
Endothelial Cell Plasma Membrane Biomechanics Mediates Effects of Pro-Inflammatory Factors on Endothelial Mechanosensors: Vicious Circle Formation in Atherogenic Inflammation

 ,
,  ,
,  , , , and
, , , and
Abstract
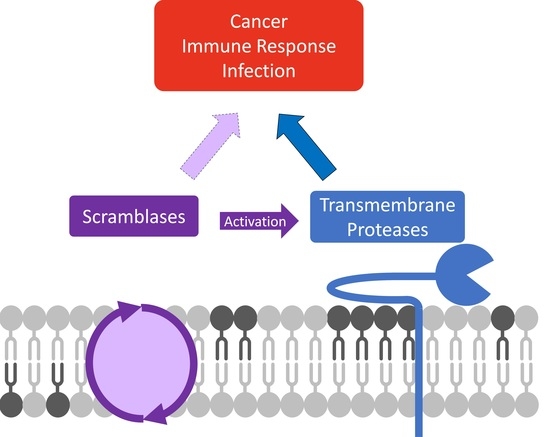
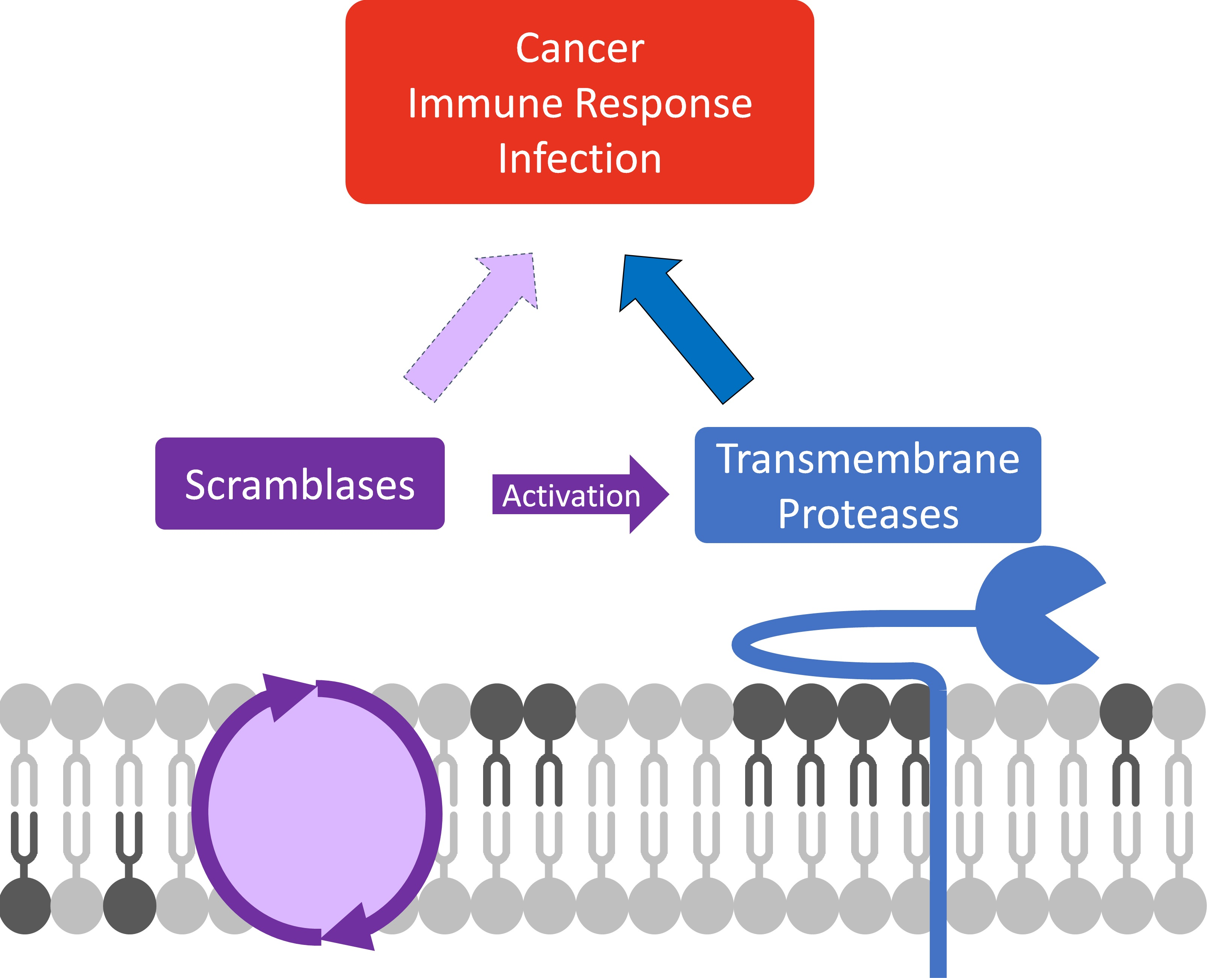
1. Introduction
1.1. Blood Flow Patterns and the Pro-Inflammatory Response of Endothelial Cells
1.2. Forces and Plasma Membrane Mechanosensors
2. What Are the Intracellular Forces Acting on Any Single Transmembrane Endothelial Mechanosensor?
2.1. From Stiffness of the Whole EC to the Mapping of Intracellular Forces Acting on Single Transmembrane Mechanosensor: From Cell- to Protein-Scale Studies
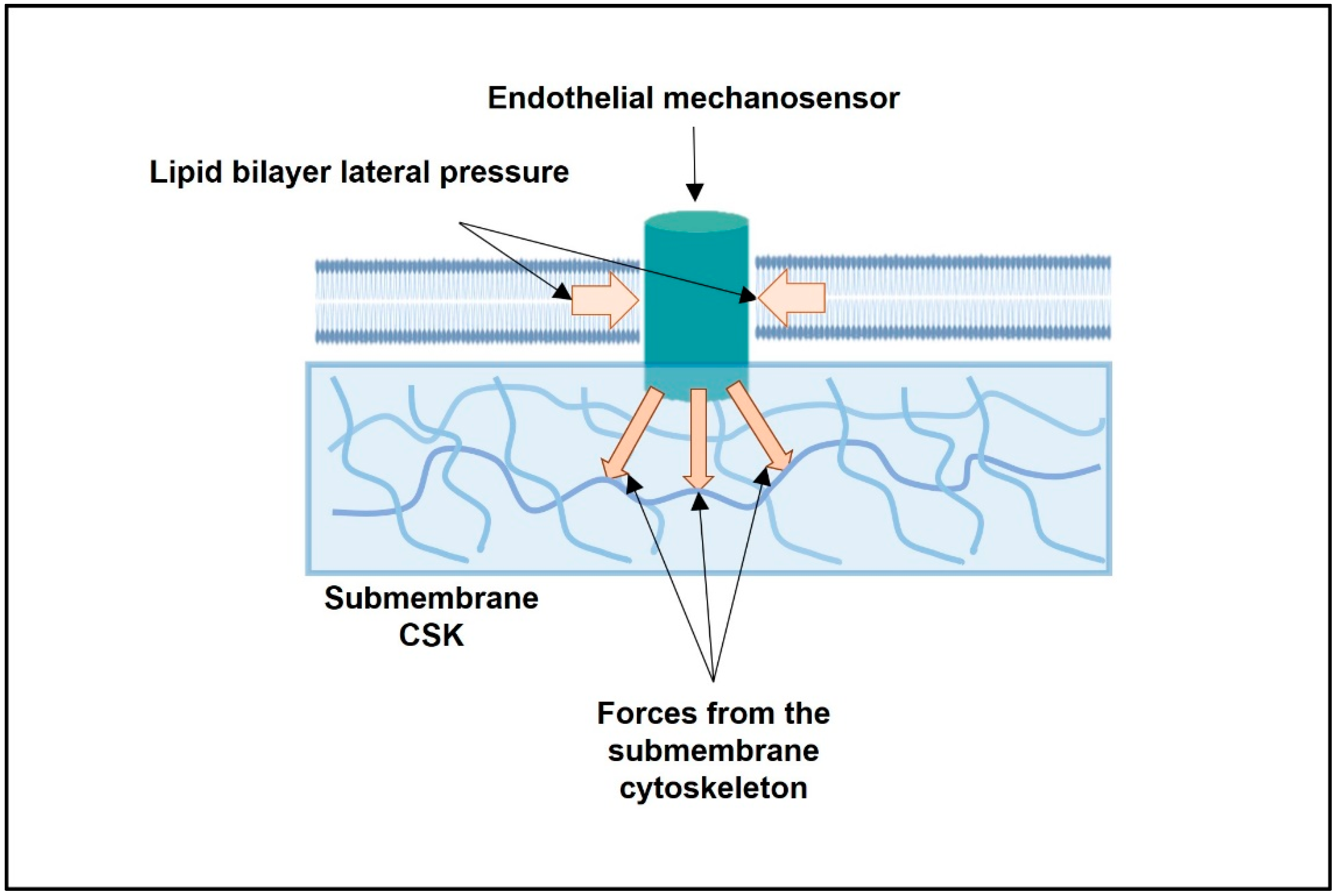
2.2. Biomechanics of the Lipid Bilayer and the Activation Energy of Mechanosensors
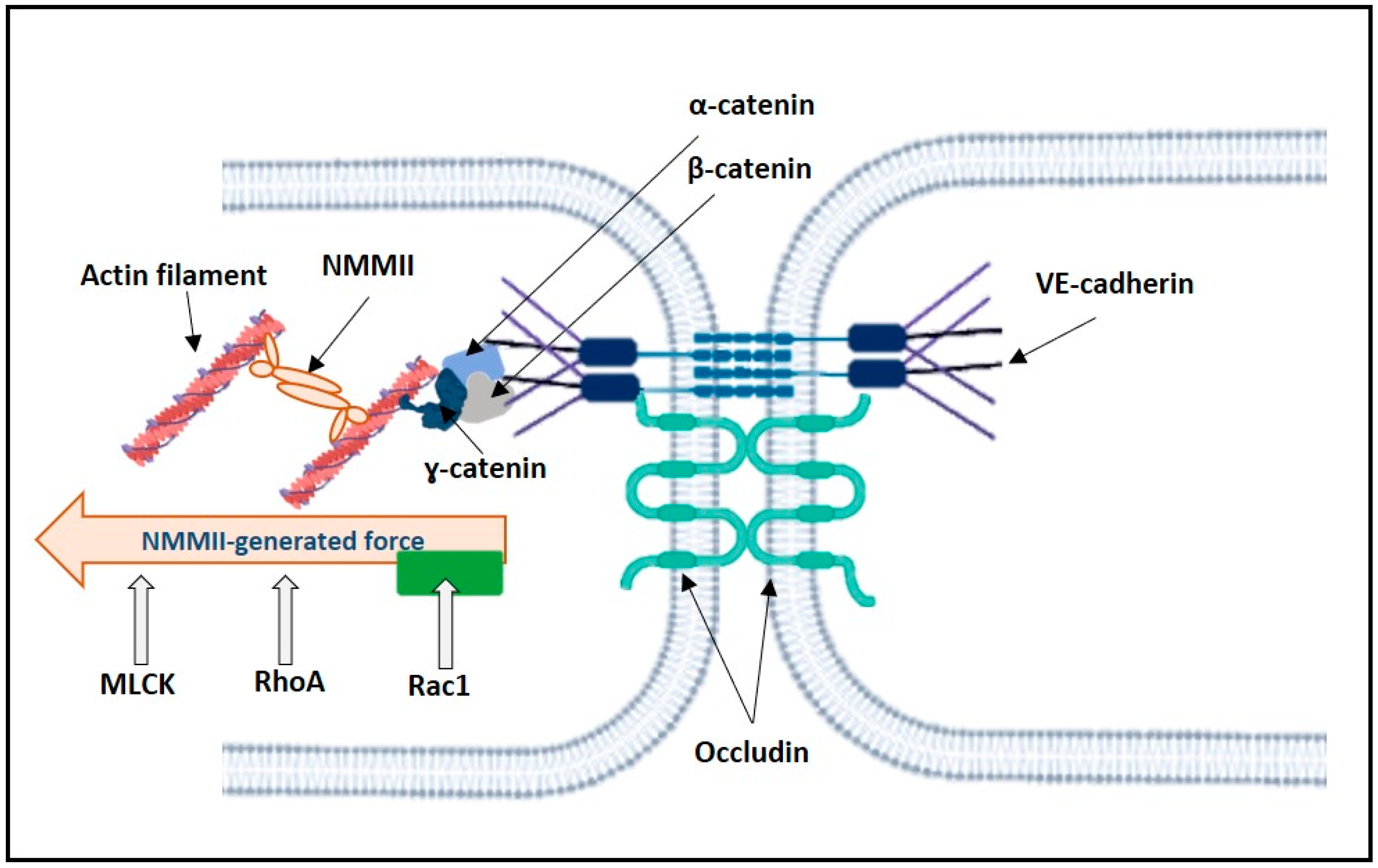
2.3. Mechanosensors and Force Generated by NMMII
2.3.1. Control of NMMII Contractility
2.3.2. Opposing Actions of RhoA and Rac1 on NMMII-Generated Pulling Force Acting on VE-Cadherin
2.3.3. Rac1 in the Regulation of Actin Polymerization Pushing Force in Lamellipodia
3. Effects of Pro-Inflammatory Stimuli on the Biomechanics of the Lipid Bilayer and Submembrane Cytoskeleton; Focus on Counterbalance between RhoA and Rac1
3.1. Pro-Inflammatory Stimuli and the Lipid Bilayer Biomechanics
3.2. Pro-Inflammatory Stimuli in RhoA and Rac1 Regulation in ECs
3.2.1. Hypoxia and Oxidative Stress in the Regulation of RhoA and Rac1 in ECs
3.2.2. Pro-Inflammatory Cytokines in RhoA and Rac1 Regulation in ECs
4. Some Mechanosensors Are Located in the Plasma Membrane of the EC; Their Sensitivity to the Lipid Bilayer and the CSK Biomechanics
4.1. Piezo1
4.2. Mechanosensory PECAM-1/VE-Cadherin/VEGFR2 Complex
4.3. Heterotrimeric G Proteins and GPCRs
4.4. Integrins
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Libby, P. Inflammation in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2045–2051. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.-J.; Chien, S. Effects of Disturbed Flow on Vascular Endothelium: Pathophysiological Basis and Clinical Perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef] [PubMed]
- Bryan, M.T.; Duckles, H.; Feng, S.; Hsiao, S.T.; Kim, H.R.; Serbanovic-Canic, J.; Evans, P.C. Mechanoresponsive Networks Controlling Vascular Inflammation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2199–2205. [Google Scholar] [CrossRef] [PubMed]
- Ramji, D.P.; Davies, T.S. Cytokines in Atherosclerosis: Key Players in All Stages of Disease and Promising Therapeutic Targets. Cytokine Growth Factor Rev. 2015, 26, 673–685. [Google Scholar] [CrossRef]
- Hirata, T.; Yamamoto, K.; Ikeda, K.; Arita, M. Functional Lipidomics of Vascular Endothelial Cells in Response to Laminar Shear Stress. FASEB J. 2021, 35, e21301. [Google Scholar] [CrossRef]
- Ishii, T.; Warabi, E.; Mann, G.E. Mechanisms Underlying Unidirectional Laminar Shear Stress-Mediated Nrf2 Activation in Endothelial Cells: Amplification of Low Shear Stress Signaling by Primary Cilia. Redox Biol. 2021, 46, 102103. [Google Scholar] [CrossRef]
- Wang, K.-C.; Yeh, Y.-T.; Nguyen, P.; Limqueco, E.; Lopez, J.; Thorossian, S.; Guan, K.-L.; Li, Y.-S.J.; Chien, S. Flow-Dependent YAP/TAZ Activities Regulate Endothelial Phenotypes and Atherosclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 11525–11530. [Google Scholar] [CrossRef]
- Tzima, E.; Irani-Tehrani, M.; Kiosses, W.B.; Dejana, E.; Schultz, D.A.; Engelhardt, B.; Cao, G.; DeLisser, H.; Schwartz, M.A. A Mechanosensory Complex That Mediates the Endothelial Cell Response to Fluid Shear Stress. Nature 2005, 437, 426–431. [Google Scholar] [CrossRef]
- Petzold, T.; Orr, A.W.; Hahn, C.; Jhaveri, K.A.; Parsons, J.T.; Schwartz, M.A. Focal Adhesion Kinase Modulates Activation of NF-ΚB by Flow in Endothelial Cells. Am. J. Physiol. Cell Physiol. 2009, 297, C814–C822. [Google Scholar] [CrossRef]
- Zhao, Q.; Ishibashi, M.; Hiasa, K.; Tan, C.; Takeshita, A.; Egashira, K. Essential Role of Vascular Endothelial Growth Factor in Angiotensin II–Induced Vascular Inflammation and Remodeling. Hypertension 2004, 44, 264–270. [Google Scholar] [CrossRef]
- Takeshita, S.; Zheng, L.P.; Brogi, E.; Kearney, M.; Pu, L.Q.; Bunting, S.; Ferrara, N.; Symes, J.F.; Isner, J.M. Therapeutic Angiogenesis. A Single Intraarterial Bolus of Vascular Endothelial Growth Factor Augments Revascularization in a Rabbit Ischemic Hind Limb Model. J. Clin. Investig. 1994, 93, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Riessen, R.; Rahimizadeh, H.; Blessing, E.; Takeshita, S.; Barry, J.J.; Isner, J.M. Arterial Gene Transfer Using Pure DNA Applied Directly to a Hydrogel-Coated Angioplasty Balloon. Hum. Gene 1993, 4, 749–758. [Google Scholar] [CrossRef]
