
Wetting-Induced Polyelectrolyte Pore Bridging

 , and
, and
Abstract
:
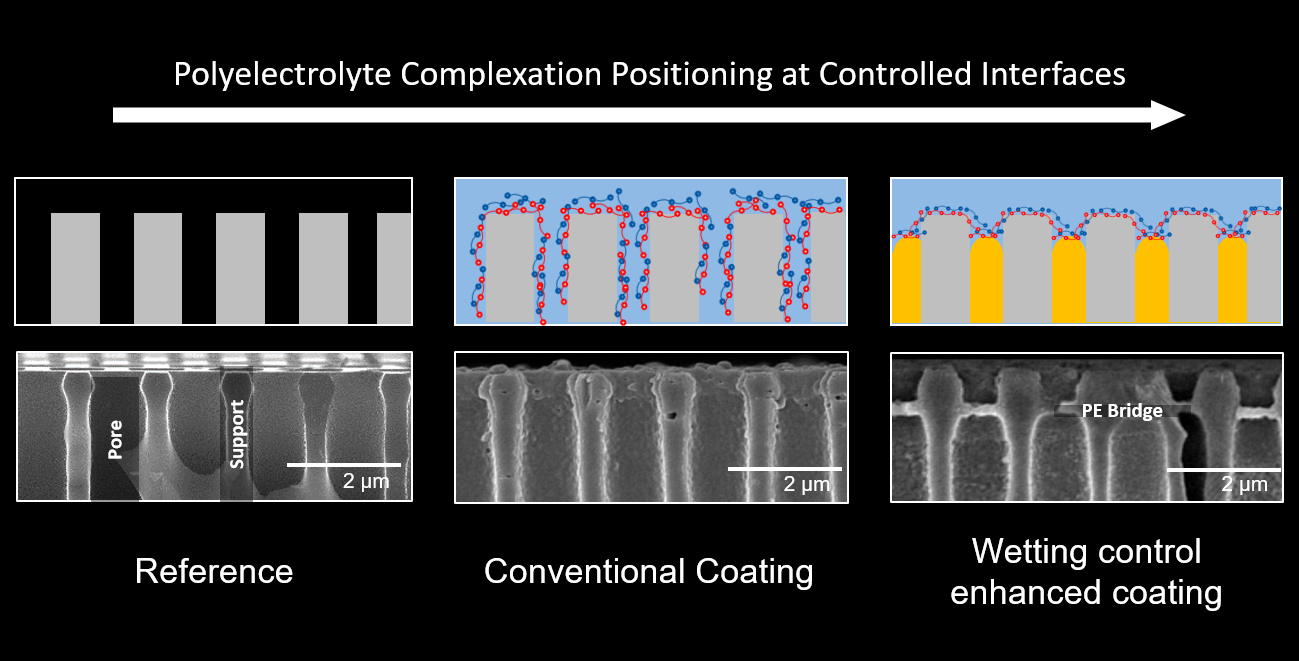{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
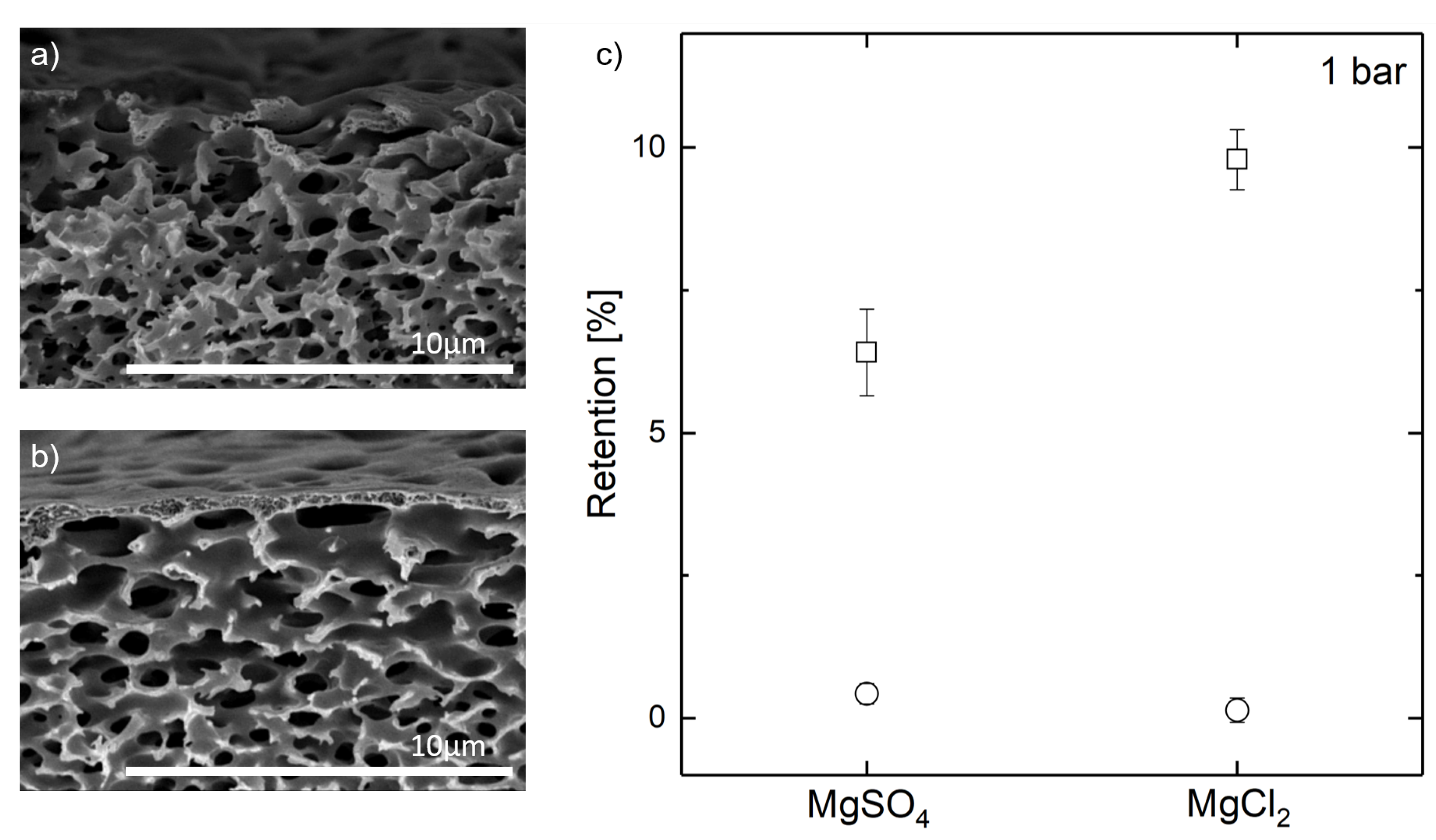
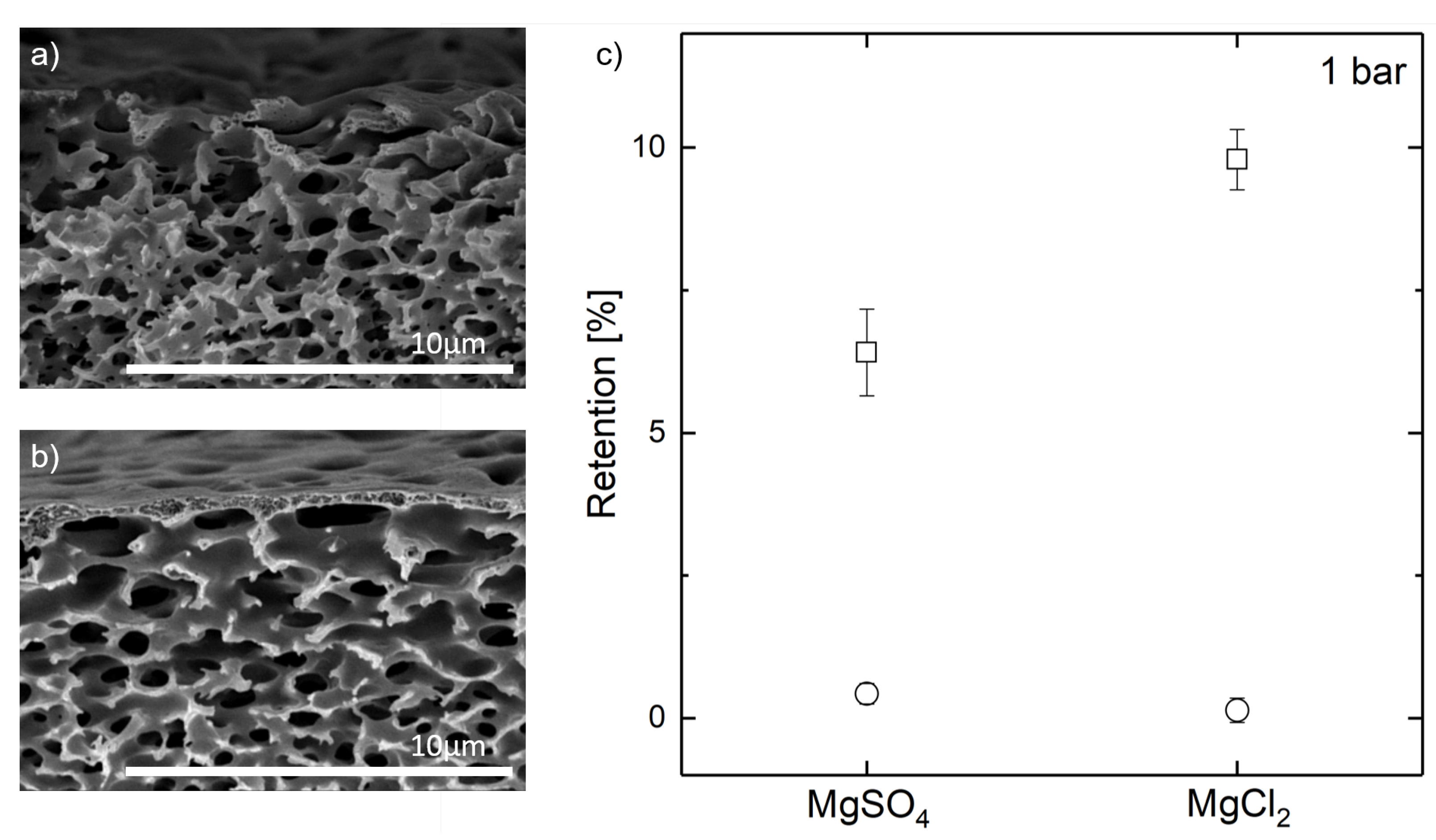{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Support Structure
2.2. Polyelectrolyte Complexation
2.3. Wetting Agents and Wetting Control
2.4. Analytical Methods
3. Results and Discussion
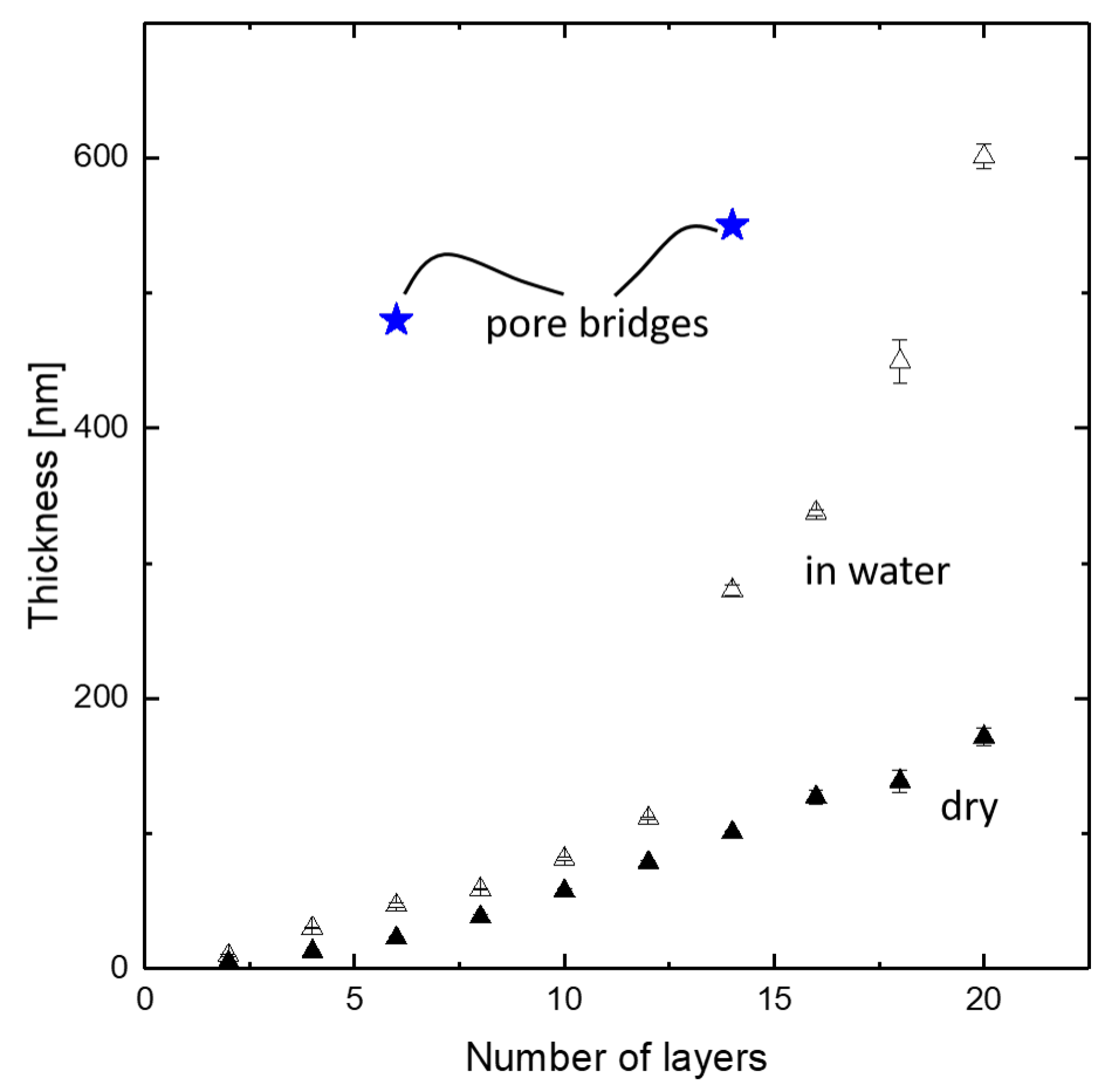
3.1. Polyelectrolyte Pore Bridging
3.2. Polyelectrolyte Multilayer Growth
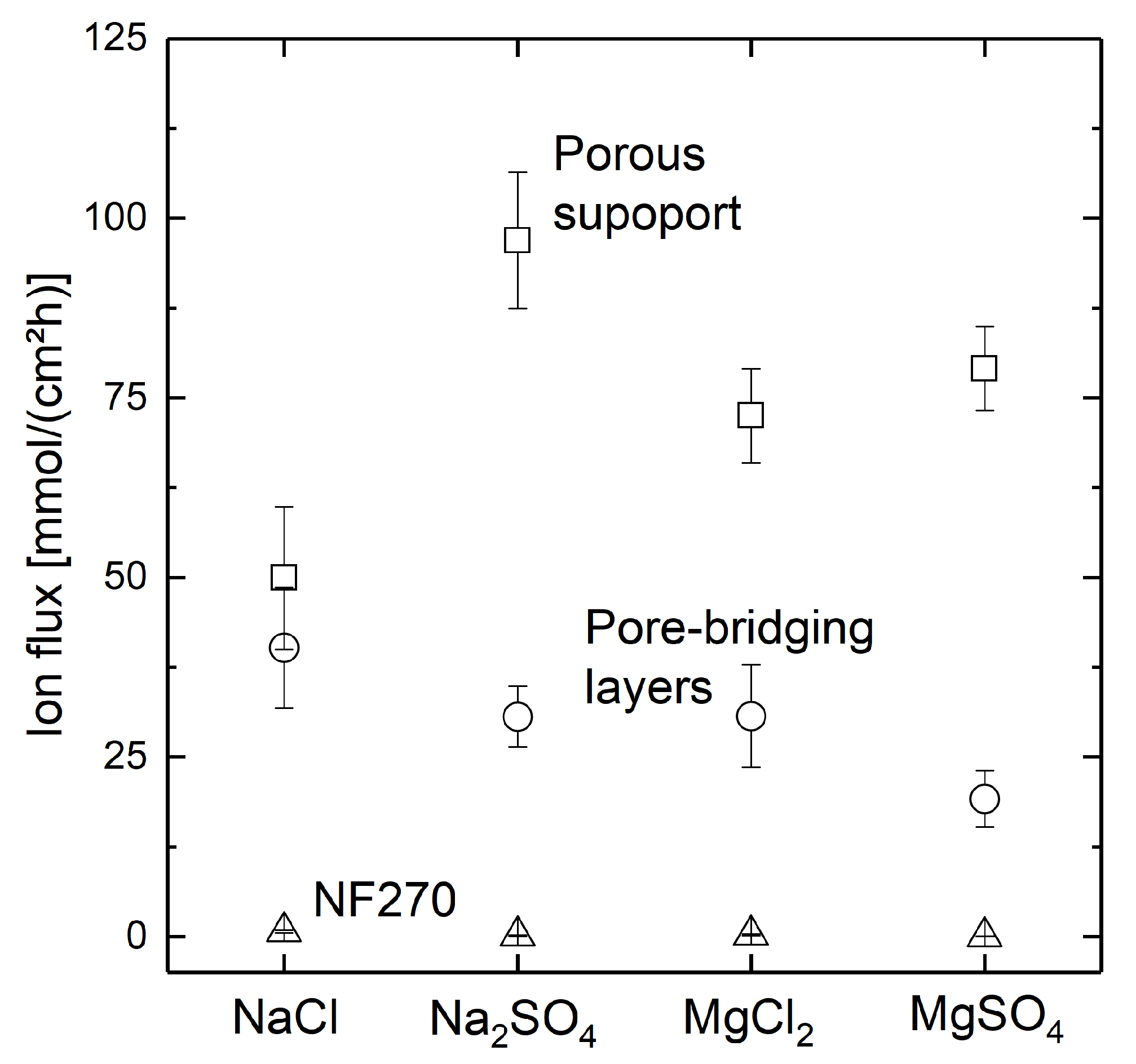
3.3. Separation Behavior
3.4. Miscible and Immiscible Wetting Agents
3.5. Heteroporous Support Structures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A | Effective membrane area |
| d | pore diameter |
| logarithmic mean concentration difference | |
| EDX | Energy Dispersive X-ray spectroscopy |
| FESEM | Field Emission Scanning Electron Microscopy |
| I | ionic flux |
| LbL | Layer by Layer |
| LMH | L/(m2 h) |
| MDPI | Multidisciplinary Digital Publishing Institute |
| n | number of ions transported |
| PAA | Polyacrylic Acid |
| PAH | Poly(allylamine hydrochloride) |
| pb | breakthrough pressure |
| PDADMAC | Poly(diallyldimethylammonium chloride) |
| PEG400 | Polyethylene Glycol 400 Da |
| PEM | Polyelectrolyte Multilayer |
| PEMM | Polyelectrolyte Multilayer Membrane |
| PES | Polyethersulphone |
| PSS | Poly(styrenesulfonate) |
| PVP | Polyvinylpyrrolidone |
| R | Retention |
| t | Time of Diffusion Dialysis |
| TMP | transmembrane pressure |
References
- Stocker, T. (Ed.) Climate Change 2013: The Physical Science Basis: Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 2014. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xu, Y.; Zhang, X.; Wu, H.; Shen, J.; Chen, R.; Xiong, Y.; Li, J.; Guo, S. Progress on the layer-by-layer assembly of multilayered polymer composites: Strategy, structural control and applications. Prog. Polym. Sci. 2019, 89, 76–107. [Google Scholar] [CrossRef]
