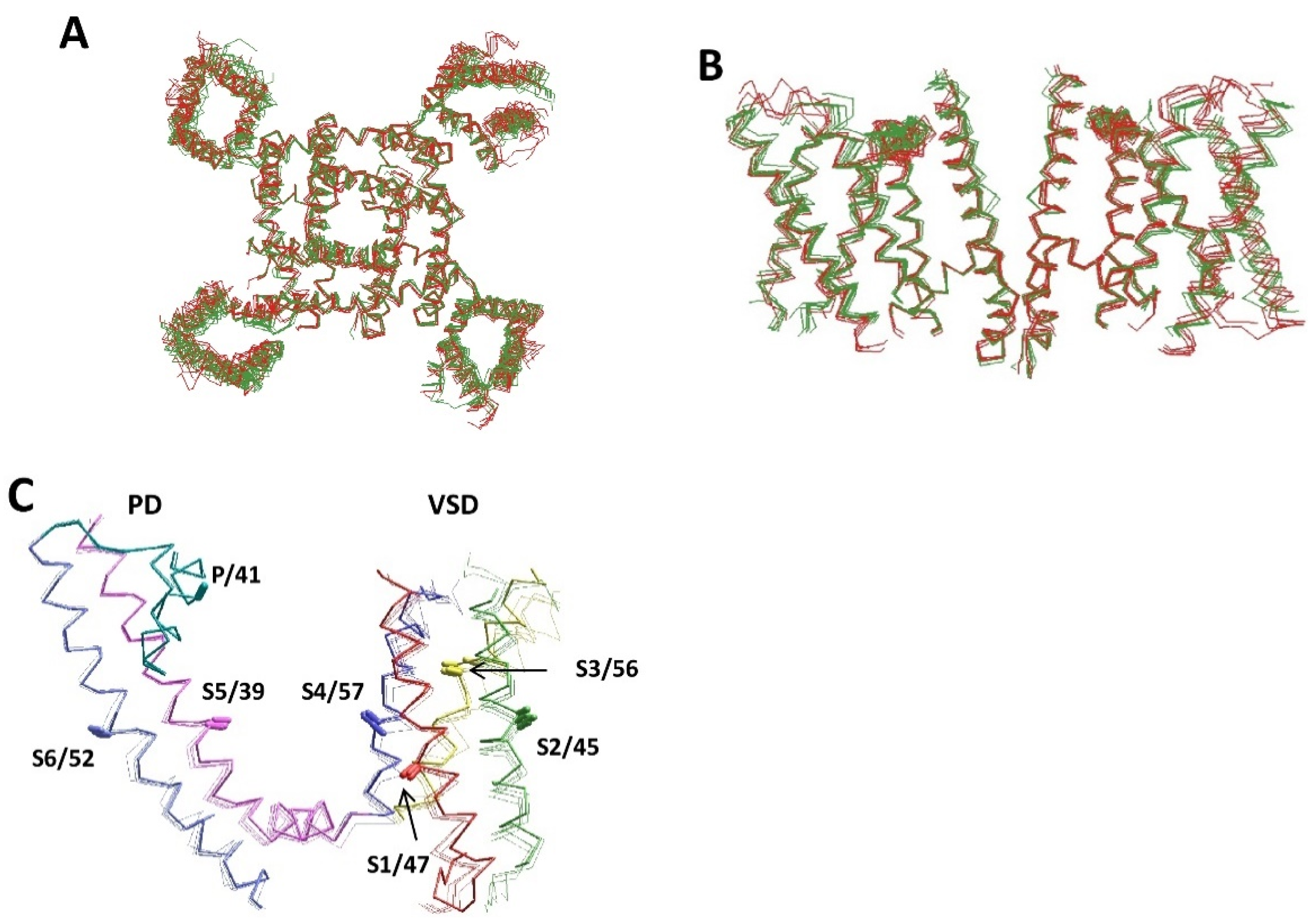
2. Comparative Structural Analysis of P-Loop Channels
3. Crystal and Cryo-EM Structures
4. Structures of P-Loop Channels with Drugs and Toxins
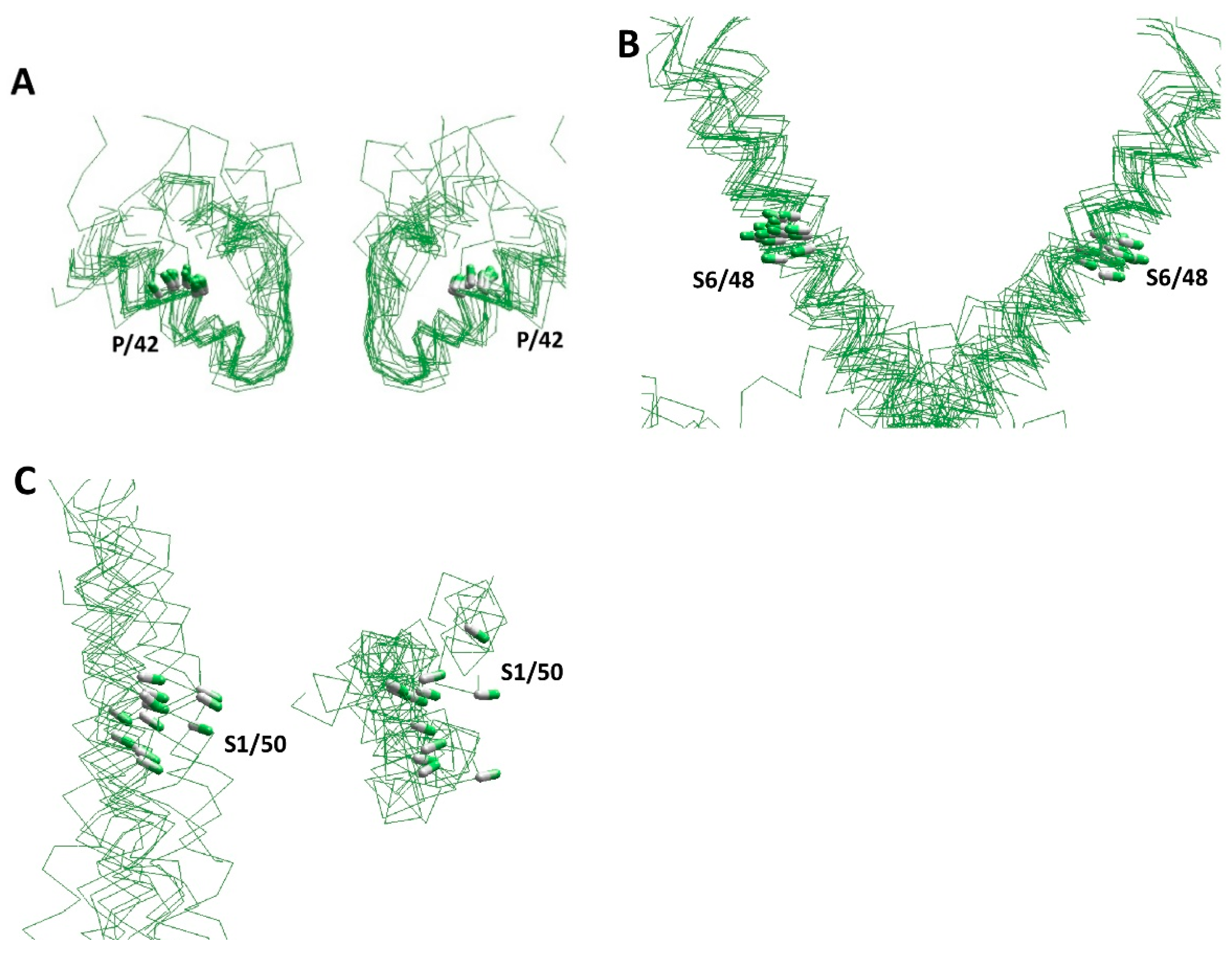
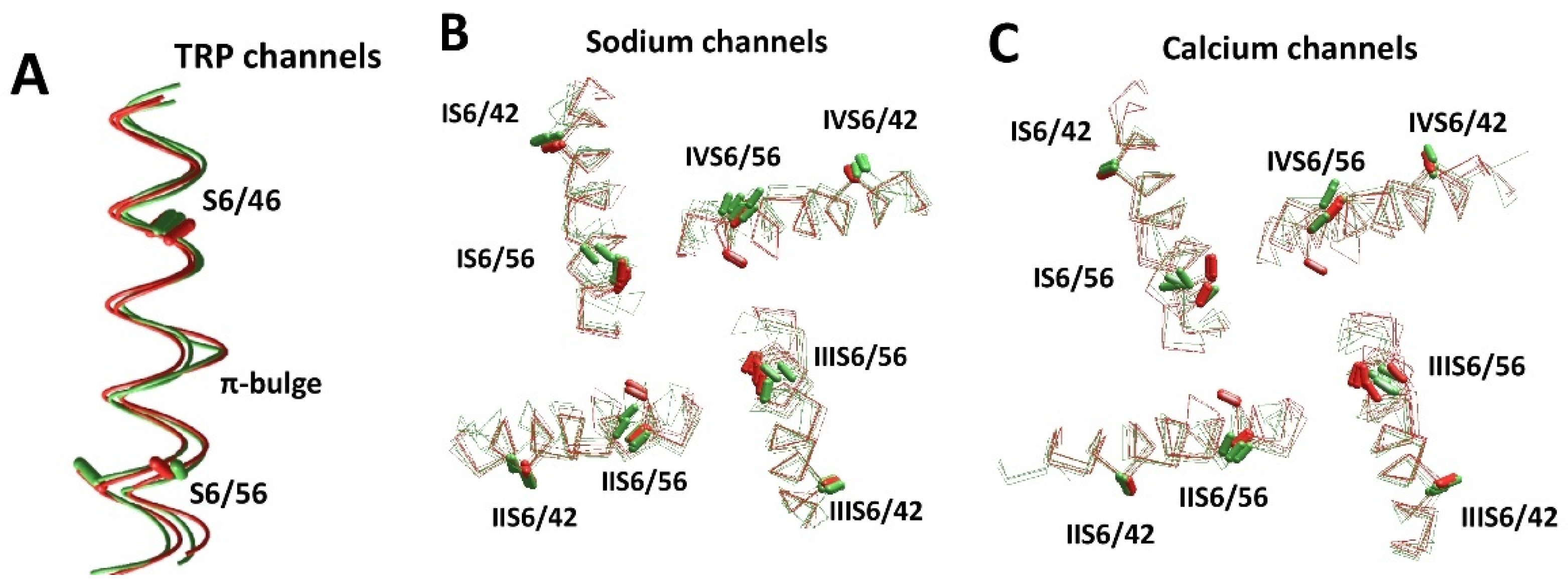
5. π-Bulges in the Inner Helices
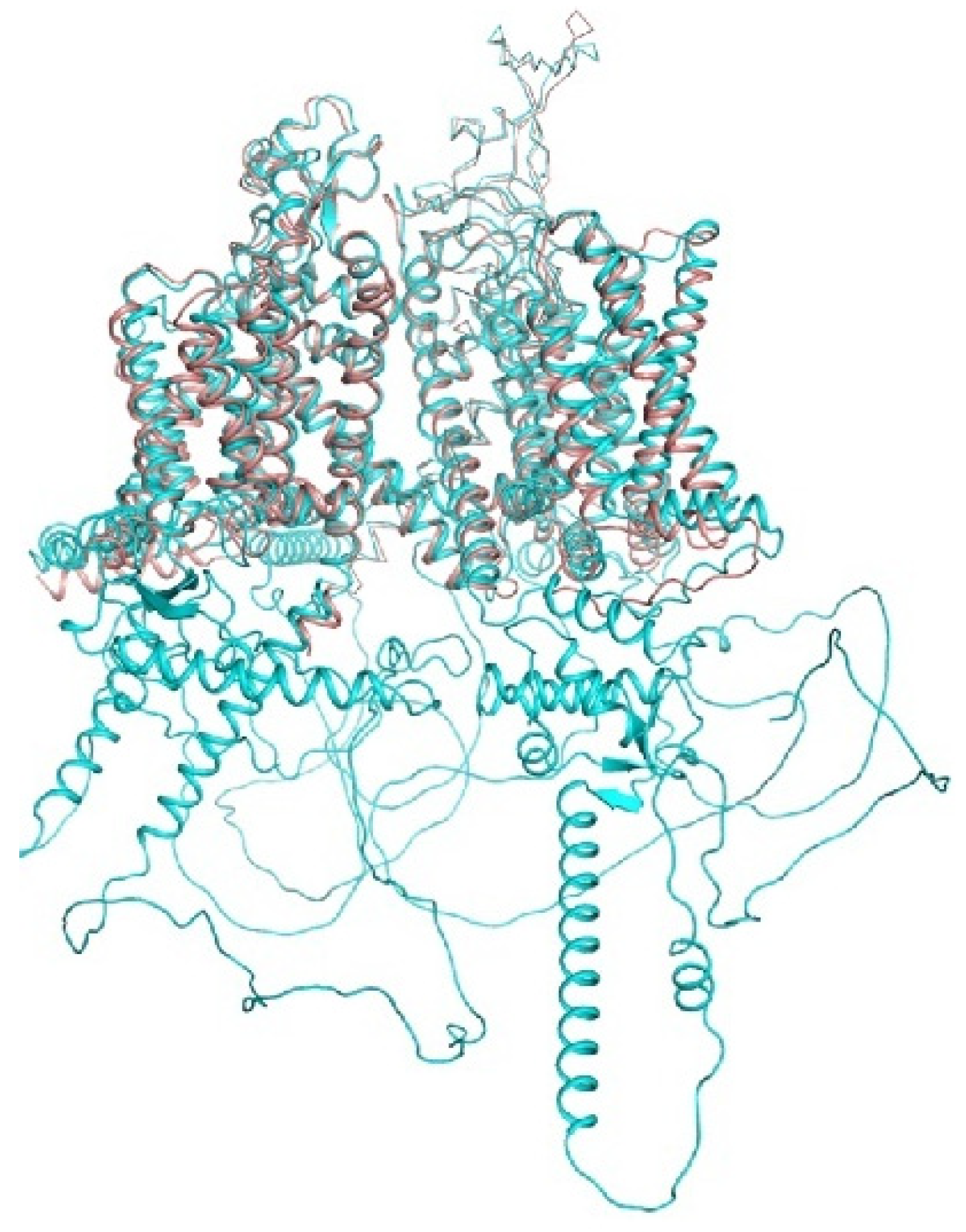
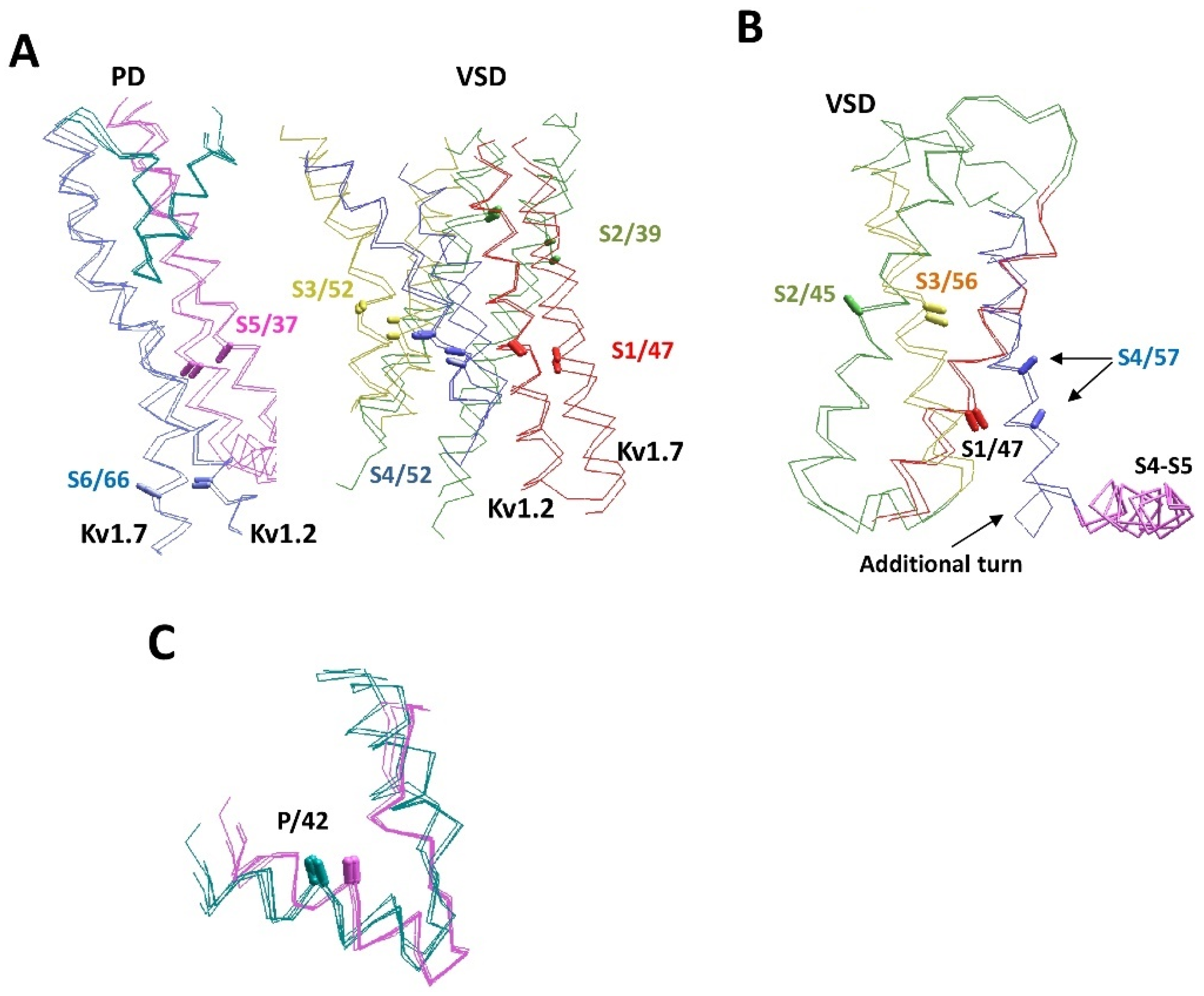
6. AlphaFold2 Models and Experimental Structures
7. Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| Cryo-EM | cryo-electron microscopy |
| DHP | dihydropyridines |
| NMDA | N-methyl-d-aspartate |
| iGluR | ionotropic glutamate receptors |
| PD | pore domain |
| PDB | protein data bank |
| PDB ID | PDB index |
| RMS | root mean square |
| S1–S6 | transmembrane helices in P-loop channels |
| SF | selectivity filter |
| TRP | transient receptor potential |
| VSD | voltage-sensing domain |
References
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Voltage-gated sodium channels at 60: Structure, function and pathophysiology. J. Physiol. 2012, 590, 2577–2589. [Google Scholar] [CrossRef] [PubMed]
- Wulff, H.; Zhorov, B.S. K+ channel modulators for the treatment of neurological disorders and autoimmune diseases. Chem. Rev. 2008, 108, 1744–1773. [Google Scholar] [CrossRef]
- MacKinnon, R. Potassium channels. FEBS Lett. 2003, 555, 62–65. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Montell, C. TRP channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Hansen, K.B.; Wollmuth, L.P.; Bowie, D.; Furukawa, H.; Menniti, F.S.; Sobolevsky, A.I.; Swanson, G.T.; Swanger, S.A.; Greger, I.H.; Nakagawa, T.; et al. Structure, Function, and Pharmacology of Glutamate Receptor Ion Channels. Pharmacol. Rev. 2021, 73, 298–487. [Google Scholar] [CrossRef]
- Noreng, S.; Li, T.; Payandeh, J. Structural Pharmacology of Voltage-Gated Sodium Channels. J. Mol. Biol. 2021, 433, 166967. [Google Scholar] [CrossRef]
- Catterall, W.A.; Lenaeus, M.J.; Gamal El-Din, T.M. Structure and Pharmacology of Voltage-Gated Sodium and Calcium Channels. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 133–154. [Google Scholar] [CrossRef]
- Huang, Y.; Fliegert, R.; Guse, A.H.; Lu, W.; Du, J. A structural overview of the ion channels of the TRPM family. Cell Calcium 2020, 85, 102111. [Google Scholar] [CrossRef]
- Mayer, M.L.; Armstrong, N. Structure and function of glutamate receptor ion channels. Annu. Rev. Physiol. 2004, 66, 161–181. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Montell, C. Forcing open TRP channels: Mechanical gating as a unifying activation mechanism. Biochem. Biophys. Res. Commun. 2015, 460, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Grizel, A.V.; Glukhov, G.S.; Sokolova, O.S. Mechanisms of activation of voltage-gated potassium channels. Acta Nat. 2014, 6, 10–26. [Google Scholar] [CrossRef]
- Bagneris, C.; Naylor, C.E.; McCusker, E.C.; Wallace, B.A. Structural model of the open-closed-inactivated cycle of prokaryotic voltage-gated sodium channels. J. Gen. Physiol. 2015, 145, 5–16. [Google Scholar] [CrossRef]
- Lipscombe, D.; Helton, T.D.; Xu, W. L-type calcium channels: The low down. J. Neurophysiol. 2004, 92, 2633–2641. [Google Scholar] [CrossRef]
- Mazzolini, M.; Marchesi, A.; Giorgetti, A.; Torre, V. Gating in CNGA1 channels. Pflug. Arch. 2010, 459, 547–555. [Google Scholar] [CrossRef]
- Enyedi, P.; Czirjak, G. Molecular background of leak K+ currents: Two-pore domain potassium channels. Physiol. Rev. 2010, 90, 559–605. [Google Scholar] [CrossRef]
- Zhorov, B.S.; Tikhonov, D.B. Potassium, sodium, calcium and glutamate-gated channels: Pore architecture and ligand action. J. Neurochem. 2004, 88, 782–799. [Google Scholar] [CrossRef]
- Wollmuth, L.P.; Sobolevsky, A.I. Structure and gating of the glutamate receptor ion channel. Trends Neurosci. 2004, 27, 321–328. [Google Scholar] [CrossRef]
- Armstrong, C.M.; Hille, B. The inner quaternary ammonium ion receptor in potassium channels of the node of Ranvier. J. Gen. Physiol. 1972, 59, 388–400. [Google Scholar] [CrossRef]