- Chen, J.; Green, J.; Yurdagul, A.; Albert, P.; McInnis, M.C.; Orr, A.W. Avβ3 Integrins Mediate Flow-Induced NF-ΚB Activation, Proinflammatory Gene Expression, and Early Atherogenic Inflammation. Am. J. Pathol. 2015, 185, 2575–2589. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-ΚB Family of Transcription Factors and Its Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Luo, J.-Y.; Li, B.; Tian, X.Y.; Chen, L.-J.; Huang, Y.; Liu, J.; Deng, D.; Lau, C.W.; Wan, S.; et al. Integrin-YAP/TAZ-JNK Cascade Mediates Atheroprotective Effect of Unidirectional Shear Flow. Nature 2016, 540, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; He, J.; Lv, H.; Liu, Y.; Lv, X.; Zhang, C.; Zhu, Y.; Ai, D. C-Abl Regulates YAPY357 Phosphorylation to Activate Endothelial Atherogenic Responses to Disturbed Flow. J. Clin. Investig. 2019, 129, 1167–1179. [Google Scholar] [CrossRef]
- Conway, D.E.; Schwartz, M.A. Flow-Dependent Cellular Mechanotransduction in Atherosclerosis. J. Cell Sci. 2013, 126, 5101–5109. [Google Scholar] [CrossRef]
- Davies, P.F.; Civelek, M.; Fang, Y.; Fleming, I. The Atherosusceptible Endothelium: Endothelial Phenotypes in Complex Haemodynamic Shear Stress Regions in Vivo. Cardiovasc. Res. 2013, 99, 315–327. [Google Scholar] [CrossRef]
- Givens, C.; Tzima, E. Endothelial Mechanosignaling: Does One Sensor Fit All? Antioxid. Redox Signal. 2016, 25, 373–388. [Google Scholar] [CrossRef]
- Baratchi, S.; Khoshmanesh, K.; Woodman, O.L.; Potocnik, S.; Peter, K.; McIntyre, P. Molecular Sensors of Blood Flow in Endothelial Cells. Trends Mol. Med. 2017, 23, 850–868. [Google Scholar] [CrossRef] [PubMed]
- Le Master, E.; Ahn, S.J.; Levitan, I. Chapter Five—Mechanisms of Endothelial Stiffening in Dyslipidemia and Aging: Oxidized Lipids and Shear Stress. In Current Topics in Membranes; Levitan, I., Trache, A., Eds.; Academic Press: London, UK, 2020; Volume 86, pp. 185–215. ISBN 1063-5823. [Google Scholar]
- Mahmoudi, M.; Farghadan, A.; McConnell, D.R.; Barker, A.J.; Wentzel, J.J.; Budoff, M.J.; Arzani, A. The Story of Wall Shear Stress in Coronary Artery Atherosclerosis: Biochemical Transport and Mechanotransduction. J. Biomech. Eng. 2020, 143, 041002. [Google Scholar] [CrossRef] [PubMed]
- Roux, E.; Bougaran, P.; Dufourcq, P.; Couffinhal, T. Fluid Shear Stress Sensing by the Endothelial Layer. Front. Physiol. 2020, 11, 861. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Joshi, D.; Timalsina, S.; Schwartz, M.A. Early Events in Endothelial Flow Sensing. Cytoskeleton 2021, 78, 217–231. [Google Scholar] [CrossRef]
- Mylvaganam, S.; Yusuf, B.; Li, R.; Lu, C.-Y.; Robinson, L.A.; Freeman, S.A.; Grinstein, S. Endothelial Integration of Mechanosensory Signals by the Spectrin Cytoskeleton. bioRxiv 2021. [Google Scholar] [CrossRef]
- Fang, Y.; Wu, D.; Birukov, K.G. Mechanosensing and Mechanoregulation of Endothelial Cell Functions. Compr. Physiol. 2019, 9, 873. [Google Scholar]
- Mack, J.J.; Mosqueiro, T.S.; Archer, B.J.; Jones, W.M.; Sunshine, H.; Faas, G.C.; Briot, A.; Aragón, R.L.; Su, T.; Romay, M.C.; et al. NOTCH1 Is a Mechanosensor in Adult Arteries. Nat. Commun. 2017, 8, 1620. [Google Scholar] [CrossRef]
- Mehta, V.; Pang, K.-L.; Rozbesky, D.; Nather, K.; Keen, A.; Lachowski, D.; Kong, Y.; Karia, D.; Ameismeier, M.; Huang, J.; et al. The Guidance Receptor Plexin D1 Is a Mechanosensor in Endothelial Cells. Nature 2020, 578, 290–295. [Google Scholar] [CrossRef]
- Yamamoto, K.; Imamura, H.; Ando, J. Shear Stress Augments Mitochondrial ATP Generation That Triggers ATP Release and Ca2+ Signaling in Vascular Endothelial Cells. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1477–H1485. [Google Scholar] [CrossRef]
- Yamamoto, K.; Nogimori, Y.; Imamura, H.; Ando, J. Shear Stress Activates Mitochondrial Oxidative Phosphorylation by Reducing Plasma Membrane Cholesterol in Vascular Endothelial Cells. Proc. Natl. Acad. Sci. USA 2020, 117, 33660. [Google Scholar] [CrossRef]
- Cox, C.D.; Bavi, N.; Martinac, B. Biophysical Principles of Ion-Channel-Mediated Mechanosensory Transduction. Cell Rep. 2019, 29, 1–12. [Google Scholar] [CrossRef]
- Hamill, O.P.; Martinac, B. Molecular Basis of Mechanotransduction in Living Cells. Physiol. Rev. 2001, 81, 685. [Google Scholar] [CrossRef]
- Hamill, O.P. Twenty Odd Years of Stretch-Sensitive Channels. Pflügers Arch. 2006, 453, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Balleza, D. Mechanical Properties of Lipid Bilayers and Regulation of Mechanosensitive Function. Channels 2012, 6, 220–233. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, A.G. How Lipids Affect the Activities of Integral Membrane Proteins. Biochim. Biophys. Acta Biomembr. 2004, 1666, 62–87. [Google Scholar] [CrossRef] [PubMed]
- Corradi, V.; Sejdiu, B.I.; Mesa-Galloso, H.; Abdizadeh, H.; Noskov, S.Y.; Marrink, S.J.; Tieleman, D.P. Emerging Diversity in Lipid–Protein Interactions. Chem. Rev. 2019, 119, 5775–5848. [Google Scholar] [CrossRef]
