
Toxicological Profile of Umbilical Cord Blood-Derived Small Extracellular Vesicles

 ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. UCB-MNC-sEV Isolation and Purification
2.2. UCB-MNC-sEV Characterization
2.3. In Vitro Toxicity
2.4. Cell Lines and Cell Culture Conditions
2.5. Cell Viability-XTT Assay
2.6. Animal Experiments
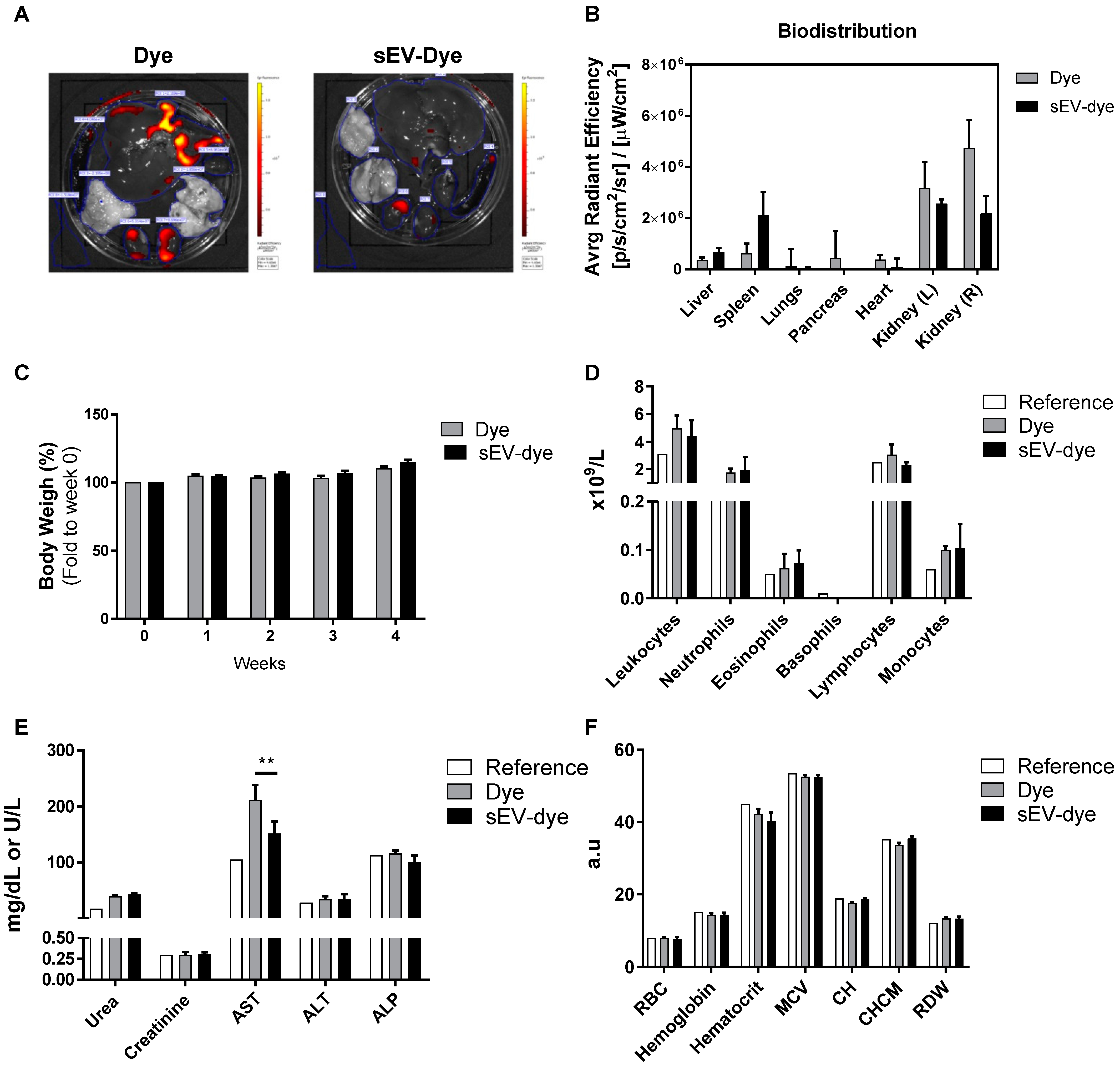
2.6.1. In Vivo Biodistribution and Toxicity: 4 Weeks
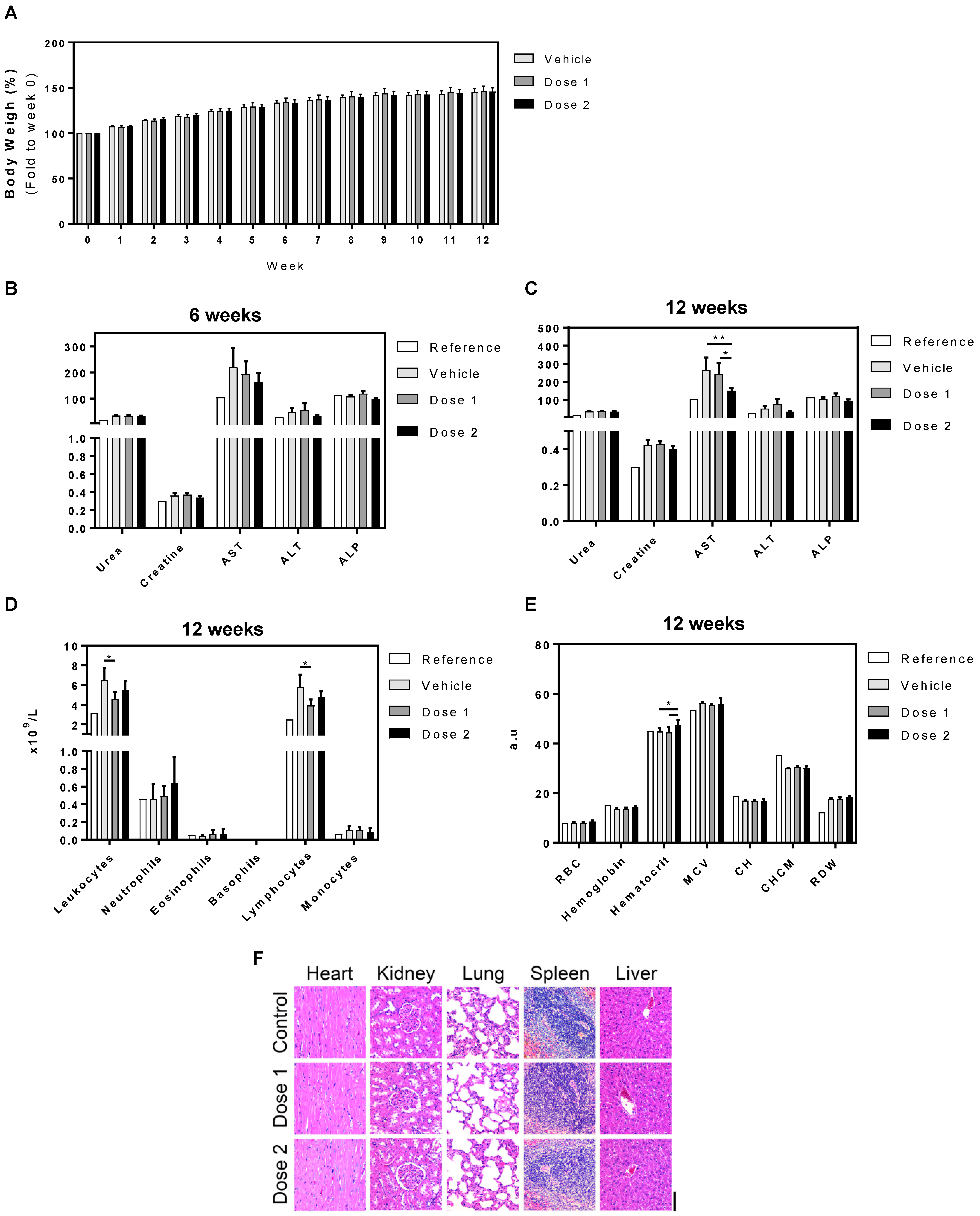
2.6.2. In Vivo Toxicity: 6 and 12 Weeks