- Heinemann, S.H.; Terlau, H.; Stuhmer, W.; Imoto, K.; Numa, S. Calcium channel characteristics conferred on the sodium channel by single mutations. Nature 1992, 356, 441–443. [Google Scholar] [CrossRef] [PubMed]
- Dudley, S.C., Jr.; Chang, N.; Hall, J.; Lipkind, G.; Fozzard, H.A.; French, R.J. μ-conotoxin GIIIA interactions with the voltage-gated Na+ channel predict a clockwise arrangement of the domains. J. Gen. Physiol. 2000, 116, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Korkosh, V.S.; Zhorov, B.S.; Tikhonov, D.B. Analysis of inter-residue contacts reveals folding stabilizers in P-loops of potassium, sodium, and TRPV channels. Eur. Biophys. J. 2016, 45, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.M.; Nimigean, C.M. Voltage-Gated Potassium Channels: A Structural Examination of Selectivity and Gating. Cold Spring Harb. Perspect. Biol. 2016, 8, a029231. [Google Scholar] [CrossRef]
- Varadi, G.; Strobeck, M.; Koch, S.; Caglioti, L.; Zucchi, C.; Palyi, G. Molecular elements of ion permeation and selectivity within calcium channels. Crit. Rev. Biochem. Mol. Biol. 1999, 34, 181–214. [Google Scholar] [CrossRef]
- Doyle, D.A.; Morais Cabral, J.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Long, S.B.; Tao, X.; Campbell, E.B.; MacKinnon, R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 2007, 450, 376–382. [Google Scholar] [CrossRef]
- Tikhonov, D.B.; Zhorov, B.S. Architecture and pore block of eukaryotic voltage-gated sodium channels in view of NavAb bacterial sodium channel structure. Mol. Pharmacol. 2012, 82, 97–104. [Google Scholar] [CrossRef]
- Wu, J.; Yan, Z.; Li, Z.; Qian, X.; Lu, S.; Dong, M.; Zhou, Q.; Yan, N. Structure of the voltage-gated calcium channel Ca(v)1.1 at 3.6 A resolution. Nature 2016, 537, 191–196. [Google Scholar] [CrossRef]
- Fan, C.; Sukomon, N.; Flood, E.; Rheinberger, J.; Allen, T.W.; Nimigean, C.M. Ball-and-chain inactivation in a calcium-gated potassium channel. Nature 2020, 580, 288–293. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, G.; Wu, J.; Wu, Q.; Gao, S.; Yan, Z.; Lei, J.; Yan, N. Molecular Basis for Ligand Modulation of a Mammalian Voltage-Gated Ca2+ Channel. Cell 2019, 177, 1495–1506.e12. [Google Scholar] [CrossRef]
- Clayton, G.M.; Altieri, S.; Heginbotham, L.; Unger, V.M.; Morais-Cabral, J.H. Structure of the transmembrane regions of a bacterial cyclic nucleotide-regulated channel. Proc. Natl. Acad. Sci. USA 2008, 105, 1511–1515. [Google Scholar] [CrossRef]
- Gao, S.; Yan, N. Structural Basis of the Modulation of the Voltage-Gated Calcium Ion Channel Cav 1.1 by Dihydropyridine Compounds. Angew. Chem. Int. Ed. Engl. 2021, 60, 3131–3137. [Google Scholar] [CrossRef]
- Sun, J.; MacKinnon, R. Structural Basis of Human KCNQ1 Modulation and Gating. Cell 2020, 180, 340–347.e9. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, G.; Wu, Q.; Wu, K.; Li, R.; Lei, J.; Pan, X.; Yan, N. Cryo-EM structures of apo and antagonist-bound human Cav3.1. Nature 2019, 576, 492–497. [Google Scholar] [CrossRef]
- Wang, W.; MacKinnon, R. Cryo-EM Structure of the Open Human Ether-a-go-go-Related K+ Channel hERG. Cell 2017, 169, 422–430.e10. [Google Scholar] [CrossRef]
- Whorton, M.R.; MacKinnon, R. X-ray structure of the mammalian GIRK2-betagamma G-protein complex. Nature 2013, 498, 190–197. [Google Scholar] [CrossRef]
- Kong, C.; Zeng, W.; Ye, S.; Chen, L.; Sauer, D.B.; Lam, Y.; Derebe, M.G.; Jiang, Y. Distinct gating mechanisms revealed by the structures of a multi-ligand gated K+ channel. Elife 2012, 1, e00184. [Google Scholar] [CrossRef]
- Tao, X.; Hite, R.K.; MacKinnon, R. Cryo-EM structure of the open high-conductance Ca2+-activated K+ channel. Nature 2017, 541, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Payandeh, J.; Scheuer, T.; Zheng, N.; Catterall, W.A. The crystal structure of a voltage-gated sodium channel. Nature 2011, 475, 353–358. [Google Scholar] [CrossRef]
- Twomey, E.C.; Yelshanskaya, M.V.; Vassilevski, A.A.; Sobolevsky, A.I. Mechanisms of Channel Block in Calcium-Permeable AMPA Receptors. Neuron 2018, 99, 956–968.e4. [Google Scholar] [CrossRef]