- Muller, M.P.; Jiang, T.; Sun, C.; Lihan, M.; Pant, S.; Mahinthichaichan, P.; Trifan, A.; Tajkhorshid, E. Characterization of Lipid–Protein Interactions and Lipid-Mediated Modulation of Membrane Protein Function through Molecular Simulation. Chem. Rev. 2019, 119, 6086–6161. [Google Scholar] [CrossRef]
- Jodaitis, L.; van Oene, T.; Martens, C. Assessing the Role of Lipids in the Molecular Mechanism of Membrane Proteins. Int. J. Mol. Sci. 2021, 22, 7267. [Google Scholar] [CrossRef]
- Overduin, M.; Trieber, C.; Prosser, R.S.; Picard, L.-P.; Sheff, J.G. Structures and Dynamics of Native-State Transmembrane Protein Targets and Bound Lipids. Membranes 2021, 11, 451. [Google Scholar] [CrossRef]
- Majeed, S.; Ahmad, A.B.; Sehar, U.; Georgieva, E.R. Lipid Membrane Mimetics in Functional and Structural Studies of Integral Membrane Proteins. Membranes 2021, 11, 685. [Google Scholar] [CrossRef]
- Renard, K.; Byrne, B. Insights into the Role of Membrane Lipids in the Structure, Function and Regulation of Integral Membrane Proteins. Int. J. Mol. Sci. 2021, 22, 9026. [Google Scholar] [CrossRef]
- Ashrafuzzaman, M.; Tuszynski, J. Regulation of Channel Function Due to Coupling with a Lipid Bilayer. J. Comput. Theor. Nanosci. 2012, 9, 564–570. [Google Scholar] [CrossRef]
- Ashrafuzzaman, M.; Tuszynski, J. Lipid Bilayer-Membrane Protein Coupling. In Membrane Biophysics; Springer: Berlin/Heidelberg, Germany, 2012; pp. 75–125. [Google Scholar]
- Huang, H.W. Deformation Free Energy of Bilayer Membrane and Its Effect on Gramicidin Channel Lifetime. Biophys. J. 1986, 50, 1061–1070. [Google Scholar] [CrossRef]
- Gruner, S.M. Intrinsic Curvature Hypothesis for Biomembrane Lipid Composition: A Role for Nonbilayer Lipids. Proc. Natl. Acad. Sci. USA 1985, 82, 3665–3669. [Google Scholar] [CrossRef] [PubMed]
- Helfrich, P.; Jakobsson, E. Calculation of Deformation Energies and Conformations in Lipid Membranes Containing Gramicidin Channels. Biophys. J. 1990, 57, 1075–1084. [Google Scholar] [CrossRef]
- Durkin, J.T.; Providence, L.L.; Koeppe, R.E., II; Andersen, O.S.; Koeppe, R.E., II; Andersen, O.S. Energetics of Heterodimer Formation among Gramicidin Analogues with an NH2-Terminal Addition or Deletion: Consequences of Missing a Residue at the Join in the Channel. J. Mol. Biol. 1993, 231, 1102–1121. [Google Scholar] [CrossRef]
- Nielsen, C.; Goulian, M.; Andersen, O.S. Energetics of Inclusion-Induced Bilayer Deformations. Biophys. J. 1998, 74, 1966–1983. [Google Scholar] [CrossRef]
- Nielsen, C.; Andersen, O.S. Inclusion-Induced Bilayer Deformations: Effects of Monolayer Equilibrium Curvature. Biophys. J. 2000, 79, 2583–2604. [Google Scholar] [CrossRef]
- Lundbæk, J.A.; Birn, P.; Hansen, A.J.; Søgaard, R.; Nielsen, C.; Girshman, J.; Bruno, M.J.; Tape, S.E.; Egebjerg, J.; Greathouse, D.V. Regulation of Sodium Channel Function by Bilayer Elasticity The Importance of Hydrophobic Coupling. Effects of Micelle-Forming Amphiphiles and Cholesterol. J. Gen. Physiol. 2004, 123, 599–621. [Google Scholar] [CrossRef]
- Lundbaek, J.; Birn, P.; Tape, S.; Toombes, G.E.; Søgaard, R.; Koeppe, R.E.; Gruner, S.M.; Hansen, A.J.; Andersen, O.S. Capsaicin Regulates Voltage-Dependent Sodium Channels by Altering Lipid Bilayer Elasticity. Mol. Pharmacol. 2005, 68, 680–689. [Google Scholar] [CrossRef]
- Ashrafuzzaman, M.; Tseng, C.-Y.; Tuszynski, J. Regulation of Channel Function Due to Physical Energetic Coupling with a Lipid Bilayer. Biochem. Biophys. Res. Commun. 2014, 445, 463–468. [Google Scholar] [CrossRef]
- Ashrafuzzaman, M.; Tseng, C.-Y.; Tuszynski, J. Charge-Based Interactions of Antimicrobial Peptides and General Drugs with Lipid Bilayers. J. Mol. Graph. Model. 2020, 95, 107502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, W.; Chen, W. Plasma Membrane Integrates Biophysical and Biochemical Regulation to Trigger Immune Receptor Functions. Front. Immunol. 2021, 12, 53. [Google Scholar] [CrossRef] [PubMed]
- Goeckeler, Z.M.; Bridgman, P.C.; Wysolmerski, R.B. Nonmuscle Myosin II Is Responsible for Maintaining Endothelial Cell Basal Tone and Stress Fiber Integrity. Am. J. Physiol. Cell Physiol. 2008, 295, C994–C1006. [Google Scholar] [CrossRef] [PubMed]
- Gordon, E.; Schimmel, L.; Frye, M. The Importance of Mechanical Forces for in Vitro Endothelial Cell Biology. Front. Physiol. 2020, 11, 684. [Google Scholar] [CrossRef]
- Ross, T.D.; Coon, B.G.; Yun, S.; Baeyens, N.; Tanaka, K.; Ouyang, M.; Schwartz, M.A. Integrins in Mechanotransduction. Curr. Opin. Cell Biol. 2013, 25, 613–618. [Google Scholar] [CrossRef]
- Silvani, G.; Romanov, V.; Cox, C.D.; Martinac, B. Biomechanical Characterization of Endothelial Cells Exposed to Shear Stress Using Acoustic Force Spectroscopy. Front. Bioeng. Biotechnol. 2021, 9, 21. [Google Scholar] [CrossRef]