- Bertrand, P.; Jonas, A.; Laschewsky, A.; Legras, R. Ultrathin polymer coatings by complexation of polyelectrolytes at interfaces: Suitable materials, structure and properties. Macromol. Rapid Commun. 2000, 21, 319–348. [Google Scholar] [CrossRef]
- Ng, L.Y.; Mohammad, A.W.; Ng, C.Y. A review on nanofiltration membrane fabrication and modification using polyelectrolytes: Effective ways to develop membrane selective barriers and rejection capability. Adv. Colloid Interface Sci. 2013, 197, 85–107. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, C.; Van der Bruggen, B. Polyelectrolytes self-assembly: Versatile membrane fabrication strategy. J. Mater. Chem. A 2020, 8, 20870–20896. [Google Scholar] [CrossRef]
- Cheng, W.; Campolongo, M.J.; Tan, S.J.; Luo, D. Freestanding ultrathin nano-membranes via self-assembly. Nano Today 2009, 4, 482–493. [Google Scholar] [CrossRef]
- Zhang, W.; Zhao, Q.; Yuan, J. Porous Polyelectrolytes: The Interplay of Charge and Pores for New Functionalities. Angew. Chem. (Int. Ed. Eng.) 2018, 57, 6754–6773. [Google Scholar] [CrossRef]
- Nabeel, F.; Rasheed, T.; Bilal, M.; Iqbal, H.M. Supramolecular membranes: A robust platform to develop separation strategies towards water-based applications. Sep. Purif. Technol. 2019, 215, 441–453. [Google Scholar] [CrossRef]
- Menne, D.; Kamp, J.; Erik Wong, J.; Wessling, M. Precise tuning of salt retention of backwashable polyelectrolyte multilayer hollow fiber nanofiltration membranes. J. Membr. Sci. 2016, 499, 396–405. [Google Scholar] [CrossRef]
- Emonds, S.; Roth, H.; Wessling, M. Chemistry in a spinneret—Formation of hollow fiber membranes with a cross-linked polyelectrolyte separation layer. J. Membr. Sci. 2020, 612, 118325. [Google Scholar] [CrossRef]