- Lenaeus, M.J.; Gamal El-Din, T.M.; Ing, C.; Ramanadane, K.; Pomes, R.; Zheng, N.; Catterall, W.A. Structures of closed and open states of a voltage-gated sodium channel. Proc. Natl. Acad. Sci. USA 2017, 114, E3051–E3060. [Google Scholar] [CrossRef] [PubMed]
- Regan, M.C.; Grant, T.; McDaniel, M.J.; Karakas, E.; Zhang, J.; Traynelis, S.F.; Grigorieff, N.; Furukawa, H. Structural Mechanism of Functional Modulation by Gene Splicing in NMDA Receptors. Neuron 2018, 98, 521–529.e3. [Google Scholar] [CrossRef]
- Wisedchaisri, G.; Tonggu, L.; McCord, E.; Gamal El-Din, T.M.; Wang, L.; Zheng, N.; Catterall, W.A. Resting-State Structure and Gating Mechanism of a Voltage-Gated Sodium Channel. Cell 2019, 178, 993–1003.e12. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Armache, J.P.; Gao, Y.; Cheng, Y.; Julius, D. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 2015, 520, 511–517. [Google Scholar] [CrossRef]
- McCusker, E.C.; Bagneris, C.; Naylor, C.E.; Cole, A.R.; D’Avanzo, N.; Nichols, C.G.; Wallace, B.A. Structure of a bacterial voltage-gated sodium channel pore reveals mechanisms of opening and closing. Nat. Commun. 2012, 3, 1102. [Google Scholar] [CrossRef]
- Cao, E.; Liao, M.; Cheng, Y.; Julius, D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 2013, 504, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ren, W.; DeCaen, P.; Yan, C.; Tao, X.; Tang, L.; Wang, J.; Hasegawa, K.; Kumasaka, T.; He, J.; et al. Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 2012, 486, 130–134. [Google Scholar] [CrossRef]
- Zubcevic, L.; Herzik, M.A., Jr.; Chung, B.C.; Liu, Z.; Lander, G.C.; Lee, S.Y. Cryo-electron microscopy structure of the TRPV2 ion channel. Nat. Struct. Mol. Biol. 2016, 23, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Li, Z.; Jiang, Y.; Pan, X.; Wu, J.; Cristofori-Armstrong, B.; Smith, J.J.; Chin, Y.K.Y.; Lei, J.; Zhou, Q.; et al. Structural basis for the modulation of voltage-gated sodium channels by animal toxins. Science 2018, 362. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; McGoldrick, L.L.; Sobolevsky, A.I. Structure and gating mechanism of the transient receptor potential channel TRPV3. Nat. Struct. Mol. Biol. 2018, 25, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Li, Z.; Huang, X.; Huang, G.; Gao, S.; Shen, H.; Liu, L.; Lei, J.; Yan, N. Molecular basis for pore blockade of human Na+ channel Nav1.2 by the μ-conotoxin KIIIA. Science 2019, 363, 1309–1313. [Google Scholar] [CrossRef]
- Zubcevic, L.; Herzik, M.A., Jr.; Wu, M.; Borschel, W.F.; Hirschi, M.; Song, A.S.; Lander, G.C.; Lee, S.Y. Conformational ensemble of the human TRPV3 ion channel. Nat. Commun. 2018, 9, 4773. [Google Scholar] [CrossRef]
- Pan, X.; Li, Z.; Zhou, Q.; Shen, H.; Wu, K.; Huang, X.; Chen, J.; Zhang, J.; Zhu, X.; Lei, J.; et al. Structure of the human voltage-gated sodium channel Nav1.4 in complex with beta1. Science 2018, 362, eaau2486. [Google Scholar] [CrossRef]
- Yan, Z.; Zhou, Q.; Wang, L.; Wu, J.; Zhao, Y.; Huang, G.; Peng, W.; Shen, H.; Lei, J.; Yan, N. Structure of the Nav1.4-beta1 Complex from Electric Eel. Cell 2017, 170, 470–482.e11. [Google Scholar] [CrossRef]
- Singh, A.K.; McGoldrick, L.L.; Demirkhanyan, L.; Leslie, M.; Zakharian, E.; Sobolevsky, A.I. Structural basis of temperature sensation by the TRP channel TRPV3. Nat. Struct. Mol. Biol. 2019, 26, 994–998. [Google Scholar] [CrossRef]
- Jiang, D.; Shi, H.; Tonggu, L.; Gamal El-Din, T.M.; Lenaeus, M.J.; Zhao, Y.; Yoshioka, C.; Zheng, N.; Catterall, W.A. Structure of the Cardiac Sodium Channel. Cell 2020, 180, 122–134.e10. [Google Scholar] [CrossRef]
- Jiang, D.; Banh, R.; Gamal El-Din, T.M.; Tonggu, L.; Lenaeus, M.J.; Pomes, R.; Zheng, N.; Catterall, W.A. Open-state structure and pore gating mechanism of the cardiac sodium channel. Cell 2021, 184, 5151–5162.e11. [Google Scholar] [CrossRef]
- Deng, Z.; Maksaev, G.; Rau, M.; Xie, Z.; Hu, H.; Fitzpatrick, J.A.J.; Yuan, P. Gating of human TRPV3 in a lipid bilayer. Nat. Struct. Mol. Biol. 2020, 27, 635–644. [Google Scholar] [CrossRef]
- Jiang, D.; Tonggu, L.; Gamal El-Din, T.M.; Banh, R.; Pomes, R.; Zheng, N.; Catterall, W.A. Structural basis for voltage-sensor trapping of the cardiac sodium channel by a deathstalker scorpion toxin. Nat. Commun. 2021, 12, 128. [Google Scholar] [CrossRef]