- Engler, A.J.; Wang, Y. Editorial: Understanding Molecular Interactions That Underpin Vascular Mechanobiology. APL Bioeng. 2021, 5, 030401. [Google Scholar] [CrossRef]
- Katsumi, A.; Milanini, J.; Kiosses, W.B.; del Pozo, M.A.; Kaunas, R.; Chien, S.; Hahn, K.M.; Schwartz, M.A. Effects of Cell Tension on the Small GTPase Rac. J. Cell Biol. 2002, 158, 153–164. [Google Scholar] [CrossRef]
- Grashoff, C.; Hoffman, B.D.; Brenner, M.D.; Zhou, R.; Parsons, M.; Yang, M.T.; McLean, M.A.; Sligar, S.G.; Chen, C.S.; Ha, T.; et al. Measuring Mechanical Tension across Vinculin Reveals Regulation of Focal Adhesion Dynamics. Nature 2010, 466, 263–266. [Google Scholar] [CrossRef]
- Conway, D.E.; Breckenridge, M.T.; Hinde, E.; Gratton, E.; Chen, C.S.; Schwartz, M.A. Fluid Shear Stress on Endothelial Cells Modulates Mechanical Tension across VE-Cadherin and PECAM-1. Curr. Biol. 2013, 23, 1024–1030. [Google Scholar] [CrossRef]
- Barvitenko, N.; Aslam, M.; Lawen, A.; Saldanha, C.; Skverchinskaya, E.; Uras, G.; Manca, A.; Pantaleo, A. Two Motors and One Spring: Hypothetic Roles of Non-Muscle Myosin II and Submembrane Actin-Based Cytoskeleton in Cell Volume Sensing. Int. J. Mol. Sci. 2021, 22, 7967. [Google Scholar] [CrossRef]
- Song, Y.; Kenworthy, A.K.; Sanders, C.R. Cholesterol as a Co-Solvent and a Ligand for Membrane Proteins. Protein Sci. 2014, 23, 1–22. [Google Scholar] [CrossRef]
- Salas-Estrada, L.A.; Leioatts, N.; Romo, T.D.; Grossfield, A. Lipids Alter Rhodopsin Function via Ligand-like and Solvent-like Interactions. Biophys. J. 2018, 114, 355–367. [Google Scholar] [CrossRef]
- Fantini, J.; Barrantes, F. How Cholesterol Interacts with Membrane Proteins: An Exploration of Cholesterol-Binding Sites Including CRAC, CARC, and Tilted Domains. Front. Physiol. 2013, 4, 31. [Google Scholar] [CrossRef]
- Lee, A.G. Interfacial Binding Sites for Cholesterol on G Protein-Coupled Receptors. Biophys. J. 2019, 116, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Jakubík, J.; El-Fakahany, E.E. Allosteric Modulation of GPCRs of Class A by Cholesterol. Int. J. Mol. Sci. 2021, 22, 1953. [Google Scholar] [CrossRef] [PubMed]
- Gudi, S.; Nolan, J.P.; Frangos, J.A. Modulation of GTPase Activity of G Proteins by Fluid Shear Stress and Phospholipid Composition. Proc. Natl. Acad. Sci. USA 1998, 95, 2515–2519. [Google Scholar] [CrossRef] [PubMed]
- Haidekker, M.A.; L’Heureux, N.; Frangos, J.A. Fluid Shear Stress Increases Membrane Fluidity in Endothelial Cells: A Study with DCVJ Fluorescence. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1401–H1406. [Google Scholar] [CrossRef] [PubMed]
- Butler, P.J.; Norwich, G.; Weinbaum, S.; Chien, S. Shear Stress Induces a Time- and Position-Dependent Increase in Endothelial Cell Membrane Fluidity. Am. J. Physiol. Cell Physiol. 2001, 280, C962–C969. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ando, J. Vascular Endothelial Cell Membranes Differentiate between Stretch and Shear Stress through Transitions in Their Lipid Phases. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1178–H1185. [Google Scholar] [CrossRef]
- Billington, N.; Wang, A.; Mao, J.; Adelstein, R.S.; Sellers, J.R. Characterization of Three Full-Length Human Nonmuscle Myosin II Paralogs. J. Biol. Chem. 2013, 288, 33398–33410. [Google Scholar] [CrossRef] [PubMed]
- Dulyaninova, N.G.; Bresnick, A.R. The Heavy Chain Has Its Day. BioArchitecture 2013, 3, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Shutova, M.S.; Svitkina, T.M. Common and Specific Functions of Nonmuscle Myosin II Paralogs in Cells. Biochemistry 2018, 83, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Sellers, J.R.; Heissler, S.M. Nonmuscle Myosin-2 Isoforms. Curr. Biol. 2019, 29, R275–R278. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Casado, M.; Asensio-Juárez, G.; Vicente-Manzanares, M. Nonmuscle Myosin II Regulation Directs Its Multiple Roles in Cell Migration and Division. Annu. Rev. Cell Dev. Biol. 2021, 37, 285–310. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Ma, X.; Conti, M.A.; Adelstein, R.S. Distinct and Redundant Roles of the Non-Muscle Myosin II Isoforms and Functional Domains. Biochem. Soc. Trans. 2011, 39, 1131–1135. [Google Scholar] [CrossRef]
- Kolega, J. Asymmetric Distribution of Myosin IIB in Migrating Endothelial Cells Is Regulated by a Rho-Dependent Kinase and Contributes to Tail Retraction. Mol. Biol. Cell 2003, 14, 4745–4757. [Google Scholar] [CrossRef]
- Kolega, J. The Role of Myosin II Motor Activity in Distributing Myosin Asymmetrically and Coupling Protrusive Activity to Cell Translocation. Mol. Biol. Cell 2006, 17, 4435–4445. [Google Scholar] [CrossRef]
- Kamm, K.E.; Stull, J.T. Dedicated Myosin Light Chain Kinases with Diverse Cellular Functions. J. Biol. Chem. 2001, 276, 4527–4530. [Google Scholar] [CrossRef]