- Decher, G. Fuzzy Nanoassemblies: Toward Layered Polymeric Multicomposites. Science 1997, 277, 1232–1237. [Google Scholar] [CrossRef]
- Decher, G.; Schlenoff, J. (Eds.) Multilayer Thin Films; Wiley VCH: Weinheim, Germany, 2012. [Google Scholar] [CrossRef]
- Jiang, C.; Tsukruk, V.V. Freestanding Nanostructures via Layer-by-Layer Assembly. Adv. Mater. 2006, 18, 829–840. [Google Scholar] [CrossRef]
- Richardson, J.J.; Cui, J.; Björnmalm, M.; Braunger, J.A.; Ejima, H.; Caruso, F. Innovation in Layer-by-Layer Assembly. Chem. Rev. 2016, 116, 14828–14867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szilagyi, I.; Trefalt, G.; Tiraferri, A.; Maroni, P.; Borkovec, M. Polyelectrolyte adsorption, interparticle forces, and colloidal aggregation. Soft Matter 2014, 10, 2479–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, J.F.; Johnston, A.P.R.; Such, G.K.; Zelikin, A.N.; Caruso, F. Next generation, sequentially assembled ultrathin films: Beyond electrostatics. Chem. Soc. Rev. 2007, 36, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, Y.; Nakamura, R.; Ito, S.; Yamamoto, M. Molecular Weight Dependence of Alternate Adsorption through Charge-Transfer Interaction. Langmuir 2001, 17, 953–956. [Google Scholar] [CrossRef]
- Elizarova, I.S.; Luckham, P.F. Layer-by-layer adsorption: Factors affecting the choice of substrates and polymers. Adv. Colloid Interface Sci. 2018, 262, 1–20. [Google Scholar] [CrossRef]
- Malaisamy, R.; Bruening, M.L. High-flux nanofiltration membranes prepared by adsorption of multilayer polyelectrolyte membranes on polymeric supports. Langmuir 2005, 21, 10587–10592. [Google Scholar] [CrossRef]
