

Structural Pharmacology of Cation-Chloride Cotransporters


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
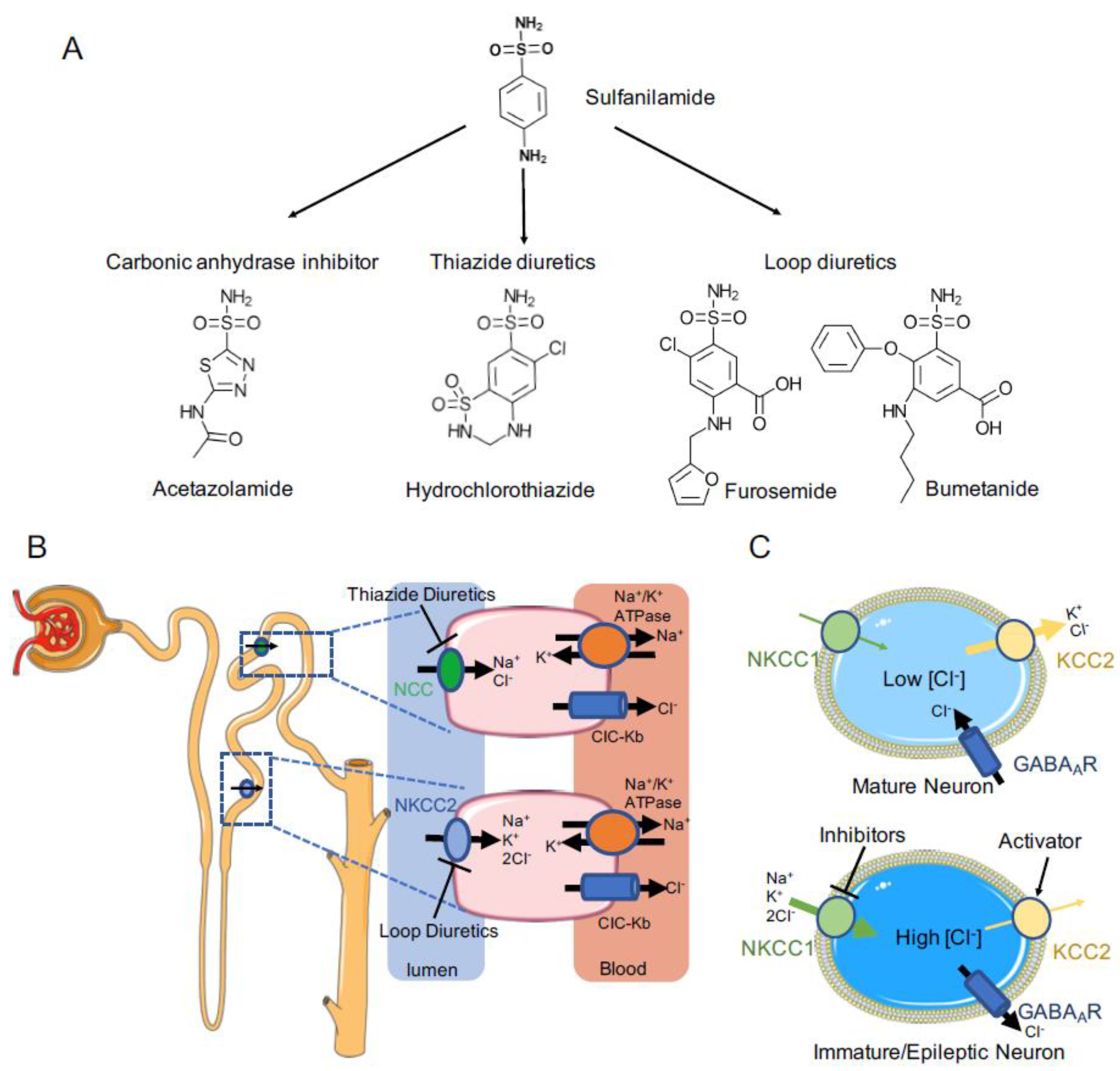
:1. Introduction
2. Structural Pharmacology of CCCs
Basic Architecture of CCCs
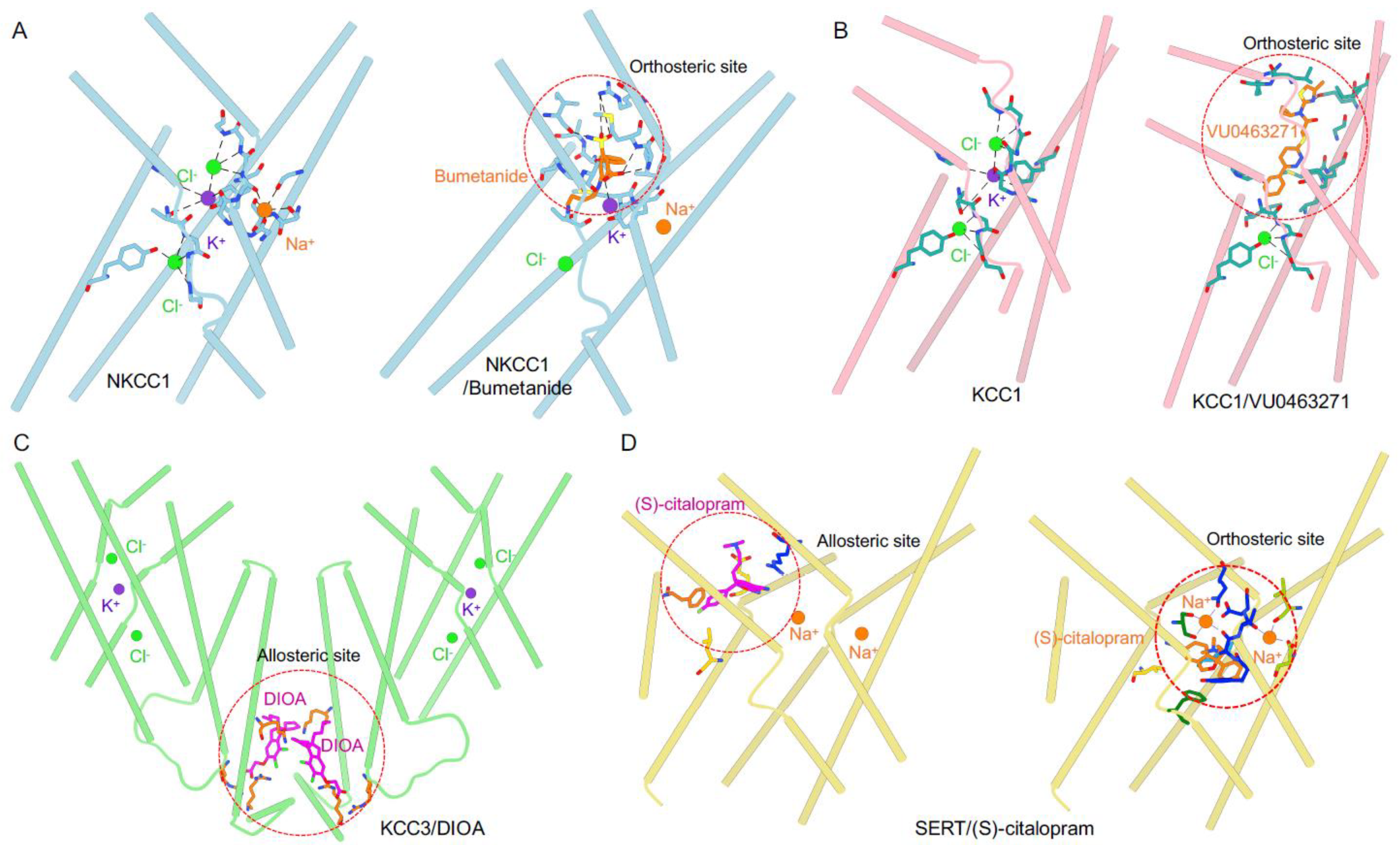
3. Extracellular Vestibule: A Hotspot for Pharmacological Inhibition of CCCs
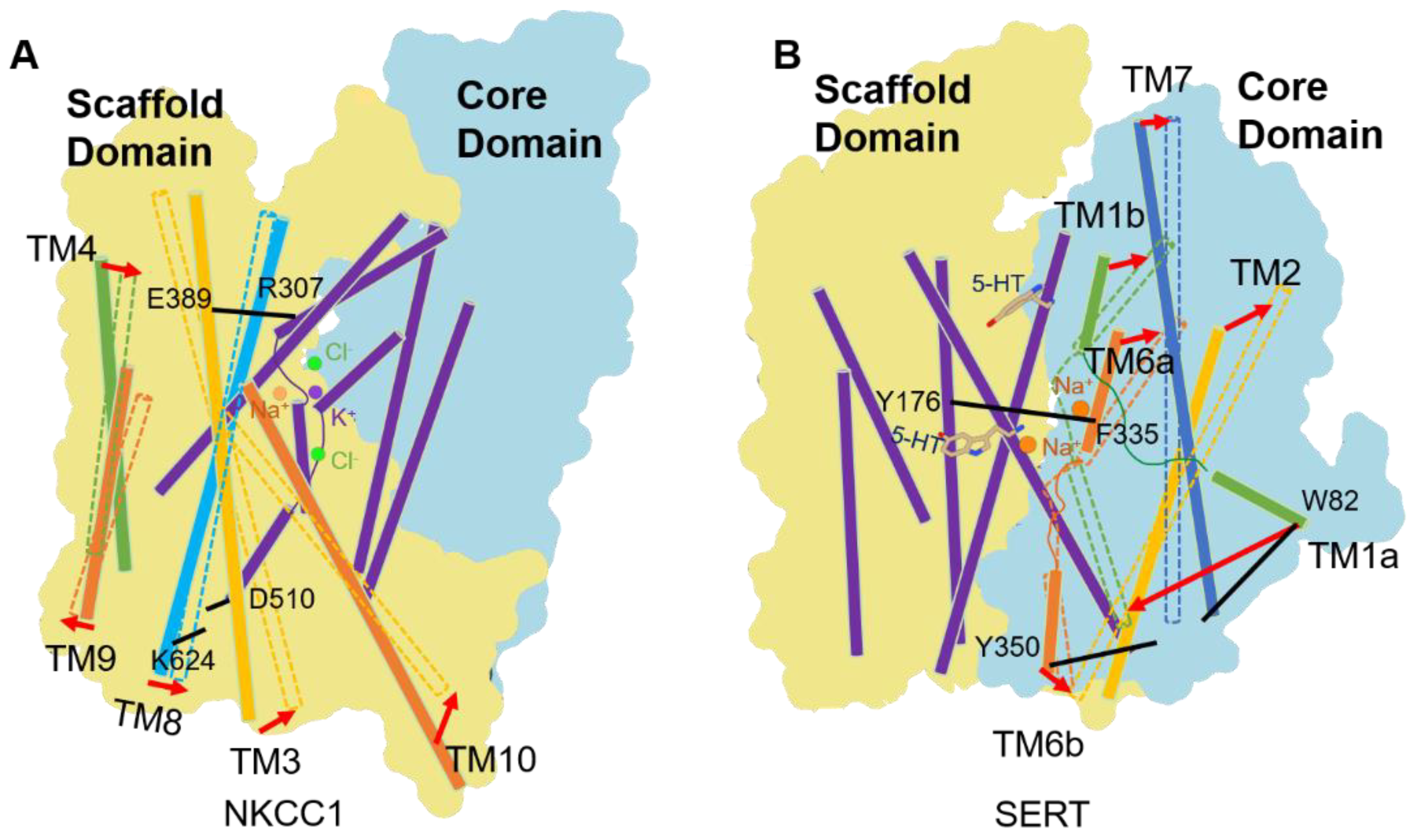
4. Alternate Access Model of CCCs
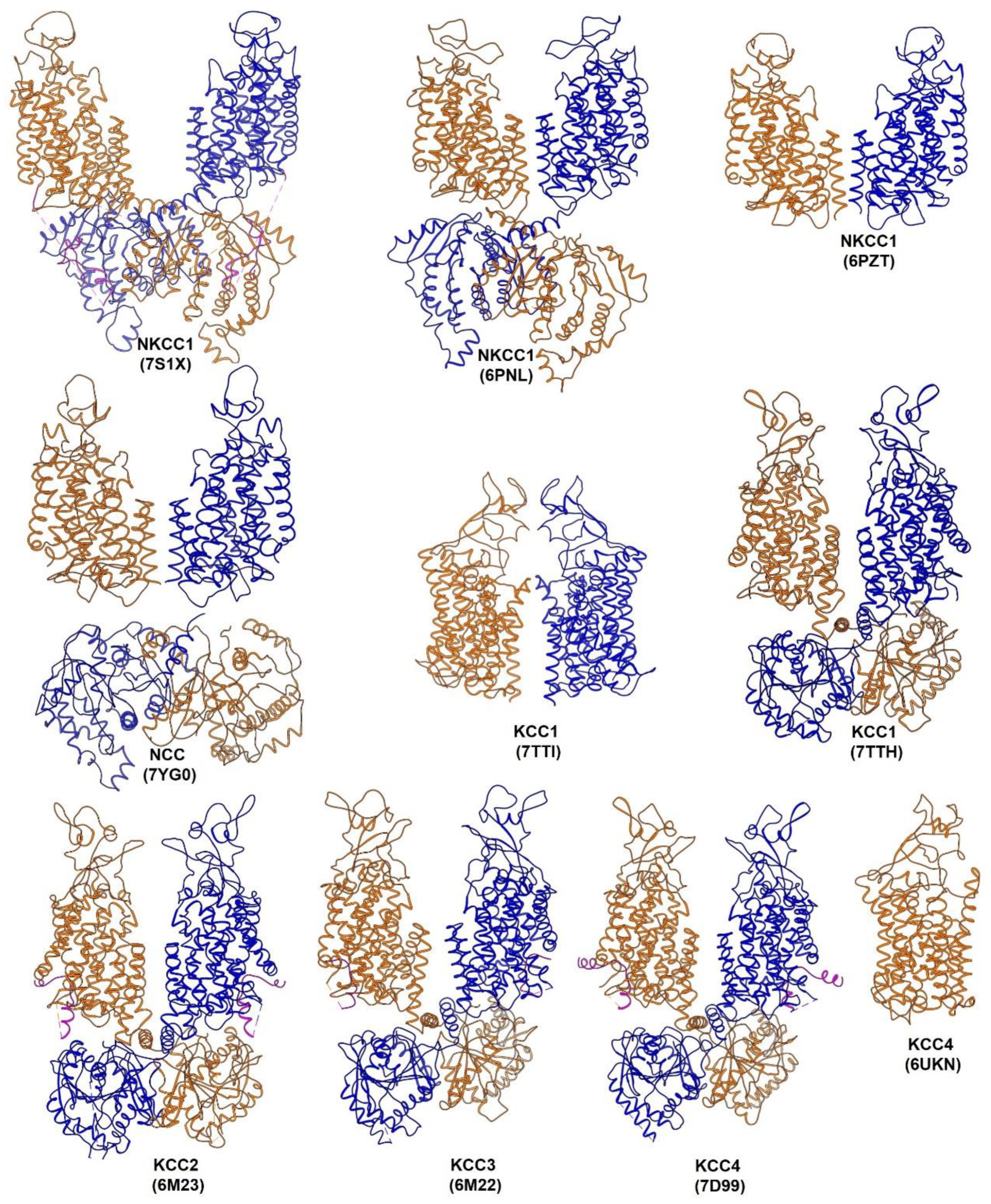
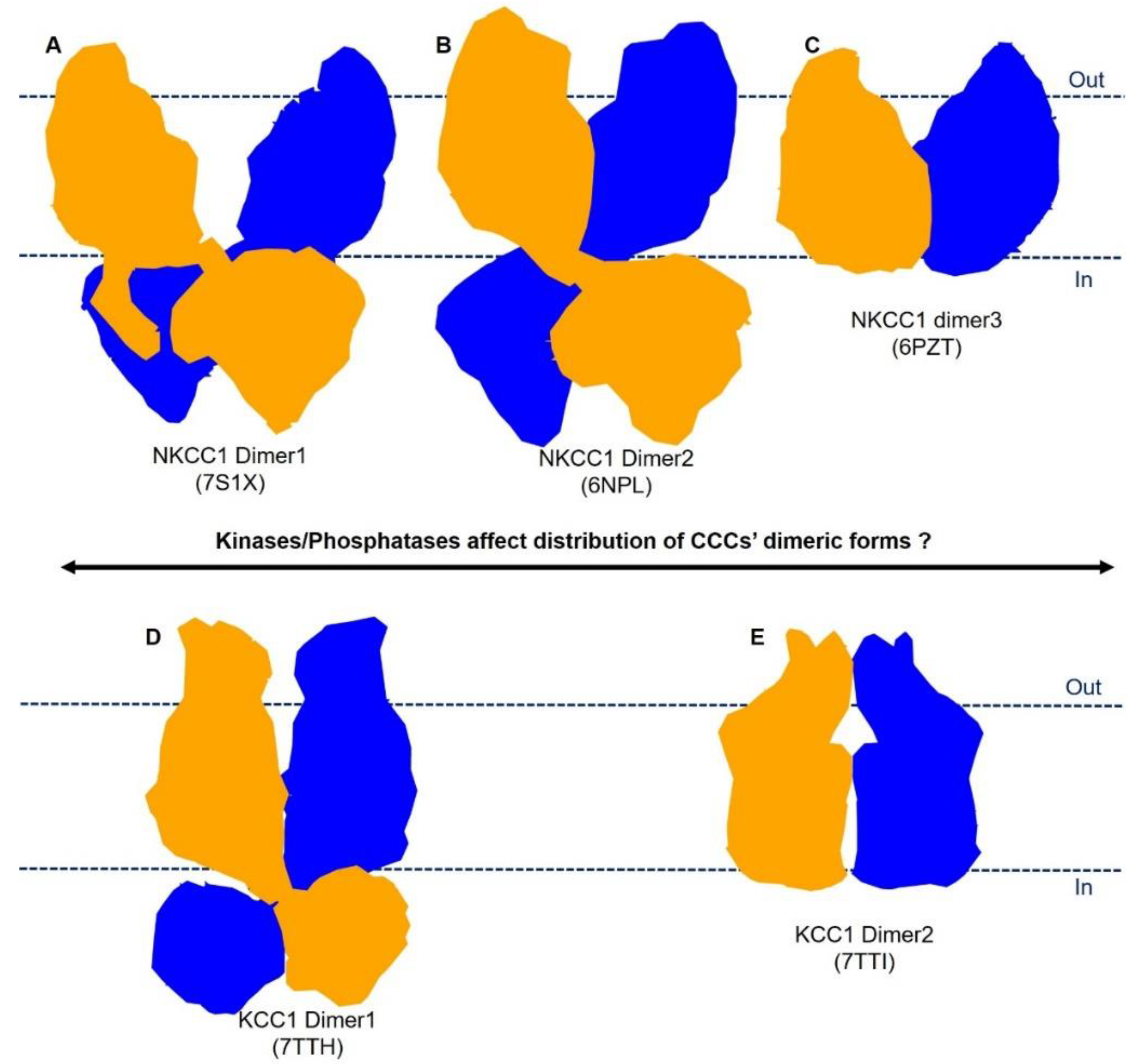
5. Plasticity in Mode of Assembly of CCC Dimers
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wile, D. Diuretics: A review. Ann. Clin. Biochem. 2012, 49, 419–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seldin, D.W.G.G.H. Diuretic Agents: Clinical Physiology and Pharmacology; Academic Press: London, UK, 1997. [Google Scholar]
- Feit, P.W. Bumetanide the Way to Its Chemical-Structure. J. Clin. Pharmacol. 1981, 21, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Hober, R. Effect of some sulfonamides on renal secretion. Proc. Soc. Exp. Biol. Med. 1942, 49, 87–90. [Google Scholar] [CrossRef]
- Maren, T.H.; Mayer, E.; Wadsworth, B.C. Carbonic anhydrase inhibition. I. The pharmacology of diamox 2-acetylamino-1,3,4-thiadiazole-5-sulfonamide. Bull. Johns. Hopkins. Hosp. 1954, 95, 199–243. [Google Scholar]
- Beyer, K.H. The mechanism of action of chlorothiazide. Ann. New York Acad. Sci. 1958, 71, 363–379. [Google Scholar] [CrossRef]
- Kleinfelder, H. Experimental Studies and Clinical Trials of a New Diuretic. Dtsch. Med. Wochenschr. 1963, 88, 1695–1702. [Google Scholar] [CrossRef] [PubMed]
- Roush, G.C.; Sica, D.A. Diuretics for Hypertension: A Review and Update. Am. J. Hypertens. 2016, 29, 1130–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokes, J.B.; Lee, I.; Damico, M. Sodium-Chloride Absorption by the Urinary-Bladder of the Winter Flounder—A Thiazide-Sensitive, Electrically Neutral Transport-System. J. Clin. Investig. 1984, 74, 7–16. [Google Scholar] [CrossRef]
- Haas, M.; Forbush, B. Photolabeling of a 150-Kda (Na+K+Cl) Cotransport Protein from Dog Kidney with a Bumetanide Analog. Am. J. Physiol. 1987, 253, C243–C252. [Google Scholar] [CrossRef]
- Xu, J.C.; Lytle, C.; Zhu, T.T.; Payne, J.A.; Benz, E., Jr.; Forbush, B., 3rd. Molecular cloning and functional expression of the bumetanide-sensitive Na-K-Cl cotransporter. Proc. Natl. Acad. Sci. USA 1994, 91, 2201–2205. [Google Scholar] [CrossRef] [Green Version]
- Gamba, G.; Saltzberg, S.N.; Lombardi, M.; Miyanoshita, A.; Lytton, J.; Hediger, M.A.; Brenner, B.M.; Hebert, S.C. Primary structure and functional expression of a cDNA encoding the thiazide-sensitive, electroneutral sodium-chloride cotransporter. Proc. Natl. Acad. Sci. USA 1993, 90, 2749–2753. [Google Scholar] [CrossRef] [PubMed]
- Gillen, C.M.; Brill, S.; Payne, J.A.; Forbush, B., 3rd. Molecular cloning and functional expression of the K-Cl cotransporter from rabbit, rat, and human. A new member of the cation-chloride cotransporter family. J. Biol. Chem. 1996, 271, 16237–16244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, J.A.; Stevenson, T.J.; Donaldson, L.F. Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J. Biol. Chem. 1996, 271, 16245–16252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Race, J.E.; Makhlouf, F.N.; Logue, P.J.; Wilson, F.H.; Dunham, P.B.; Holtzman, E.J. Molecular cloning and functional characterization of KCC3, a new K-Cl cotransporter. Am. J. Physiol. 