- Hartshorne, D.J.; Ito, M.; Erdodi, F. Myosin Light Chain Phosphatase: Subunit Composition, Interactions and Regulation. J. Muscle Res. Cell Motil. 1998, 19, 325–341. [Google Scholar] [CrossRef]
- Kiss, A.; Erdődi, F.; Lontay, B. Myosin Phosphatase: Unexpected Functions of a Long-Known Enzyme. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Malik, A.B. Signaling Mechanisms Regulating Endothelial Permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef] [PubMed]
- Rigor, R.R.; Shen, Q.; Pivetti, C.D.; Wu, M.H.; Yuan, S.Y. Myosin Light Chain Kinase Signaling in Endothelial Barrier Dysfunction. Med. Res. Rev. 2013, 33, 911–933. [Google Scholar] [CrossRef]
- Nobes, C.D.; Hall, A. Rho, Rac, and Cdc42 GTPases Regulate the Assembly of Multimolecular Focal Complexes Associated with Actin Stress Fibers, Lamellipodia, and Filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho GTPase Signalling in Cell Migration. Curr. Opin. Cell Biol. 2015, 36, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Flentje, A.; Kalsi, R.; Monahan, T.S. Small GTPases and Their Role in Vascular Disease. Int. J. Mol. Sci. 2019, 20, 917. [Google Scholar] [CrossRef]
- Tzima, E.; del Pozo, M.A.; Shattil, S.J.; Chien, S.; Schwartz, M.A. Activation of Integrins in Endothelial Cells by Fluid Shear Stress Mediates Rho-Dependent Cytoskeletal Alignment. EMBO J. 2001, 20, 4639–4647. [Google Scholar] [CrossRef] [PubMed]
- Tzima, E.; Del Pozo, M.A.; Kiosses, W.B.; Mohamed, S.A.; Li, S.; Chien, S.; Schwartz, M.A. Activation of Rac1 by Shear Stress in Endothelial Cells Mediates Both Cytoskeletal Reorganization and Effects on Gene Expression. EMBO J. 2002, 21, 6791–6800. [Google Scholar] [CrossRef]
- Birukov, K.G. Small GTPases in Mechanosensitive Regulation of Endothelial Barrier. Microvasc. Res. 2009, 77, 46–52. [Google Scholar] [CrossRef]
- Aslam, M.; Schluter, K.-D.; Rohrbach, S.; Rafiq, A.; Nazli, S.; Piper, H.M.; Noll, T.; Schulz, R.; Gündüz, D. Hypoxia–Reoxygenation-Induced Endothelial Barrier Failure: Role of RhoA, Rac1 and Myosin Light Chain Kinase. J. Physiol. 2013, 591, 461–473. [Google Scholar] [CrossRef]
- Komarova, Y.A.; Kruse, K.; Mehta, D.; Malik, A.B. Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ. Res. 2017, 120, 179–206. [Google Scholar] [CrossRef]
- Rho, S.-S.; Ando, K.; Fukuhara, S. Dynamic Regulation of Vascular Permeability by Vascular Endothelial Cadherin-Mediated Endothelial Cell-Cell Junctions. J. Nippon Med. Sch. 2017, 84, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Amano, M.; Ito, M.; Kimura, K.; Fukata, Y.; Chihara, K.; Nakano, T.; Matsuura, Y.; Kaibuchi, K. Phosphorylation and Activation of Myosin by Rho-Associated Kinase (Rho-Kinase). J. Biol. Chem. 1996, 271, 20246–20249. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Ito, M.; Amano, M.; Chihara, K.; Fukata, Y.; Nakafuku, M.; Yamamori, B.; Feng, J.; Nakano, T.; Okawa, K. Regulation of Myosin Phosphatase by Rho and Rho-Associated Kinase (Rho-Kinase). Science 1996, 273, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Noma, K.; Oyama, N.; Liao, J.K. Physiological Role of ROCKs in the Cardiovascular System. Am. J. Physiol. Cell Physiol. 2006, 290, C661–C668. [Google Scholar] [CrossRef] [PubMed]
- Sanders, L.C.; Matsumura, F.; Bokoch, G.M.; De Lanerolle, P. Inhibition of Myosin Light Chain Kinase by P21-Activated Kinase. Science 1999, 283, 2083–2085. [Google Scholar] [CrossRef] [PubMed]
- Borghi, N.; Sorokina, M.; Shcherbakova, O.G.; Weis, W.I.; Pruitt, B.L.; Nelson, W.J.; Dunn, A.R. E-Cadherin Is under Constitutive Actomyosin-Generated Tension That Is Increased at Cell–Cell Contacts upon Externally Applied Stretch. Proc. Natl. Acad. Sci. USA 2012, 109, 12568. [Google Scholar] [CrossRef]
- Daneshjou, N.; Sieracki, N.; van Nieuw Amerongen, G.P.; Conway, D.E.; Schwartz, M.A.; Komarova, Y.A.; Malik, A.B. Rac1 Functions as a Reversible Tension Modulator to Stabilize VE-Cadherin Trans-Interaction. J. Cell Biol. 2015, 208, 23–32. [Google Scholar] [CrossRef]
- Efimova, N.; Svitkina, T.M. Branched Actin Networks Push against Each Other at Adherens Junctions to Maintain Cell–Cell Adhesion. J. Cell Biol. 2018, 217, 1827–1845. [Google Scholar] [CrossRef]
- Morimura, S.; Suzuki, K.; Takahashi, K. Nonmuscle Myosin IIA Is Required for Lamellipodia Formation through Binding to WAVE2 and Phosphatidylinositol 3, 4, 5-Triphosphate. Biochem. Biophys. Res. Commun. 2011, 404, 834–840. [Google Scholar] [CrossRef]
- Barvitenko, N.N.; Aslam, M.; Lawen, A.; Pantaleo, A.; Saldanha, C.; Matteucci, E. Effects of Oxygen Depletion on Transmembrane Protein Activities. Curr. Org. Chem. 2015, 19, 2002–2010. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive Oxygen Species Generated at Mitochondrial Complex III Stabilize Hypoxia-Inducible Factor-1α during Hypoxia: A Mechanism of O2 Sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed]