- Kamp, J.; Emonds, S.; Wessling, M. Designing tubular composite membranes of polyelectrolyte multilayer on ceramic supports with nanofiltration and reverse osmosis transport properties. J. Membr. Sci. 2021, 620, 118851. [Google Scholar] [CrossRef]
- Kamp, J.; Emonds, S.; Seidenfaden, M.; Papenheim, P.; Kryschewski, M.; Rubner, J.; Wessling, M. Tuning the excess charge and inverting the salt rejection hierarchy of polyelectrolyte multilayer membranes. J. Membr. Sci. 2021, 277, 119636. [Google Scholar] [CrossRef]
- Mamedov, A.A.; Kotov, N.A. Free-Standing Layer-by-Layer Assembled Films of Magnetite Nanoparticles. Langmuir 2000, 16, 5530–5533. [Google Scholar] [CrossRef]
- Penfold, J.; Thomas, R.K.; Taylor, D. Polyelectrolyte/surfactant mixtures at the air–solution interface. Curr. Opin. Colloid Interface Sci. 2006, 11, 337–344. [Google Scholar] [CrossRef]
- Ariga, K.; Yamauchi, Y.; Rydzek, G.; Ji, Q.; Yonamine, Y.; Wu, K.C.W.; Hill, J.P. Layer-by-layer Nanoarchitectonics: Invention, Innovation, and Evolution. Chem. Lett. 2014, 43, 36–68. [Google Scholar] [CrossRef]
- Ariga, K. Nano-architectonics for coordination assemblies at interfacial media. In Nanoscale Coordination Chemistry; Advances in Inorganic Chemistry; Elsevier: Amsterdam, The Netherlands, 2020; Volume 76, pp. 239–268. [Google Scholar] [CrossRef]
- Guo, S.; Zhang, H.; Chen, X.; Feng, S.; Wan, Y.; Luo, J. Fabrication of Antiswelling Loose Nanofiltration Membranes via a “Selective-Etching-Induced Reinforcing” Strategy for Bioseparation. Acs Appl. Mater. Interfaces 2021, 13, 19312–19323. [Google Scholar] [CrossRef]
- Kamp, J.; Emonds, S.; Borowec, J.; Restrepo Toro, M.A.; Wessling, M. On the organic solvent free preparation of ultrafiltration and nanofiltration membranes using polyelectrolyte complexation in an all aqueous phase inversion process. J. Membr. Sci. 2021, 618, 118632. [Google Scholar] [CrossRef]