1999, 277, C1210–C1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mount, D.B.; Mercado, A.; Song, L.; Xu, J.; George, A.L., Jr.; Delpire, E.; Gamba, G. Cloning and characterization of KCC3 and KCC4, new members of the cation-chloride cotransporter gene family. J. Biol. Chem. 1999, 274, 16355–16362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, J.A.; Forbush, B. Alternatively Spliced Isoforms of the Putative Renal Na-K-Cl Cotransporter Are Differentially Distributed within the Rabbit Kidney. Proc. Natl. Acad. Sci. USA 1994, 91, 4544–4548. [Google Scholar] [CrossRef] [Green Version]
- Russell, J.M. Sodium-potassium-chloride cotransport. Physiol. Rev. 2000, 80, 211–276. [Google Scholar] [CrossRef] [Green Version]
- Gamba, G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol. Rev. 2005, 85, 423–493. [Google Scholar] [CrossRef]
- Gagnon, K.B.; Delpire, E. Physiology of SLC12 transporters: Lessons from inherited human genetic mutations and genetically engineered mouse knockouts. Am. J. Physiol.-Cell Physiol. 2013, 304, C693–C714. [Google Scholar] [CrossRef] [Green Version]
- Daigle, N.D.; Carpentier, G.A.; Frenette-Cotton, R.; Simard, M.G.; Lefoll, M.H.; Noel, M.; Caron, L.; Noel, J.; Isenring, P. Molecular characterization of a human cation-Cl- cotransporter (SLC12A8A, CCC9A) that promotes polyamine and amino acid transport. J. Cell. Physiol. 2009, 220, 680–689. [Google Scholar] [CrossRef]
- Grozio, A.; Mills, K.F.; Yoshino, J.; Bruzzone, S.; Sociali, G.; Tokizane, K.; Lei, H.C.; Cunningham, R.; Sasaki, Y.; Migaud, M.E.; et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab. 2019, 1, 743. [Google Scholar] [CrossRef] [PubMed]
- Grozio, A.; Mills, K.; Yoshino, J.; Bruzzone, S.; Sociali, G.; Tokizane, K.; Lei, H.C.; Sasaki, Y.; Migaud, M.; Imai, S. Reply to: Absence of evidence that Slc12a8 encodes a nicotinamide mononucleotide transporter. Nat. Metab. 2019, 1, 662–665. [Google Scholar] [CrossRef] [PubMed]
- Caron, L.; Rousseau, F.; Gagnon, E.; Isenring, P. Cloning and functional characterization of a cation-Cl- cotransporter-interacting protein. J. Biol. Chem. 2000, 275, 32027–32036. [Google Scholar] [CrossRef] [Green Version]
- Wenz, M.; Hartmann, A.M.; Friauf, E.; Nothwang, H.G. CIP1 is an activator of the K+-Cl- cotransporter KCC2. Biochem. Biophys. Res. Commun. 2009, 381, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Delpire, E.; Mount, D.B. Human and murine phenotypes associated with defects in cation-chloride cotransport. Annu. Rev. Physiol. 2002, 64, 803–843. [Google Scholar] [CrossRef] [PubMed]
- Meor Azlan, N.F.; Koeners, M.P.; Zhang, J. Regulatory control of the Na-Cl co-transporter NCC and its therapeutic potential for hypertension. Acta. Pharm. Sin. B 2021, 11, 1117–1128. [Google Scholar] [CrossRef]
- Castrop, H.; Schiessl, I.M. Physiology and pathophysiology of the renal Na-K-2Cl cotransporter (NKCC2). Am. J. Physiol. Ren. Physiol. 2014, 307, F991–F1002. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.B.; Karet, F.E.; Hamdan, J.M.; DiPietro, A.; Sanjad, S.A.; Lifton, R.P. Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat. Genet. 1996, 13, 183–188. [Google Scholar] [CrossRef]