- Shimada, H.; Kusakizako, T.; Dung Nguyen, T.H.; Nishizawa, T.; Hino, T.; Tominaga, M.; Nureki, O. The structure of lipid nanodisc-reconstituted TRPV3 reveals the gating mechanism. Nat. Struct. Mol. Biol. 2020, 27, 645–652. [Google Scholar] [CrossRef]
- Li, Z.; Jin, X.; Wu, T.; Huang, G.; Wu, K.; Lei, J.; Pan, X.; Yan, N. Structural Basis for Pore Blockade of the Human Cardiac Sodium Channel Nav 1.5 by the Antiarrhythmic Drug Quinidine. Angew. Chem. Int. Ed. Engl. 2021, 60, 11474–11480. [Google Scholar] [CrossRef]
- Shen, H.; Liu, D.; Wu, K.; Lei, J.; Yan, N. Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science 2019, 363, 1303–1308. [Google Scholar] [CrossRef]
- Xu, H.; Li, T.; Rohou, A.; Arthur, C.P.; Tzakoniati, F.; Wong, E.; Estevez, A.; Kugel, C.; Franke, Y.; Chen, J.; et al. Structural Basis of Nav1.7 Inhibition by a Gating-Modifier Spider Toxin. Cell 2019, 176, 1238–1239. [Google Scholar] [CrossRef]
- Saotome, K.; Singh, A.K.; Yelshanskaya, M.V.; Sobolevsky, A.I. Crystal structure of the epithelial calcium channel TRPV6. Nature 2016, 534, 506–511. [Google Scholar] [CrossRef]
- Singh, A.K.; McGoldrick, L.L.; Twomey, E.C.; Sobolevsky, A.I. Mechanism of calmodulin inactivation of the calcium-selective TRP channel TRPV6. Sci. Adv. 2018, 4, eaau6088. [Google Scholar] [CrossRef]
- Wang, L.; Fu, T.M.; Zhou, Y.; Xia, S.; Greka, A.; Wu, H. Structures and gating mechanism of human TRPM2. Science 2018, 362, eaav4809. [Google Scholar] [CrossRef]
- She, J.; Guo, J.; Chen, Q.; Zeng, W.; Jiang, Y.; Bai, X.C. Structural insights into the voltage and phospholipid activation of the mammalian TPC1 channel. Nature 2018, 556, 130–134. [Google Scholar] [CrossRef]
- McGoldrick, L.L.; Singh, A.K.; Saotome, K.; Yelshanskaya, M.V.; Twomey, E.C.; Grassucci, R.A.; Sobolevsky, A.I. Opening of the human epithelial calcium channel TRPV6. Nature 2018, 553, 233–237. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Tikhonov, D.B.; Zhorov, B.S.; Magazanik, L.G. Intersegment hydrogen bonds as possible structural determinants of the N/Q/R site in glutamate receptors. Biophys. J. 1999, 77, 1914–1926. [Google Scholar] [CrossRef]
- Lipkind, G.M.; Fozzard, H.A. Molecular modeling of interactions of dihydropyridines and phenylalkylamines with the inner pore of the L-type Ca2+ channel. Mol. Pharmacol. 2003, 63, 499–511. [Google Scholar] [CrossRef]
- Lipkind, G.M.; Fozzard, H.A. Molecular modeling of local anesthetic drug binding by voltage-gated sodium channels. Mol. Pharmacol. 2005, 68, 1611–1622. [Google Scholar] [CrossRef]
- Tikhonov, D.B.; Zhorov, B.S. Modeling P-loops domain of sodium channel: Homology with potassium channels and interaction with ligands. Biophys. J. 2005, 88, 184–197. [Google Scholar] [CrossRef]
- O’Reilly, A.O.; Khambay, B.P.; Williamson, M.S.; Field, L.M.; Wallace, B.A.; Davies, T.G. Modelling insecticide-binding sites in the voltage-gated sodium channel. Biochem. J. 2006, 396, 255–263. [Google Scholar] [CrossRef]
- Huber, I.; Wappl, E.; Herzog, A.; Mitterdorfer, J.; Glossmann, H.; Langer, T.; Striessnig, J. Conserved Ca2+-antagonist-binding properties and putative folding structure of a recombinant high-affinity dihydropyridine-binding domain. Biochem. J. 2000, 347 Pt 3, 829–836. [Google Scholar] [CrossRef]
- Corry, B.; Vora, T.; Chung, S.H. Electrostatic basis of valence selectivity in cationic channels. Biochim. Biophys. Acta 2005, 1711, 72–86. [Google Scholar] [CrossRef][Green Version]
- Cosconati, S.; Marinelli, L.; Lavecchia, A.; Novellino, E. Characterizing the 1,4-dihydropyridines binding interactions in the L-type Ca2+ channel: Model construction and docking calculations. J. Med. Chem. 2007, 50, 1504–1513. [Google Scholar] [CrossRef]
- Catterall, W.A.; Striessnig, J. Receptor sites for Ca2+ channel antagonists. Trends Pharmacol. Sci. 1992, 13, 256–262. [Google Scholar] [CrossRef]
- Hockerman, G.H.; Peterson, B.Z.; Johnson, B.D.; Catterall, W.A. Molecular determinants of drug binding and action on L-type calcium channels. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 361–396. [Google Scholar] [CrossRef]