- Emonds, S.; Kamp, J.; Borowec, J.; Roth, H.; Wessling, M. Polyelectrolyte Complex Tubular Membranes via a Salt Dilution Induced Phase Inversion Process. Adv. Eng. Mater. 2021, 23, 2001401. [Google Scholar] [CrossRef]
- Fujie, T. Development of free-standing polymer nanosheets for advanced medical and health-care applications. Polym. J. 2016, 48, 773–780. [Google Scholar] [CrossRef]
- Buck, M.E.; Lynn, D.M. Free-Standing and Reactive Thin Films Fabricated by Covalent Layer-by-Layer Assembly and Subsequent Lift-Off of Azlactone-Containing Polymer Multilayers. Langmuir ACS J. Surfaces Colloids 2010, 26, 16134–16140. [Google Scholar] [CrossRef] [Green Version]
- Magerl, A.; Goedel, W.A. Porous polymer membranes via selectively wetted surfaces. Langmuir ACS J. Surfaces Colloids 2012, 28, 5622–5632. [Google Scholar] [CrossRef]
- Yan, F.; Ding, A.; Gironès, M.; Lammertink, R.G.H.; Wessling, M.; Börger, L.; Vilsmeier, K.; Goedel, W.A. Hierarchically structured assembly of polymer microsieves, made by a combination of phase separation micromolding and float-casting. Adv. Mater. 2012, 24, 1551–1557. [Google Scholar] [CrossRef]
- Xie, J.; Han, X.; Ji, H.; Wang, J.; Zhao, J.; Lu, C. Self-Supported Crack-Free Conducting Polymer Films with Stabilized Wrinkling Patterns and Their Applications. Sci. Rep. 2016, 6, 36686. [Google Scholar] [CrossRef]
- Zhang, J.; Yuan, B.; Wang, Z.; Chen, T. Fabrication of free-standing multilayer films by using pH-responsive microgels as sacrificial layers. Colloid Polym. Sci. 2014, 292, 1235–1240. [Google Scholar] [CrossRef]
- Nolte, M.; Schoeler, B.; Peyratout, C.S.; Kurth, D.G.; Fery, A. Filled Microcavity Arrays Produced by Polyelectrolyte Multilayer Membrane Transfer. Adv. Mater. 2005, 17, 1665–1669. [Google Scholar] [CrossRef]