- Zmysłowski, A.; Szterk, A. Current Knowledge on the Mechanism of Atherosclerosis and Pro-Atherosclerotic Properties of Oxysterols. Lipids Health Dis. 2017, 16, 188. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, Inflammation and Innate Immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Pederiva, C.; Capra, M.E.; Viggiano, C.; Rovelli, V.; Banderali, G.; Biasucci, G. Early Prevention of Atherosclerosis: Detection and Management of Hypercholesterolaemia in Children and Adolescents. Life 2021, 11, 345. [Google Scholar] [CrossRef]
- Wang, H.H.; Garruti, G.; Liu, M.; Portincasa, P.; Wang, D.Q.-H. Cholesterol and Lipoprotein Metabolism and Atherosclerosis: Recent Advances in Reverse Cholesterol Transport. Ann. Hepatol. 2017, 16, S27–S42. [Google Scholar] [CrossRef]
- Berliner, J.A.; Leitinger, N.; Tsimikas, S. The Role of Oxidized Phospholipids in Atherosclerosis. J. Lipid Res. 2009, 50, S207–S212. [Google Scholar] [CrossRef]
- Jurkiewicz, P.; Olżyńska, A.; Cwiklik, L.; Conte, E.; Jungwirth, P.; Megli, F.M.; Hof, M. Biophysics of Lipid Bilayers Containing Oxidatively Modified Phospholipids: Insights from Fluorescence and EPR Experiments and from MD Simulations. Biochim. Biophys. Acta Biomembr. 2012, 1818, 2388–2402. [Google Scholar] [CrossRef]
- Yadav, D.K.; Kumar, S.; Choi, E.-H.; Chaudhary, S.; Kim, M.-H. Molecular Dynamic Simulations of Oxidized Skin Lipid Bilayer and Permeability of Reactive Oxygen Species. Sci. Rep. 2019, 9, 4496. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Wu, J.H.Y. (N-3) Fatty Acids and Cardiovascular Health: Are Effects of EPA and DHA Shared or Complementary? J. Nutr. 2012, 142, 614S–625S. [Google Scholar] [CrossRef]
- Yamagata, K. Docosahexaenoic Acid Regulates Vascular Endothelial Cell Function and Prevents Cardiovascular Disease. Lipids Health Dis. 2017, 16, 118. [Google Scholar] [CrossRef] [PubMed]
- Innes, J.K.; Calder, P.C. Marine Omega-3 (N-3) Fatty Acids for Cardiovascular Health: An Update for 2020. Int. J. Mol. Sci. 2020, 21, 1362. [Google Scholar] [CrossRef] [PubMed]
- Wojciak-Stothard, B.; Tsang, L.Y.F.; Paleolog, E.; Hall, S.M.; Haworth, S.G. Rac1 and RhoA as Regulators of Endothelial Phenotype and Barrier Function in Hypoxia-Induced Neonatal Pulmonary Hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L1173–L1182. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chi, A.Y.; Waypa, G.B.; Mungai, P.T.; Schumacker, P.T. Prolonged Hypoxia Increases ROS Signaling and RhoA Activation in Pulmonary Artery Smooth Muscle and Endothelial Cells. Antioxid. Redox Signal. 2010, 12, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Kalwa, H.; Sartoretto, J.L.; Sartoretto, S.M.; Michel, T. Angiotensin-II and MARCKS: A Hydrogen Peroxide- and Rac1-Dependent Signaling Pathway in Vascular Endothelium. J. Biol. Chem. 2012, 287, 29147–29158. [Google Scholar] [CrossRef]
- Fatima, N.; Patel, S.N.; Hussain, T. Angiotensin II Type 2 Receptor: A Target for Protection Against Hypertension, Metabolic Dysfunction, and Organ Remodeling. Hypertension 2021, 77, 1845–1856. [Google Scholar] [CrossRef]
- Terenzi, R.; Manetti, M.; Rosa, I.; Romano, E.; Galluccio, F.; Guiducci, S.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Angiotensin II Type 2 Receptor (AT2R) as a Novel Modulator of Inflammation in Rheumatoid Arthritis Synovium. Sci. Rep. 2017, 7, 13293. [Google Scholar] [CrossRef]
- Patel, S.N.; Fatima, N.; Ali, R.; Hussain, T. Emerging Role of Angiotensin AT2 Receptor in Anti-Inflammation: An Update. Curr. Pharm. Des. 2020, 26, 492–500. [Google Scholar] [CrossRef]
- Shatanawi, A.; Romero, M.J.; Iddings, J.A.; Chandra, S.; Umapathy, N.S.; Verin, A.D.; Caldwell, R.B.; Caldwell, R.W. Angiotensin II-Induced Vascular Endothelial Dysfunction through RhoA/Rho Kinase/P38 Mitogen-Activated Protein Kinase/Arginase Pathway. Am. J. Physiol. Cell Physiol. 2011, 300, C1181–C1192. [Google Scholar] [CrossRef]
- Savoia, C.; Tabet, F.; Yao, G.; Schiffrin, E.L.; Touyz, R.M. Negative Regulation of RhoA/Rho Kinase by Angiotensin II Type 2 Receptor in Vascular Smooth Muscle Cells: Role in Angiotensin II-Induced Vasodilation in Stroke-Prone Spontaneously Hypertensive Rats. J. Hypertens. 2005, 23, 1037–1045. [Google Scholar] [CrossRef]
- Nakakuki, T.; Ito, M.; Iwasaki, H.; Kureishi, Y.; Okamoto, R.; Moriki, N.; Kongo, M.; Kato, S.; Yamada, N.; Isaka, N.; et al. Rho/Rho-Kinase Pathway Contributes to C-Reactive Protein–Induced Plasminogen Activator Inhibitor-1 Expression in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2088–2093. [Google Scholar] [CrossRef] [PubMed]
- Kurano, M.; Yatomi, Y. Sphingosine 1-Phosphate and Atherosclerosis. J. Atheroscler. Thromb. 2018, 25, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.; Mehta, D. Post-Translational Modifications of S1PR1 and Endothelial Barrier Regulation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158760. [Google Scholar] [CrossRef]