- Simon, D.B.; NelsonWilliams, C.; Bia, M.J.; Ellison, D.; Karet, F.E.; Molina, A.M.; Vaara, I.; Iwata, F.; Cushner, H.M.; Koolen, M.; et al. Gitelman’s variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat. Genet. 1996, 12, 24–30. [Google Scholar] [CrossRef]
- Trepiccione, F.; Zacchia, M.; Capasso, G. The role of the kidney in salt-sensitive hypertension. Clin. Exp. Nephrol. 2012, 16, 68–72. [Google Scholar] [CrossRef]
- Anglani, F.; Salviati, L.; Cassina, M.; Rigato, M.; Gobbi, L.; Calo, L.A. Genotype-phenotype correlation in Gordon’s syndrome: Report of two cases carrying novel heterozygous mutations. J. Nephrol. 2022, 35, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Kahle, K.T.; Ring, A.M.; Lifton, R.P. Molecular physiology of the WNK kinases. Annu. Rev. Physiol. 2008, 70, 329–355. [Google Scholar] [CrossRef] [PubMed]
- Boyden, L.M.; Choi, M.; Choate, K.A.; Nelson-Williams, C.J.; Farhi, A.; Toka, H.R.; Tikhonova, I.R.; Bjornson, R.; Mane, S.M.; Colussi, G.; et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 2012, 482, 98–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, D.A.; Carter, B.; Cushman, W.; Hamm, L. Thiazide and Loop Diuretics. J. Clin. Hypertens. 2011, 13, 639–643. [Google Scholar] [CrossRef]
- Blaesse, P.; Airaksinen, M.S.; Rivera, C.; Kaila, K. Cation-Chloride Cotransporters and Neuronal Function. Neuron 2009, 61, 820–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaila, K.; Price, T.J.; Payne, J.A.; Puskarjov, M.; Voipio, J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 2014, 15, 637–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Tornberg, J.; Kaila, K.; Airaksinen, M.S.; Rivera, C. Patterns of cation-chloride cotransporter expression during embryonic rodent CNS development. Eur. J. Neurosci. 2002, 16, 2358–2370. [Google Scholar] [CrossRef]
- Balakrishnan, V.; Becker, M.; Lohrke, S.; Nothwang, H.G.; Guresir, E.; Friauf, E. Expression and function of chloride transporters during development of inhibitory neurotransmission in the auditory brainstem. J. Neurosci. 2003, 23, 4134–4145. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Zhang, J.; Mansuri, M.S.; Duan, J.; Karimy, J.K.; Delpire, E.; Alper, S.L.; Lifton, R.P.; Fukuda, A.; Kahle, K.T. Developmentally regulated KCC2 phosphorylation is essential for dynamic GABA-mediated inhibition and survival. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Sedmak, G.; Jovanov-Milosevic, N.; Puskarjov, M.; Ulamec, M.; Kruslin, B.; Kaila, K.; Judas, M. Developmental Expression Patterns of KCC2 and Functionally Associated Molecules in the Human Brain. Cereb. Cortex. 2016, 26, 4574–4589. [Google Scholar] [CrossRef] [Green Version]
- Rivera, C.; Voipio, J.; Payne, J.A.; Ruusuvuori, E.; Lahtinen, H.; Lamsa, K.; Pirvola, U.; Saarma, M.; Kaila, K. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 1999, 397, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Duy, P.Q.; David, W.B.; Kahle, K.T. Identification of KCC2 Mutations in Human Epilepsy Suggests Strategies for Therapeutic Transporter Modulation. Front. Cell Neurosci. 2019, 13, 515. [Google Scholar] [CrossRef] [PubMed]
- Stodberg, T.; McTague, A.; Ruiz, A.J.; Hirata, H.; Zhen, J.; Long, P.; Farabella, I.; Meyer, E.; Kawahara, A.; Vassallo, G.; et al. Mutations in SLC12A5 in epilepsy of infancy with migrating focal seizures. Nat. Commun. 2015, 6, 8038. [Google Scholar] [CrossRef] [PubMed]