- Buck, M.E.; Lynn, D.M. Reactive Layer-by-Layer Assembly of Suspended Thin Films and Semipermeable Membranes at Interfaces Created Between Aqueous and Organic Phases. Adv. Mater. 2010, 22, 994–998. [Google Scholar] [CrossRef] [Green Version]
- Shchukin, D.G.; Köhler, K.; Möhwald, H.; Sukhorukov, G.B. Gas-Filled Polyelectrolyte Capsules. Angew. Chem. (Int. Ed. Eng.) 2005, 44, 3310–3314. [Google Scholar] [CrossRef]
- Lehmann, P.; Kurth, D.G.; Brezesinski, G.; Symietz, C. Structural Analysis of a Metallosupramolecular Polyelectrolyte-Amphiphile Complex at the Air/Water Interface. Chemistry 2001, 7, 1646–1651. [Google Scholar] [CrossRef]
- Hu, D.; Yang, Z.; Chou, K.C. Interactions of Polyelectrolytes with Water and Ions at Air/Water Interfaces Studied by Phase-Sensitive Sum Frequency Generation Vibrational Spectroscopy. J. Phys. Chem. C 2013, 117, 15698–15703. [Google Scholar] [CrossRef]
- He, J.; Kanjanaboos, P.; Frazer, N.L.; Weis, A.; Lin, X.M.; Jaeger, H.M. Fabrication and Mechanical Properties of Large-Scale Freestanding Nanoparticle Membranes. Small 2010, 6, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski, M.; Borme, J.; Carbó-Argibay, E.; Sharma, D.; Nicoara, N.; Sadewasser, S.; Petrovykh, D.Y.; Rodríguez-Abreu, C.; Kolen’ko, Y.V. Template-directed self-organization of colloidal PbTe nanocrystals into pillars, conformal coatings, and self-supported membranes. Nanoscale Adv. 2019, 1, 3049–3055. [Google Scholar] [CrossRef] [Green Version]
- Mallwitz, F.; Laschewsky, A. Direct Access to Stable, Freestanding Polymer Membranes by Layer-by-Layer Assembly of Polyelectrolytes. Adv. Mater. 2005, 17, 1296–1299. [Google Scholar] [CrossRef]
- Armstrong, J.A.; Bernal, E.E.L.; Yaroshchuk, A.; Bruening, M.L. Separation of ions using polyelectrolyte-modified nanoporous track-etched membranes. Langmuir 2013, 29, 10287–10296. [Google Scholar] [CrossRef] [PubMed]