- Alpert, L.A.; Fozzard, H.A.; Hanck, D.A.; Makielski, J.C. Is there a second external lidocaine binding site on mammalian cardiac cells? Am. J. Physiol. 1989, 257, H79–H84. [Google Scholar] [CrossRef]
- Jiang, Y.; Lee, A.; Chen, J.; Cadene, M.; Chait, B.T.; MacKinnon, R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature 2002, 417, 515–522. [Google Scholar] [CrossRef]
- Long, S.B.; Campbell, E.B.; Mackinnon, R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 2005, 309, 897–903. [Google Scholar] [CrossRef]
- Cordero-Morales, J.F.; Cuello, L.G.; Zhao, Y.; Jogini, V.; Cortes, D.M.; Roux, B.; Perozo, E. Molecular determinants of gating at the potassium-channel selectivity filter. Nat. Struct. Mol. Biol. 2006, 13, 311–318. [Google Scholar] [CrossRef]
- Cuello, L.G.; Jogini, V.; Cortes, D.M.; Pan, A.C.; Gagnon, D.G.; Dalmas, O.; Cordero-Morales, J.F.; Chakrapani, S.; Roux, B.; Perozo, E. Structural basis for the coupling between activation and inactivation gates in K+ channels. Nature 2010, 466, 272–275. [Google Scholar] [CrossRef]
- Gibor, G.; Yakubovich, D.; Rosenhouse-Dantsker, A.; Peretz, A.; Schottelndreier, H.; Seebohm, G.; Dascal, N.; Logothetis, D.E.; Paas, Y.; Attali, B. An inactivation gate in the selectivity filter of KCNQ1 potassium channels. Biophys. J. 2007, 93, 4159–4172. [Google Scholar] [CrossRef]
- Xiong, W.; Li, R.A.; Tian, Y.; Tomaselli, G.F. Molecular motions of the outer ring of charge of the sodium channel: Do they couple to slow inactivation? J. Gen. Physiol. 2003, 122, 323–332. [Google Scholar] [CrossRef]
- Abderemane-Ali, F.; Findeisen, F.; Rossen, N.D.; Minor, D.L., Jr. A Selectivity Filter Gate Controls Voltage-Gated Calcium Channel Calcium-Dependent Inactivation. Neuron 2019, 101, 1134–1149.e3. [Google Scholar] [CrossRef]
- Twomey, E.C.; Yelshanskaya, M.V.; Grassucci, R.A.; Frank, J.; Sobolevsky, A.I. Channel opening and gating mechanism in AMPA-subtype glutamate receptors. Nature 2017, 549, 60–65. [Google Scholar] [CrossRef]
- Ahern, C.A.; Payandeh, J.; Bosmans, F.; Chanda, B. The hitchhiker’s guide to the voltage-gated sodium channel galaxy. J. Gen. Physiol. 2016, 147, 1–24. [Google Scholar] [CrossRef]
- Koivisto, A.P.; Belvisi, M.G.; Gaudet, R.; Szallasi, A. Advances in TRP channel drug discovery: From target validation to clinical studies. Nat. Rev. Drug Discov. 2022, 21, 41–59. [Google Scholar] [CrossRef]
- Korkosh, V.S.; Zaytseva, A.K.; Kostareva, A.A.; Zhorov, B.S. Intersegment Contacts of Potentially Damaging Variants of Cardiac Sodium Channel. Front. Pharmacol. 2021, 12, 3055. [Google Scholar] [CrossRef]
- Korkosh, V.S.; Kiselev, A.M.; Mikhaylov, E.N.; Kostareva, A.A.; Zhorov, B.S. Atomic Mechanisms of Timothy Syndrome-Associated Mutations in Calcium Channel Cav1.2. Front. Physiol. 2019, 10, 335. [Google Scholar] [CrossRef]
- Zaytseva, A.K.; Boitsov, A.S.; Kostareva, A.A.; Zhorov, B.S. Possible Interactions of Extracellular Loop IVP2-S6 with Voltage-Sensing Domain III in Cardiac Sodium Channel. Front. Pharmacol. 2021, 12, 742508. [Google Scholar] [CrossRef]
- Ulmschneider, M.B.; Bagneris, C.; McCusker, E.C.; Decaen, P.G.; Delling, M.; Clapham, D.E.; Ulmschneider, J.P.; Wallace, B.A. Molecular dynamics of ion transport through the open conformation of a bacterial voltage-gated sodium channel. Proc. Natl. Acad. Sci. USA 2013, 110, 6364–6369. [Google Scholar] [CrossRef]
- Berneche, S.; Roux, B. Molecular dynamics of the KcsA K+ channel in a bilayer membrane. Biophys. J. 2000, 78, 2900–2917. [Google Scholar] [CrossRef]
- Shrivastava, I.H.; Sansom, M.S. Simulations of ion permeation through a potassium channel: Molecular dynamics of KcsA in a phospholipid bilayer. Biophys. J. 2000, 78, 557–570. [Google Scholar] [CrossRef]
- Jensen, M.O.; Jogini, V.; Borhani, D.W.; Leffler, A.E.; Dror, R.O.; Shaw, D.E. Mechanism of voltage gating in potassium channels. Science 2012, 336, 229–233. [Google Scholar] [CrossRef]
- Noskov, S.Y.; Berneche, S.; Roux, B. Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature 2004, 431, 830–834. [Google Scholar] [CrossRef]