- Uyanik, G. Novel truncating and missense mutations of the KCC3 gene associated with Andermann syndrome. Neurology 2006, 67, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- Portioli, C.; Ruiz Munevar, M.J.; De Vivo, M.; Cancedda, L. Cation-coupled chloride cotransporters: Chemical insights and disease implications. Trends. Chem. 2021, 3, 832–849. [Google Scholar] [CrossRef]
- Savardi, A.; Borgogno, M.; De Vivo, M.; Cancedda, L. Pharmacological tools to target NKCC1 in brain disorders. Trends Pharmacol. Sci. 2021, 42, 1009–1034. [Google Scholar] [CrossRef]
- Kharod, S.C.; Kang, S.K.; Kadam, S.D. Off-Label Use of Bumetanide for Brain Disorders: An Overview. Front. Neurosci.-Switz. 2019, 13, 310. [Google Scholar] [CrossRef] [Green Version]
- Loscher, W.; Kaila, K. CNS pharmacology of NKCC1 inhibitors*. Neuropharmacology 2022, 205, 108910. [Google Scholar] [CrossRef]
- Hampel, P.; Romermann, K.; Gramer, M.; Loscher, W. The search for brain-permeant NKCC1 inhibitors for the treatment of seizures: Pharmacokinetic-pharmacodynamic modelling of NKCC1 inhibition by azosemide, torasemide, and bumetanide in mouse brain. Epilepsy Behav. 2021, 114, 107616. [Google Scholar] [CrossRef]
- Taubes, A.; Nova, P.; Zalocusky, K.A.; Kosti, I.; Bicak, M.; Zilberter, M.Y.; Hao, Y.; Yoon, S.Y.; Oskotsky, T.; Pineda, S.; et al. Experimental and real-world evidence supporting the computational repurposing of bumetanide for APOE4-related Alzheimer’s disease. Nat. Aging 2021, 1, 932–947. [Google Scholar] [CrossRef]
- Puskarjov, M.; Kahle, K.T.; Ruusuvuori, E.; Kaila, K. Pharmacotherapeutic targeting of cation-chloride cotransporters in neonatal seizures. Epilepsia 2014, 55, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Delpire, E. Advances in the development of novel compounds targeting cation-chloride cotransporter physiology. Am. J. Physiol.-Cell Physiol. 2021, 320, C324–C340. [Google Scholar] [CrossRef] [PubMed]
- Savardi, A.; Borgogno, M.; Narducci, R.; La Sala, G.; Ortega, J.A.; Summa, M.; Armirotti, A.; Bertorelli, R.; Contestabile, A.; De Vivo, M.; et al. Discovery of a Small Molecule Drug Candidate for Selective NKCC1 Inhibition in Brain Disorders. Chem 2020, 6, 2073–2096. [Google Scholar] [CrossRef] [PubMed]
- Delpire, E.; Days, E.; Lewis, L.M.; Mi, D.H.; Kim, K.; Lindsley, C.W.; Weaver, C.D. Small-molecule screen identifies inhibitors of the neuronal K-Cl cotransporter KCC2. Proc. Natl. Acad. Sci. USA 2009, 106, 5383–5388. [Google Scholar] [CrossRef] [Green Version]
- Gagnon, M.; Bergeron, M.J.; Lavertu, G.; Castonguay, A.; Tripathy, S.; Bonin, R.P.; Perez-Sanchez, J.; Boudreau, D.; Wang, B.; Dumas, L.; et al. Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat. Med. 2013, 19, 1524–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiner, A.A.; Levine, J.; Russo, J.L.; Zagala, J. Treatment of congestive heart failure and hypertension: Clinical experience with hydrochlorothiazide. Am. Pract. Dig. Treat. 1960, 11, 224–226. [Google Scholar]
- Ingram, T.T. Today’s Drugs. Frusemide. Br. Med. J. 1964, 2, 1640–1641. [Google Scholar]
- Asbury, M.J.; Gatenby, P.B.; O’Sullivan, S.; Bourke, E. Bumetanide: Potent new “loop” diuretic. Br. Med. J. 1972, 1, 211–213. [Google Scholar] [CrossRef]
- Marsh, J.D.; Smith, T.W. Piretanide: A loop-active diuretic. Pharmacology, therapeutic efficacy and adverse effects. Pharmacotherapy 1984, 4, 170–180. [Google Scholar] [CrossRef]