- de Grooth, J.; Oborný, R.; Potreck, J.; Nijmeijer, K.; de Vos, W.M. The role of ionic strength and odd–even effects on the properties of polyelectrolyte multilayer nanofiltration membranes. J. Membr. Sci. 2015, 475, 311–319. [Google Scholar] [CrossRef]
- te Brinke, E.; Achterhuis, I.; Reurink, D.M.; de Grooth, J.; de Vos, W.M. Multiple Approaches to the Buildup of Asymmetric Polyelectrolyte Multilayer Membranes for Efficient Water Purification. ACS Appl. Polym. Mater. 2020, 2, 715–724. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Xue, S.; Wang, P.; Wu, C.; Wu, Y. Diffusion dialysis for NaCl and NaAc recovery using polyelectrolyte complexes/PVA membranes. Sep. Purif. Technol. 2017, 172, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Porcel, C.; Lavalle, P.; Ball, V.; Decher, G.; Senger, B.; Voegel, J.C.; Schaaf, P. From exponential to linear growth in polyelectrolyte multilayers. Langmuir ACS J. Surfaces Colloids 2006, 22, 4376–4383. [Google Scholar] [CrossRef]
- Zhao, Q.; Qian, J.; An, Q.; Du, B. Speedy fabrication of free-standing layer-by-layer multilayer films by using polyelectrolyte complex particles as building blocks. J. Mater. Chem. 2009, 19, 8448. [Google Scholar] [CrossRef]
- Madaboosi, N.; Uhlig, K.; Jäger, M.S.; Möhwald, H.; Duschl, C.; Volodkin, D.V. Microfluidics as A Tool to Understand the Build-Up Mechanism of Exponential-Like Growing Films. Macromol. Rapid Commun. 2012, 33, 1775–1779. [Google Scholar] [CrossRef]
- Guzmán, E.; Rubio, R.G.; Ortega, F. A closer physico-chemical look to the Layer-by-Layer electrostatic self-assembly of polyelectrolyte multilayers. Adv. Colloid Interface Sci. 2020, 282, 102197. [Google Scholar] [CrossRef]
- Yu, L.; Yuan, W.; Liu, X.; Xu, X.; Ruan, S. Asymmetry of the free-standing polyelectrolyte multilayers. Appl. Surf. Sci. 2017, 422, 46–55. [Google Scholar] [CrossRef]