- Obinata, H.; Hla, T. Sphingosine 1-Phosphate and Inflammation. Int. Immunol. 2019, 31, 617–625. [Google Scholar] [CrossRef]
- Chen, D.; Dorling, A. Critical Roles for Thrombin in Acute and Chronic Inflammation. J. Thromb. Haemost. 2009, 7, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.; Pfeil, U.; Gündüz, D.; Rafiq, A.; Kummer, W.; Piper, H.; Noll, T. Intermedin (Adrenomedullin2) Stabilizes the Endothelial Barrier and Antagonizes Thrombin-Induced Barrier Failure in Endothelial Cell Monolayers. Br. J. Pharmacol. 2012, 165, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.; Tanislav, C.; Troidl, C.; Schulz, R.; Hamm, C.; Gündüz, D. CAMP Controls the Restoration of Endothelial Barrier Function after Thrombin-Induced Hyperpermeability via Rac1 Activation. Physiol Rep. 2014, 2, e12175. [Google Scholar] [CrossRef]
- Li, J.; Hou, B.; Tumova, S.; Muraki, K.; Bruns, A.; Ludlow, M.J.; Sedo, A.; Hyman, A.J.; McKeown, L.; Young, R.S.; et al. Piezo1 Integration of Vascular Architecture with Physiological Force. Nature 2014, 515, 279–282. [Google Scholar] [CrossRef]
- Coste, B.; Xiao, B.; Santos, J.S.; Syeda, R.; Grandl, J.; Spencer, K.S.; Kim, S.E.; Schmidt, M.; Mathur, J.; Dubin, A.E.; et al. Piezo Proteins Are Pore-Forming Subunits of Mechanically Activated Channels. Nature 2012, 483, 176–181. [Google Scholar] [CrossRef]
- Ge, J.; Li, W.; Zhao, Q.; Li, N.; Chen, M.; Zhi, P.; Li, R.; Gao, N.; Xiao, B.; Yang, M. Architecture of the Mammalian Mechanosensitive Piezo1 Channel. Nature 2015, 527, 64–69. [Google Scholar] [CrossRef]
- Cox, C.D.; Bae, C.; Ziegler, L.; Hartley, S.; Nikolova-Krstevski, V.; Rohde, P.R.; Ng, C.-A.; Sachs, F.; Gottlieb, P.A.; Martinac, B. Removal of the Mechanoprotective Influence of the Cytoskeleton Reveals PIEZO1 Is Gated by Bilayer Tension. Nat. Commun. 2016, 7, 10366. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.H.; Grandl, J. Mechanical Sensitivity of Piezo1 Ion Channels Can Be Tuned by Cellular Membrane Tension. eLife 2015, 4, e12088. [Google Scholar] [CrossRef] [PubMed]
- Coste, B.; Mathur, J.; Schmidt, M.; Earley, T.J.; Ranade, S.; Petrus, M.J.; Dubin, A.E.; Patapoutian, A. Piezo1 and Piezo2 Are Essential Components of Distinct Mechanically Activated Cation Channels. Science 2010, 330, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Parpaite, T.; Coste, B. Piezo Channels. Curr. Biol. 2017, 27, R250–R252. [Google Scholar] [CrossRef] [PubMed]
- Shinge, S.A.U.; Zhang, D.; Muluh, T.A.; Nie, Y.; Yu, F. Mechanosensitive Piezo1 Channel Evoked-Mechanical Signals in Atherosclerosis. J. Inflamm. Res. 2021, 14, 3621. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Hong, Z.; Zhong, M.; Klomp, J.; Bayless, K.J.; Mehta, D.; Karginov, A.V.; Hu, G.; Malik, A.B. Piezo1 Mediates Angiogenesis through Activation of MT1-MMP Signaling. Am. J. Physiol. Cell Physiol. 2019, 316, C92–C103. [Google Scholar] [CrossRef]
- Albarrán-Juárez, J.; Iring, A.; Wang, S.; Joseph, S.; Grimm, M.; Strilic, B.; Wettschureck, N.; Althoff, T.F.; Offermanns, S. Piezo1 and Gq/G11 Promote Endothelial Inflammation Depending on Flow Pattern and Integrin Activation. J. Exp. Med. 2018, 215, 2655–2672. [Google Scholar] [CrossRef]
- Wang, S.; Chennupati, R.; Kaur, H.; Iring, A.; Wettschureck, N.; Offermanns, S. Endothelial Cation Channel PIEZO1 Controls Blood Pressure by Mediating Flow-Induced ATP Release. J. Clin. Investig. 2016, 126, 4527–4536. [Google Scholar] [CrossRef]
- Otte, L.A.; Bell, K.S.; Loufrani, L.; Yeh, J.-C.; Melchior, B.; Dao, D.N.; Stevens, H.Y.; White, C.R.; Frangos, J.A. Rapid Changes in Shear Stress Induce Dissociation of a Gαq/11–Platelet Endothelial Cell Adhesion Molecule-1 Complex. J. Physiol. 2009, 587, 2365–2373. [Google Scholar] [CrossRef]
- Dela Paz, N.G.; Frangos, J.A. Rapid Flow-Induced Activation of Gαq/11 Is Independent of Piezo1 Activation. Am. J. Physiol. Cell Physiol. 2019, 316, C741–C752. [Google Scholar] [CrossRef]
- Zabroski, I.O.; Nugent, M.A. Lipid Raft Association Stabilizes VEGF Receptor 2 in Endothelial Cells. Int. J. Mol. Sci. 2021, 22, 798. [Google Scholar] [CrossRef]
- Coon, B.G.; Baeyens, N.; Han, J.; Budatha, M.; Ross, T.D.; Fang, J.S.; Yun, S.; Thomas, J.-L.; Schwartz, M.A. Intramembrane Binding of VE-Cadherin to VEGFR2 and VEGFR3 Assembles the Endothelial Mechanosensory Complex. J. Cell Biol. 2015, 208, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Ladoux, B.; Nelson, W.J.; Yan, J.; Mège, R.M. The Mechanotransduction Machinery at Work at Adherens Junctions. Integr. Biol. 2015, 7, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Gudi, S.R.P.; Clark, C.B.; Frangos, J.A. Fluid Flow Rapidly Activates G Proteins in Human Endothelial Cells. Circ. Res. 1996, 79, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Dela Paz, N.G.; Melchior, B.; Frangos, J.A. Shear Stress Induces Gαq/11 Activation Independently of G Protein-Coupled Receptor Activation in Endothelial Cells. Am. J. Physiol. Cell Physiol. 2017, 312, C428–C437. [Google Scholar] [CrossRef]