- Allen, T.W.; Kuyucak, S.; Chung, S.H. Molecular dynamics study of the KcsA potassium channel. Biophys. J. 1999, 77, 2502–2516. [Google Scholar] [CrossRef]
- Biggin, P.C.; Smith, G.R.; Shrivastava, I.; Choe, S.; Sansom, M.S. Potassium and sodium ions in a potassium channel studied by molecular dynamics simulations. Biochim. Biophys. Acta 2001, 1510, 1–9. [Google Scholar] [CrossRef]
- Zhorov, B.S. Possible Mechanism of Ion Selectivity in Eukaryotic Voltage-Gated Sodium Channels. J. Phys. Chem. B 2021, 125, 2074–2088. [Google Scholar] [CrossRef]
- Corry, B.; Thomas, M. Mechanism of ion permeation and selectivity in a voltage gated sodium channel. J. Am. Chem. Soc. 2012, 134, 1840–1846. [Google Scholar] [CrossRef]
- Flood, E.; Boiteux, C.; Allen, T.W. Selective ion permeation involves complexation with carboxylates and lysine in a model human sodium channel. PLoS Comput. Biol. 2018, 14, e1006398. [Google Scholar] [CrossRef]
- Chakrabarti, N.; Ing, C.; Payandeh, J.; Zheng, N.; Catterall, W.A.; Pomes, R. Catalysis of Na+ permeation in the bacterial sodium channel Na(V)Ab. Proc. Natl. Acad. Sci. USA 2013, 110, 11331–11336. [Google Scholar] [CrossRef]
- Gomez-Lagunas, F.; Armstrong, C.M. The relation between ion permeation and recovery from inactivation of ShakerB K+ channels. Biophys. J. 1994, 67, 1806–1815. [Google Scholar] [CrossRef]
- Hoshi, T.; Zagotta, W.N.; Aldrich, R.W. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 1990, 250, 533–538. [Google Scholar] [CrossRef]
- Zagotta, W.N.; Hoshi, T.; Aldrich, R.W. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science 1990, 250, 568–571. [Google Scholar] [CrossRef]
- Tikhonov, D.B.; Zhorov, B.S. The pore domain in glutamate-gated ion channels: Structure, drug binding and similarity with potassium channels. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183401. [Google Scholar] [CrossRef]
- Stevens, M.; Peigneur, S.; Tytgat, J. Neurotoxins and their binding areas on voltage-gated sodium channels. Front. Pharmacol. 2011, 2, 71. [Google Scholar] [CrossRef]
- Catterall, W.A.; Swanson, T.M. Structural Basis for Pharmacology of Voltage-Gated Sodium and Calcium Channels. Mol. Pharmacol. 2015, 88, 141–150. [Google Scholar] [CrossRef]
- Catterall, W.A. Sodium channels, inherited epilepsy, and antiepileptic drugs. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 317–338. [Google Scholar] [CrossRef]
- Silver, K.S.; Du, Y.; Nomura, Y.; Oliveira, E.E.; Salgado, V.L.; Zhorov, B.S.; Dong, K. Voltage-Gated Sodium Channels as Insecticide Targets. Adv. Insect Phys. 2014, 46, 389–433. [Google Scholar] [CrossRef]
- Korkosh, V.S.; Zhorov, B.S.; Tikhonov, D.B. Folding similarity of the outer pore region in prokaryotic and eukaryotic sodium channels revealed by docking of conotoxins GIIIA, PIIIA, and KIIIA in a NavAb-based model of Nav1.4. J. Gen. Physiol. 2014, 144, 231–244. [Google Scholar] [CrossRef]
- Tomasic, T.; Hartzoulakis, B.; Zidar, N.; Chan, F.; Kirby, R.W.; Madge, D.J.; Peigneur, S.; Tytgat, J.; Kikelj, D. Ligand- and structure-based virtual screening for clathrodin-derived human voltage-gated sodium channel modulators. J. Chem. Inf. Model. 2013, 53, 3223–3232. [Google Scholar] [CrossRef]
- Palestro, P.H.; Enrique, N.; Goicoechea, S.; Villalba, M.L.; Sabatier, L.L.; Martin, P.; Milesi, V.; Bruno Blanch, L.E.; Gavernet, L. Searching for New Leads to Treat Epilepsy: Target-Based Virtual Screening for the Discovery of Anticonvulsant Agents. J. Chem. Inf. Model. 2018, 58, 1331–1342. [Google Scholar] [CrossRef]
- Tikhonov, D.B.; Bruhova, I.; Zhorov, B.S. Atomic determinants of state-dependent block of sodium channels by charged local anesthetics and benzocaine. FEBS Lett. 2006, 580, 6027–6032. [Google Scholar] [CrossRef][Green Version]
- Bruhova, I.; Tikhonov, D.B.; Zhorov, B.S. Access and binding of local anesthetics in the closed sodium channel. Mol. Pharmacol. 2008, 74, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Hille, B. Local anesthetics: Hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 1977, 69, 497–515. [Google Scholar] [CrossRef] [PubMed]