- Brater, D.C.; Leinfelder, J.; Anderson, S.A. Clinical pharmacology of torasemide, a new loop diuretic. Clin. Pharmacol. Ther. 1987, 42, 187–192. [Google Scholar] [CrossRef]
- Krumlovsky, F.A.; del Greco, F. Diuretic agents. Mechanisms of action and clinical uses. Postgrad. Med. 1976, 59, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Eraly, S.A.; Vallon, V.; Vaughn, D.A.; Gangoiti, J.A.; Richter, K.; Nagle, M.; Monte, J.C.; Rieg, T.; Truong, D.M.; Long, J.M.; et al. Decreased renal organic anion secretion and plasma accumulation of endogenous organic anions in OAT1 knock-out mice. J Biol. Chem. 2006, 281, 5072–5083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallon, V.; Rieg, T.; Ahn, S.Y.; Wu, W.; Eraly, S.A.; Nigam, S.K. Overlapping in vitro and in vivo specificities of the organic anion transporters OAT1 and OAT3 for loop and thiazide diuretics. Am. J. Physiol.-Ren. 2008, 294, F867–F873. [Google Scholar] [CrossRef] [PubMed]
- Rybak, L.P. Ototoxicity of loop diuretics. Otolaryngol. Clin. N. Am. 1993, 26, 829–844. [Google Scholar] [CrossRef]
- Delpire, E.; Guo, J. Cryo-EM structures of DrNKCC1 and hKCC1: A new milestone in the physiology of cation-chloride cotransporters. Am. J. Physiol. Cell. Physiol. 2020, 318, C225–C237. [Google Scholar] [CrossRef]
- Chew, T.A.; Zhang, J.R.; Feng, L. High-Resolution Views and Transport Mechanisms of the NKCC1 and KCC Transporters. J. Mol. Biol. 2021, 433, 167056. [Google Scholar] [CrossRef]
- Orlov, S.N.; Koltsova, S.V.; Kapilevich, L.V.; Gusakova, S.V.; Dulin, N.O. NKCC1 and NKCC2: The pathogenetic role of cation-chloride cotransporters in hypertension. Genes Dis. 2015, 2, 186–196. [Google Scholar] [CrossRef] [Green Version]
- Payne, J.A. Functional characterization of the neuronal-specific K-Cl cotransporter: Implications for [K+]o regulation. Am. J. Physiol. 1997, 273, C1516–C1525. [Google Scholar] [CrossRef]
- Mercado, A.; Song, L.; Vazquez, N.; Mount, D.B.; Gamba, G. Functional comparison of the K+-Cl- cotransporters KCC1 and KCC4. J. Biol. Chem. 2000, 275, 30326–30334. [Google Scholar] [CrossRef] [Green Version]
- Warmuth, S.; Zimmermann, I.; Dutzler, R. X-ray Structure of the C-Terminal Domain of a Prokaryotic Cation-Chloride Cotransporter. Structure 2009, 17, 538–546. [Google Scholar] [CrossRef] [Green Version]
- Chew, T.A.; Orlando, B.J.; Zhang, J.R.; Latorraca, N.R.; Wang, A.; Hollingsworth, S.A.; Chen, D.H.; Dror, R.O.; Liao, M.F.; Feng, L. Structure and mechanism of the cation-chloride cotransporter NKCC1. Nature 2019, 572, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Y.; Wang, Q.Z.; Cao, E.R. Structure of the human cation-chloride cotransporter NKCC1 determined by single-particle electron cryo-microscopy. Nat. Commun. 2020, 11, 1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.S.; Zhou, J.; Zhang, Y.B.; Liu, T.Y.; Friedel, P.; Zhuo, W.; Somasekharan, S.; Roy, K.; Zhang, L.X.; Liu, Y.; et al. The structural basis of function and regulation of neuronal cotransporters NKCC1 and KCC2. Commun. Biol. 2021, 4, 226. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.X.; Roy, K.; Vidossich, P.; Cancedda, L.; De Vivo, M.; Forbush, B.; Cao, E.H. Structural basis for inhibition of the Cation-chloride cotransporter NKCC1 by the diuretic drug bumetanide. Nat. Commun. 2022, 13, 2747. [Google Scholar] [CrossRef]