- Krasemann, L.; Tieke, B. Selective Ion Transport across Self-Assembled Alternating Multilayers of Cationic and Anionic Polyelectrolytes. Langmuir 2000, 16, 287–290. [Google Scholar] [CrossRef]
- Jin, W.; Toutianoush, A.; Tieke, B. Size- and charge-selective transport of aromatic compounds across polyelectrolyte multilayer membranes. Appl. Surf. Sci. 2005, 246, 444–450. [Google Scholar] [CrossRef]
- Hong, S.U.; Ouyang, L.; Bruening, M.L. Recovery of phosphate using multilayer polyelectrolyte nanofiltration membranes. J. Membr. Sci. 2009, 327, 2–5. [Google Scholar] [CrossRef]
- Yaroshchuk, A.E. Non-steric mechanisms of nanofiltration: Superposition of Donnan and dielectric exclusion. Sep. Purif. Technol. 2001, 22, 143–158. [Google Scholar] [CrossRef]
- Evdochenko, E.; Kamp, J.; Femmer, R.; Xu, Y.; Nikonenko, V.; Wessling, M. Unraveling the effect of charge distribution in a polyelectrolyte multilayer nanofiltration membrane on its ion transport properties. J. Membr. Sci. 2020, 611, 118045. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalde, A.; Kamp, J.; Evdochenko, E.; Linkhorst, J.; Wessling, M. Wetting-Induced Polyelectrolyte Pore Bridging. Membranes 2021, 11, 671. https://doi.org/10.3390/membranes11090671
Kalde A, Kamp J, Evdochenko E, Linkhorst J, Wessling M. Wetting-Induced Polyelectrolyte Pore Bridging. Membranes. 2021; 11(9):671. https://doi.org/10.3390/membranes11090671
Chicago/Turabian StyleKalde, Anna, Johannes Kamp, Elizaveta Evdochenko, John Linkhorst, and Matthias Wessling. 2021. "Wetting-Induced Polyelectrolyte Pore Bridging" Membranes 11, no. 9: 671. https://doi.org/10.3390/membranes11090671
APA StyleKalde, A., Kamp, J., Evdochenko, E., Linkhorst, J., & Wessling, M. (2021). Wetting-Induced Polyelectrolyte Pore Bridging. Membranes, 11(9), 671. https://doi.org/10.3390/membranes11090671