- Buyan, A.; Sun, D.; Corry, B. Protonation state of inhibitors determines interaction sites within voltage-gated sodium channels. Proc. Natl. Acad. Sci. USA 2018, 115, E3135–E3144. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Corry, B. Locating the route of entry and binding sites of benzocaine and phenytoin in a bacterial voltage gated sodium channel. PLoS Comput. Biol. 2014, 10, e1003688. [Google Scholar] [CrossRef] [PubMed]
- Tikhonov, D.B.; Zhorov, B.S. Mechanism of sodium channel block by local anesthetics, antiarrhythmics, and anticonvulsants. J. Gen. Physiol. 2017, 149, 465–481. [Google Scholar] [CrossRef]
- Nguyen, P.T.; DeMarco, K.R.; Vorobyov, I.; Clancy, C.E.; Yarov-Yarovoy, V. Structural basis for antiarrhythmic drug interactions with the human cardiac sodium channel. Proc. Natl. Acad. Sci. USA 2019, 116, 2945–2954. [Google Scholar] [CrossRef]
- Fozzard, H.A.; Lipkind, G.M. The tetrodotoxin binding site is within the outer vestibule of the sodium channel. Mar. Drugs 2010, 8, 219–234. [Google Scholar] [CrossRef]
- Lipkind, G.M.; Fozzard, H.A. A structural model of the tetrodotoxin and saxitoxin binding site of the Na+ channel. Biophys. J. 1994, 66, 1–13. [Google Scholar] [CrossRef]
- Xu, L.; Li, D.; Ding, J.; Pan, L.; Ding, X. Insight into tetrodotoxin blockade and resistance mechanisms of Nav 1.2 sodium channel by theoretical approaches. Chem. Biol. Drug Des. 2018, 92, 1445–1457. [Google Scholar] [CrossRef]
- Chen, R.; Chung, S.H. Binding modes of μ-conotoxin to the bacterial sodium channel (NaVAb). Biophys. J. 2012, 102, 483–488. [Google Scholar] [CrossRef]
- Mahdavi, S.; Kuyucak, S. Molecular dynamics study of binding of micro-conotoxin GIIIA to the voltage-gated sodium channel Na(v)1.4. PLoS ONE 2014, 9, e105300. [Google Scholar] [CrossRef]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef]
- Godfraind, T. Discovery and Development of Calcium Channel Blockers. Front. Pharmacol. 2017, 8, 286. [Google Scholar] [CrossRef]
- Cheng, R.C.; Tikhonov, D.B.; Zhorov, B.S. Structural model for phenylalkylamine binding to L-type calcium channels. J. Biol. Chem. 2009, 284, 28332–28342. [Google Scholar] [CrossRef] [PubMed]
- Tikhonov, D.B.; Zhorov, B.S. Structural Model for Dihydropyridine Binding to L-type Calcium Channels. J. Biol. Chem. 2009, 284, 19006–19017. [Google Scholar] [CrossRef]
- Tikhonov, D.B.; Zhorov, B.S. Molecular modeling of benzothiazepine binding in the L-type calcium channel. J. Biol. Chem. 2008, 283, 17594–17604. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shi, G. How CaV1.2-bound verapamil blocks Ca2+ influx into cardiomyocyte: Atomic level views. Pharmacol. Res. 2019, 139, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Tikhonov, D.B.; Bruhova, I.; Garden, D.P.; Zhorov, B.S. State-dependent inter-repeat contacts of exceptionally conserved asparagines in the inner helices of sodium and calcium channels. Pflug. Arch. 2015, 467, 253–266. [Google Scholar] [CrossRef]
- Du, Y.Z.; Tikhonov, D.B.; Nomura, Y.; Dong, K.; Zhorov, B.S. Mutational analysis of state-dependent contacts in the pore module of eukaryotic sodium channels. Arch. Biochem. Biophys. 2018, 652, 59–70. [Google Scholar] [CrossRef]
- Tikhonov, D.B.; Zhorov, B.S. Conservation and variability of the pore-lining helices in P-loop channels. Channels 2017, 11, 660–672. [Google Scholar] [CrossRef][Green Version]
- Yelshanskaya, M.V.; Nadezhdin, K.D.; Kurnikova, M.G.; Sobolevsky, A.I. Structure and function of the calcium-selective TRP channel TRPV6. J. Physiol. 2021, 599, 2673–2697. [Google Scholar] [CrossRef]
- Zubcevic, L.; Lee, S.Y. The role of pi-helices in TRP channel gating. Curr. Opin. Struct. Biol. 2019, 58, 314–323. [Google Scholar] [CrossRef]
- Kokubun, S.; Prod’hom, B.; Becker, C.; Porzig, H.; Reuter, H. Studies on Ca channels in intact cardiac cells: Voltage-dependent effects and cooperative interactions of dihydropyridine enantiomers. Mol. Pharmacol. 1986, 30, 571–584. [Google Scholar]
- Correa, A.M.; Bezanilla, F.; Latorre, R. Gating kinetics of batrachotoxin-modified Na+ channels in the squid giant axon. Voltage and temperature effects. Biophys. J. 1992, 61, 1332–1352. [Google Scholar] [CrossRef]