- Liu, S.; Chang, S.H.; Han, B.M.; Xu, L.Y.; Zhang, M.F.; Zhao, C.; Yang, W.; Wang, F.; Li, J.Y.; Delpire, E.; et al. Cryo-EM structures of the human cation-chloride cotransporter KCC1. Science 2019, 366, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Chi, G.M.; Ebenhoch, R.; Man, H.; Tang, H.P.; Tremblay, L.E.; Reggiano, G.; Qiu, X.Y.; Bohstedt, T.; Liko, I.; Almeida, F.G.; et al. Phospho-regulation, nucleotide binding and ion access control in potassium-chloride cotransporters. EMBO J. 2021, 40, e107294. [Google Scholar] [CrossRef]
- Zhao, Y.; Shen, J.; Wang, Q.; Ruiz Munevar, M.J.; Vidossich, P.; De Vivo, M.; Zhou, M.; Cao, E. Structure of the human cation-chloride cotransport KCC1 in an outward-open state. Proc. Natl. Acad. Sci. USA 2022, 119, e2109083119. [Google Scholar] [CrossRef]
- Chi, X.M.; Li, X.R.; Chen, Y.; Zhang, Y.Y.; Su, Q.; Zhou, Q. Cryo-EM structures of the full-length human KCC2 and KCC3 cation-chloride cotransporters. Cell Res. 2021, 31, 941. [Google Scholar] [CrossRef]
- Xie, Y.; Chang, S.H.; Zhao, C.; Wang, F.; Liu, S.; Wang, J.; Delpire, E.; Ye, S.; Guo, J.T. Structures and an activation mechanism of human potassium-chloride cotransporters. Sci. Adv. 2020, 6, eabc5883. [Google Scholar] [CrossRef]
- Reid, M.S.; Kern, D.M.; Brohawn, S.G. Cryo-EM structure of the potassium-chloride cotransporter KCC4 in lipid nanodiscs. Elife 2020, 9, e52505. [Google Scholar] [CrossRef] [Green Version]
- Nan, J.; Yuan, Y.; Yang, X.; Shan, Z.; Liu, H.; Wei, F.; Zhang, W.; Zhang, Y. Cryo-EM structure of the human sodium-chloride cotransporter NCC. Sci. Adv. 2022, 8, eadd7176. [Google Scholar] [CrossRef] [PubMed]
- Moseng, M.A.; Su, C.C.; Rios, K.; Cui, M.; Lyu, M.; Glaza, P.; Klenotic, P.A.; Delpire, E.; Yu, E.W. Inhibition mechanism of NKCC1 involves the carboxyl terminus and long-range conformational coupling. Sci. Adv. 2022, 8, eabq0952. [Google Scholar] [CrossRef]
- Zhao, Y.; Shen, J.; Wang, Q.; Zhou, M.; Cao, E. Inhibitory and Transport Mechanisms of the Human Cation-Chloride Cotransport KCC1. bioRxiv 2020, 221770. [Google Scholar] [CrossRef]
- Moore-Hoon, M.L.; Turner, R.J. The structural unit of the secretory Na+-K+-2Cl(-) cotransporter (NKCCl) is a homodimer. Biochemistry 2000, 39, 3718–3724. [Google Scholar] [CrossRef] [PubMed]
- Simard, C.F.; Bergeron, M.J.; Frenette-Cotton, R.; Carpentier, G.A.; Pelchat, M.E.; Caron, L.; Isenring, P. Homooligomeric and heterooligomeric associations between K+-Cl- cotransporter isoforms and between K+-Cl- and Na+-K+-Cl- cotransporters. J Biol. Chem. 2007, 282, 18083–18093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monette, M.Y.; Forbush, B. Regulatory activation is accompanied by movements in the C-terminus of the Na-K-Cl cotransporter (NKCC1). J. Biol. Chem. 2012, 287, 2210–2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, C.; Rosenbaek, L.L.; Flygaard, R.K.; Habeck, M.; Karlsen, J.L.; Wang, Y.; Lindorff-Larsen, K.; Gad, H.H.; Hartmann, R.; Lyons, J.A.; et al. Cryo-EM structure of the human NKCC1 transporter reveals mechanisms of ion coupling and specificity. Embo. J. 2022, e110169. [Google Scholar] [CrossRef]
- Krishnamurthy, H.; Piscitelli, C.L.; Gouaux, E. Unlocking the molecular secrets of sodium-coupled transporters. Nature 2009, 459, 347–355. [Google Scholar] [CrossRef]