- Chachisvilis, M.; Zhang, Y.-L.; Frangos, J.A. G Protein-Coupled Receptors Sense Fluid Shear Stress in Endothelial Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15463–15468. [Google Scholar] [CrossRef]
- Candelario, J.; Chachisvilis, M. Activity of Bradykinin B2 Receptor Is Regulated by Long-Chain Polyunsaturated Fatty Acids. PLoS ONE 2013, 8, e68151. [Google Scholar] [CrossRef]
- Erdogmus, S.; Storch, U.; Danner, L.; Becker, J.; Winter, M.; Ziegler, N.; Wirth, A.; Offermanns, S.; Hoffmann, C.; Gudermann, T.; et al. Helix 8 Is the Essential Structural Motif of Mechanosensitive GPCRs. Nat. Commun. 2019, 10, 5784. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, Allosteric Signaling Machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Finney, A.C.; Stokes, K.Y.; Pattillo, C.B.; Orr, A.W. Integrin Signaling in Atherosclerosis. Cell. Mol. Life Sci. 2017, 74, 2263–2282. [Google Scholar] [CrossRef]
- Mo, F.-E. Shear-Regulated Extracellular Microenvironments and Endothelial Cell Surface Integrin Receptors Intertwine in Atherosclerosis. Front. Cell Dev. Biol. 2021, 9, 832. [Google Scholar] [CrossRef] [PubMed]
- Gaus, K.; Le Lay, S.; Balasubramanian, N.; Schwartz, M.A. Integrin-Mediated Adhesion Regulates Membrane Order. J. Cell Biol. 2006, 174, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Fu, Y.; Gu, M.; Zhang, L.; Li, D.; Li, H.; Chien, S.; Shyy, J.Y.-J.; Zhu, Y. Activation of Integrin A5 Mediated by Flow Requires Its Translocation to Membrane Lipid Rafts in Vascular Endothelial Cells. Proc. Natl. Acad. Sci. USA 2016, 113, 769. [Google Scholar] [CrossRef] [PubMed]
- Son, S.; Moroney, G.J.; Butler, P.J. Β1-Integrin-Mediated Adhesion Is Lipid-Bilayer Dependent. Biophys. J. 2017, 113, 1080–1092. [Google Scholar] [CrossRef]
- Lietha, D.; Izard, T. Roles of Membrane Domains in Integrin-Mediated Cell Adhesion. Int. J. Mol. Sci. 2020, 21, 5531. [Google Scholar] [CrossRef]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell Adhesion: Integrating Cytoskeletal Dynamics and Cellular Tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Tension across Protein | NMMII Involvement | Cell Type | Reference |
|---|---|---|---|---|
| E-cadherin 1 | 1–2 pN | + | MDCK epithelial cells | [100] |
| PECAM-1 in static cells | negligible | BAECs | [63] | |
| PECAM-1 in cells under SS | 2.0 pN/molecule | Vimentin is involved | BAECs | [63] |
| VE-cadherin in static cells | 2.4 nN/molecule | + | BAECs | [63] |
| VE-cadherin in cells under SS | 1.8 nN/molecule | + | BAECs | [63] |
| Vinculin | ~2.5 | + | BAECs | [62] |
| Stress Factor | Effect on RhoA or Rac1 | EC Type | Reference |
|---|---|---|---|
| Hypoxia | ↑ RhoA | Porcine AECs 1 | [93] |
| ↑ RhoA | Piglet PAEC | [116] | |
| ↑ RhoA | Rat PAECs | [117] | |
| ↓ Rac1 | Porcine -AECs | [93] | |
| ↓ Rac1 | Piglet PAECs | [116] | |
| Oxidative stress | ↑ RhoA | Rat PAECs | [117] |
| ↑ Rac1 | BAECs | [118] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barvitenko, N.; Ashrafuzzaman, M.; Lawen, A.; Skverchinskaya, E.; Saldanha, C.; Manca, A.; Uras, G.; Aslam, M.; Pantaleo, A. Endothelial Cell Plasma Membrane Biomechanics Mediates Effects of Pro-Inflammatory Factors on Endothelial Mechanosensors: Vicious Circle Formation in Atherogenic Inflammation. Membranes 2022, 12, 205. https://doi.org/10.3390/membranes12020205
Barvitenko N, Ashrafuzzaman M, Lawen A, Skverchinskaya E, Saldanha C, Manca A, Uras G, Aslam M, Pantaleo A. Endothelial Cell Plasma Membrane Biomechanics Mediates Effects of Pro-Inflammatory Factors on Endothelial Mechanosensors: Vicious Circle Formation in Atherogenic Inflammation. Membranes. 2022; 12(2):205. https://doi.org/10.3390/membranes12020205
Chicago/Turabian StyleBarvitenko, Nadezhda, Mohammad Ashrafuzzaman, Alfons Lawen, Elisaveta Skverchinskaya, Carlota Saldanha, Alessia Manca, Giuseppe Uras, Muhammad Aslam, and Antonella Pantaleo. 2022. "Endothelial Cell Plasma Membrane Biomechanics Mediates Effects of Pro-Inflammatory Factors on Endothelial Mechanosensors: Vicious Circle Formation in Atherogenic Inflammation" Membranes 12, no. 2: 205. https://doi.org/10.3390/membranes12020205
APA StyleBarvitenko, N., Ashrafuzzaman, M., Lawen, A., Skverchinskaya, E., Saldanha, C., Manca, A., Uras, G., Aslam, M., & Pantaleo, A. (2022). Endothelial Cell Plasma Membrane Biomechanics Mediates Effects of Pro-Inflammatory Factors on Endothelial Mechanosensors: Vicious Circle Formation in Atherogenic Inflammation. Membranes, 12(2), 205. https://doi.org/10.3390/membranes12020205