- Quandt, F.N.; Narahashi, T. Modification of single Na+ channels by batrachotoxin. Proc. Natl. Acad. Sci. USA 1982, 79, 6732–6736. [Google Scholar] [CrossRef]
- Garber, S.S.; Miller, C. Single Na+ channels activated by veratridine and batrachotoxin. J. Gen. Physiol. 1987, 89, 459–480. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Zidek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Buel, G.R.; Walters, K.J. Can AlphaFold2 predict the impact of missense mutations on structure? Nat. Struct. Mol. Biol. 2022, 29, 1–2. [Google Scholar] [CrossRef]
- Meadows, L.S.; Isom, L.L. Sodium channels as macromolecular complexes: Implications for inherited arrhythmia syndromes. Cardiovasc. Res. 2005, 67, 448–458. [Google Scholar] [CrossRef]
- Haworth, A.S.; Brackenbury, W.J. Emerging roles for multifunctional ion channel auxiliary subunits in cancer. Cell Calcium 2019, 80, 125–140. [Google Scholar] [CrossRef]
- Dolphin, A.C. Voltage-gated calcium channels and their auxiliary subunits: Physiology and pathophysiology and pharmacology. J. Physiol. 2016, 594, 5369–5390. [Google Scholar] [CrossRef]
- Gonzalez-Perez, V.; Lingle, C.J. Regulation of BK Channels by Beta and Gamma Subunits. Annu. Rev. Physiol. 2019, 81, 113–137. [Google Scholar] [CrossRef]
- Zhorov, B.S.; Du, Y.; Song, W.; Luo, N.; Gordon, D.; Gurevitz, M.; Dong, K. Mapping the interaction surface of scorpion beta-toxins with an insect sodium channel. Biochem. J. 2021, 478, 2843–2869. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Channel | PDB ID | Ref. | AF a | Channel | PDB ID | Ref. | AF a |
|---|---|---|---|---|---|---|---|
| Potassium | Calcium | ||||||
| KcsA | 1bl8 | [26] | rbCav1.1 | 5gjv | [30] | ||
| MthK | 6u6e | [31] | 6jpa | [32] | |||
| MlotiK1 | 3beh | [33] | 6jpb | [32] | |||
| Kv1.2/Kv2.1 | 2r9r | [28] | 7jpk | [34] | |||
| hKv1.2 | P16389 | 7jpw | [34] | ||||
| hKv1.6 | P17658 | 6jp5 | [32] | ||||
| hKv2.1 | Q14721 | hCav1.1 | Q13698 | ||||
| hKv3.1 | P48547 | hCav1.3 | Q01668 | ||||
| hKv7.1 | 6uzz | [35] | P51787 | hCav3.1 | 6kzo | [36] | O43497 |
| hERG | 5va2 | [37] | 6kzp | [36] | |||
| Kir3.2 | 4kfm | [38] | hCav3.2 | O95180 | |||
| GsuK | 4gx5 | [39] | hCav1.4 | O60840 | |||
| Slo | 5tj6 | [40] | |||||
| Sodium | iGluR | ||||||
| NavAb | 3rvy | [41] | AMPA | 6dm0 | [42] | ||
| 5vb2 | [43] | NMDA | 6cna | [44] | |||
| 5vb8 | [43] | ||||||
| 6p6x | [45] | TRP | |||||
| 6pwp | [45] | TRPA1 | 3j9p | [46] | O75762 | ||
| NavMs | 4f4l | [47] | TRPV1 | 3j5r | [48] | Q8NER1 | |
| NavRh | 4dxw | [49] | TRPV2 | 6oo7 | [50] | Q9Y5S1 | |
| NavPaS | 6a95 | [51] | TRPV3 | 6dvy | [52] | Q8NET8 | |
| Nav1.2 | 6j8e | [53] | Q01118 | 6mhw | [54] | ||
| hNav1.4 | 6agf | [55] | P35499 | 6mho | [54] | ||
| EeNav1.4 | 5xsy | [56] | 6pvl | [57] | |||
| rNav1.5 | 6uz3 | [58] | P15389 | 6dvw | [52] | ||
| 7fbs | [59] | 6uw9 | [60] | ||||
| 7k18 | [61] | 6lgp | [62] | ||||
| hNav1.5 | 6lqa | [63] | Q14524 | 6uw4 | [60] | ||
| hNav1.7 | 6j8j | [64] | 6mhs | [54] | |||
| 6n4r | [65] | TRPV6 | 5iwk | [66] | |||
| hNav1.9 | Q9UI33 | 6e2g | [67] | Q9H1D0 | |||
| hNav2.1 | Q01118 | TRPM2 | 6mj2 | [68] | O94759 | ||
| TPC1 | 6c96 | [69] | TRPM6 | 6Bo9 | [70] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tikhonov, D.B.; Zhorov, B.S. P-Loop Channels: Experimental Structures, and Physics-Based and Neural Networks-Based Models. Membranes 2022, 12, 229. https://doi.org/10.3390/membranes12020229
Tikhonov DB, Zhorov BS. P-Loop Channels: Experimental Structures, and Physics-Based and Neural Networks-Based Models. Membranes. 2022; 12(2):229. https://doi.org/10.3390/membranes12020229
Chicago/Turabian StyleTikhonov, Denis B., and Boris S. Zhorov. 2022. "P-Loop Channels: Experimental Structures, and Physics-Based and Neural Networks-Based Models" Membranes 12, no. 2: 229. https://doi.org/10.3390/membranes12020229
APA StyleTikhonov, D. B., & Zhorov, B. S. (2022). P-Loop Channels: Experimental Structures, and Physics-Based and Neural Networks-Based Models. Membranes, 12(2), 229. https://doi.org/10.3390/membranes12020229