- Faham, S.; Watanabe, A.; Besserer, G.M.; Cascio, D.; Specht, A.; Hirayama, B.A.; Wright, E.M.; Abramson, J. The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na(+)/sugar symport. Science 2008, 321, 810–814. [Google Scholar] [CrossRef] [Green Version]
- Weyand, S.; Shimamura, T.; Yajima, S.; Suzuki, S.; Mirza, O.; Krusong, K.; Carpenter, E.P.; Rutherford, N.G.; Hadden, J.M.; O’Reilly, J.; et al. Structure and Molecular Mechanism of a Nucleobase-Cation-Symport-1 Family Transporter. Science 2008, 322, 709–713. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Li, Y.; Yu, B.; Zhang, Z.; Brommer, B.; Williams, P.R.; Liu, Y.; Hegarty, S.V.; Zhou, S.; Zhu, J.; et al. Reactivation of Dormant Relay Pathways in Injured Spinal Cord by KCC2 Manipulations. Cell 2018, 174, 521–535.e513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardarelli, R.A.; Jones, K.; Pisella, L.I.; Wobst, H.J.; McWilliams, L.J.; Sharpe, P.M.; Burnham, M.P.; Baker, D.J.; Chudotvorova, I.; Guyot, J.; et al. The small molecule CLP257 does not modify activity of the K+-Cl- co-transporter KCC2 but does potentiate GABA(A) receptor activity. Nat. Med. 2017, 23, 1394–1396. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Forbush, B. [H-3] Bumetanide Binding to Duck Red-Cells-Correlation with Inhibition of (Na + K + 2cl) Cotransport. J. Biol. Chem. 1986, 261, 8434–8441. [Google Scholar] [CrossRef]
- Lykke, K.; Tollner, K.; Feit, P.W.; Erker, T.; MacAulay, N.; Loscher, W. The search for NKCC1-selective drugs for the treatment of epilepsy: Structure-function relationship of bumetanide and various bumetanide derivatives in inhibiting the human cation-chloride cotransporter NKCC1A. Epilepsy Behav. 2016, 59, 42–49. [Google Scholar] [CrossRef]
- Navratna, V.; Gouaux, E. Insights into the mechanism and pharmacology of neurotransmitter sodium symporters. Curr. Opin. Struc. Biol. 2019, 54, 161–170. [Google Scholar] [CrossRef]
- Claxton, D.P.; Quick, M.; Shi, L.; de Carvalho, F.D.; Weinstein, H.; Javitch, J.A.; Mchaourab, H.S. Ion/substrate-dependent conformational dynamics of a bacterial homolog of neurotransmitter: Sodium symporters. Nat. Struct. Mol. Biol. 2010, 17, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Penmatsa, A.; Gouaux, E. How LeuT shapes our understanding of the mechanisms of sodium-coupled neurotransmitter transporters. J. Physiol.-Lond. 2014, 592, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, H.; Gouaux, E. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 2012, 481, U469–U480. [Google Scholar] [CrossRef] [Green Version]
- Coleman, J.A.; Yang, D.X.; Zhao, Z.Y.; Wen, P.C.; Yoshioka, C.; Tajkhorshid, E.; Gouaux, E. Serotonin transporter-ibogaine complexes illuminate mechanisms of inhibition and transport. Nature 2019, 569, 141–145. [Google Scholar] [CrossRef]
- Somasekharan, S.; Tanis, J.; Forbush, B. Loop Diuretic and Ion-binding Residues Revealed by Scanning Mutagenesis of Transmembrane Helix 3 (TM3) of Na-K-Cl Cotransporter (NKCC1). J. Biol. Chem. 2012, 287, 17308–17317. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Ueno, S.; Fukuda, A. Interaction of neuron-specific K+-Cl- cotransporter, KCC2, with brain-type creatine kinase. FEBS Lett. 2004, 564, 131–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, K.; Yamada, J.; Ueno, S.; Fukuda, A. Brain-type creatine kinase activates neuron-specific K+-Cl- co-transporter KCC2. J. Neurochem. 2006, 96, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Ivakine, E.A.; Acton, B.A.; Mahadevan, V.; Ormond, J.; Tang, M.; Pressey, J.C.; Huang, M.Y.; Ng, D.; Delpire, E.; Salter, M.W.; et al. Neto2 is a KCC2 interacting protein required for neuronal Cl- regulation in hippocampal neurons. Proc. Natl. Acad. Sci. USA 2013, 110, 3561–3566. [Google Scholar] [CrossRef] [PubMed]
- Al Awabdh, S.; Donneger, F.; Goutierre, M.; Seveno, M.; Vigy, O.; Weinzettl, P.; Russeau, M.; Moutkine, I.; Levi, S.; Marin, P.; et al. Gephyrin Interacts with the K-Cl Cotransporter KCC2 to Regulate Its Surface Expression and Function in Cortical Neurons. J. Neurosci. 2022, 42, 166–182. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Zhang, J.W.; Khanna, A.; Hochdorfer, T.; Shang, Y.Z.; Kahle, K.T. The WNK-SPAK/OSR1 pathway: Master regulator of cation-chloride cotransporters. Sci. Signal 2014, 7, re3. [Google Scholar] [CrossRef]
- Gagnon, K.B.; Delpire, E. Multiple pathways for protein phosphatase 1 (PP1) regulation of Na-K-2Cl cotransporter (NKCC1) function: The N-terminal tail of the Na-K-2Cl cotransporter serves as a regulatory scaffold for Ste20-related proline/alanine-rich kinase (SPAK) AND PP1. J. Biol. Chem. 2010, 285, 14115–14121. [Google Scholar] [CrossRef] [Green Version]
- Rinehart, J.; Maksimova, Y.D.; Tanis, J.E.; Stone, K.L.; Hodson, C.A.; Zhang, J.H.; Risinger, M.; Pan, W.J.; Wu, D.Q.; Colangelo, C.M.; et al. Sites of Regulated Phosphorylation that Control K-Cl Cotransporter Activity. Cell 2009, 138, 525–536. [Google Scholar] [CrossRef] [Green Version]
- Darman, R.B.; Forbush, B. A regulatory locus of phosphorylation in the N terminus of the Na-K-Cl cotransporter, NKCC1. J. Biol. Chem. 2002, 277, 37542–37550. [Google Scholar] [CrossRef] [Green Version]
- Pacheco-Alvarez, D.; Cristobal, P.S.; Meade, P.; Moreno, E.; Vazquez, N.; Munoz, E.; Diaz, A.; Juarez, M.E.; Gimenez, I.; Gamba, G. The Na+: Cl- cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J. Biol. Chem. 2006, 281, 28755–28763. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.; Hartmann, A.M.; Beyer, T.; Ripperger, A.; Nothwang, H.G. A novel regulatory locus of phosphorylation in the C terminus of the potassium chloride cotransporter KCC2 that interferes with N-ethylmaleimide or staurosporine-mediated activation. J. Biol. Chem. 2014, 289, 18668–18679. [Google Scholar] [CrossRef] [Green Version]
- Cordshagen, A.; Busch, W.; Winklhofer, M.; Nothwang, H.G.; Hartmann, A.M. Phosphoregulation of the intracellular termini of K(+)-Cl(-) cotransporter 2 (KCC2) enables flexible control of its activity. J. Biol. Chem. 2018, 293, 16984–16993. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Cao, E. Structural Pharmacology of Cation-Chloride Cotransporters. Membranes 2022, 12, 1206. https://doi.org/10.3390/membranes12121206
Zhao Y, Cao E. Structural Pharmacology of Cation-Chloride Cotransporters. Membranes. 2022; 12(12):1206. https://doi.org/10.3390/membranes12121206
Chicago/Turabian StyleZhao, Yongxiang, and Erhu Cao. 2022. "Structural Pharmacology of Cation-Chloride Cotransporters" Membranes 12, no. 12: 1206. https://doi.org/10.3390/membranes12121206
APA StyleZhao, Y., & Cao, E. (2022). Structural Pharmacology of Cation-Chloride Cotransporters. Membranes, 12(12), 1206. https://doi.org/10.3390/membranes12121206