2.6.3. Histological Examination
2.7. Statistical Analysis
3. Results
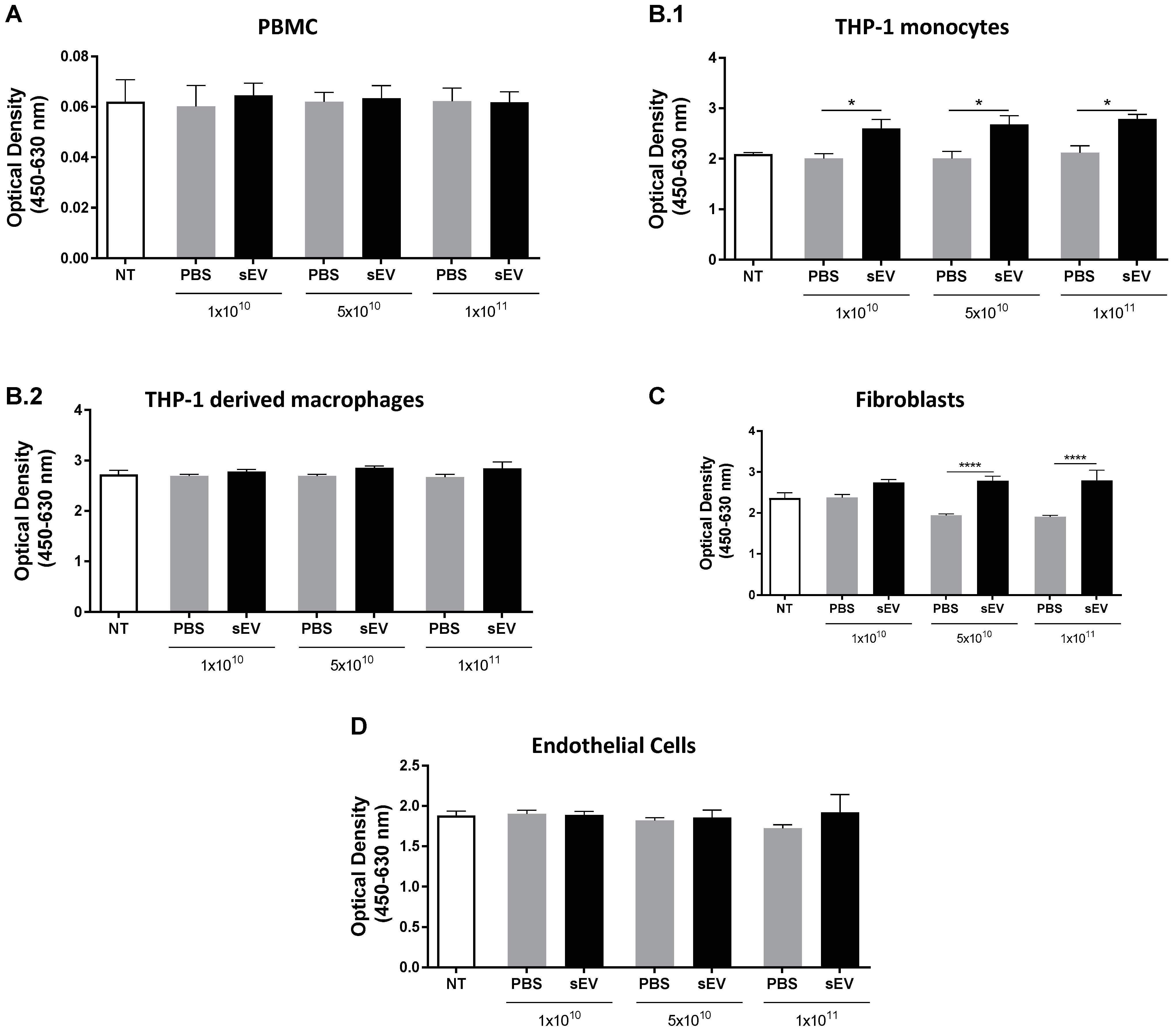
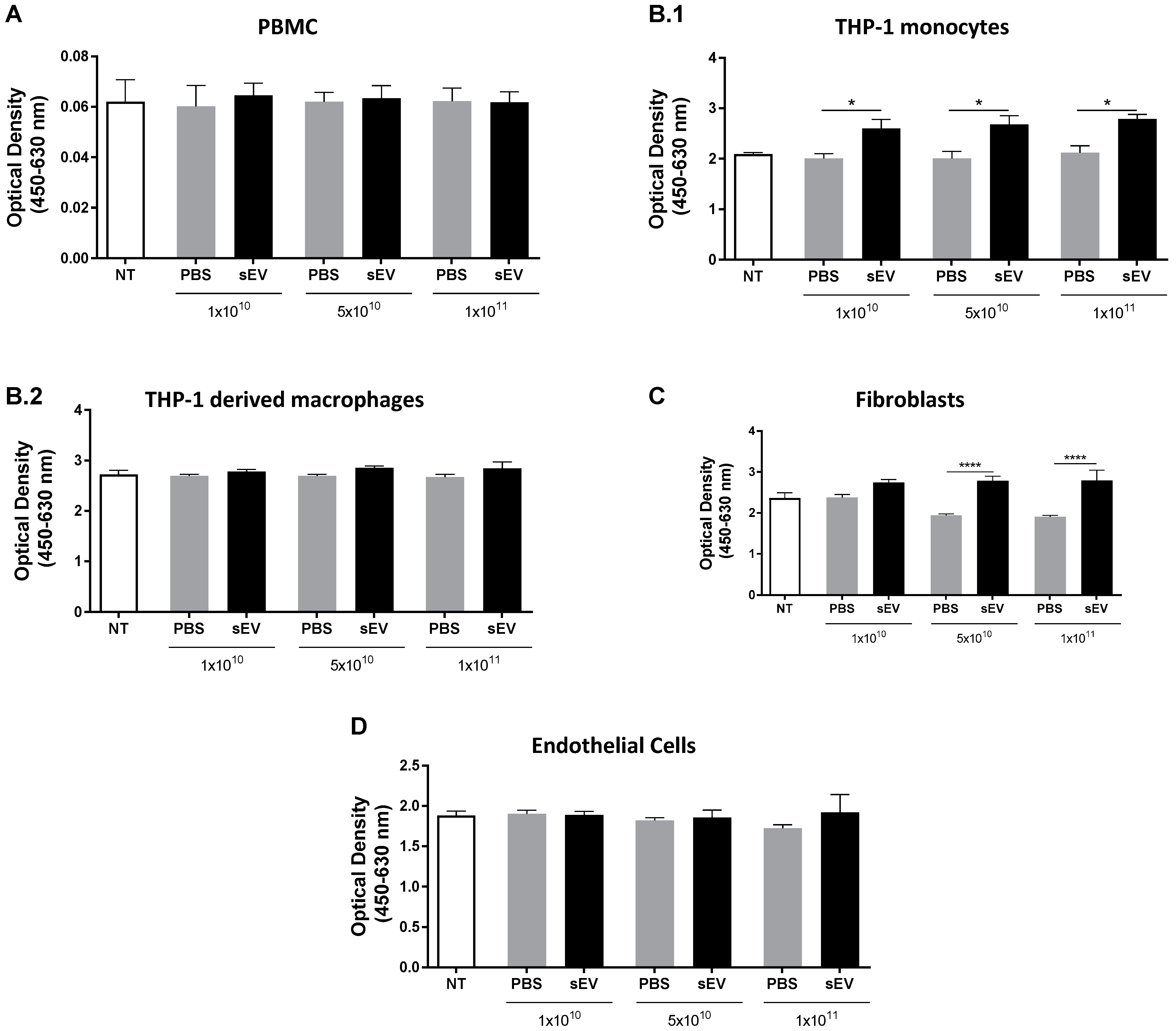
3.1. In Vitro Toxicity
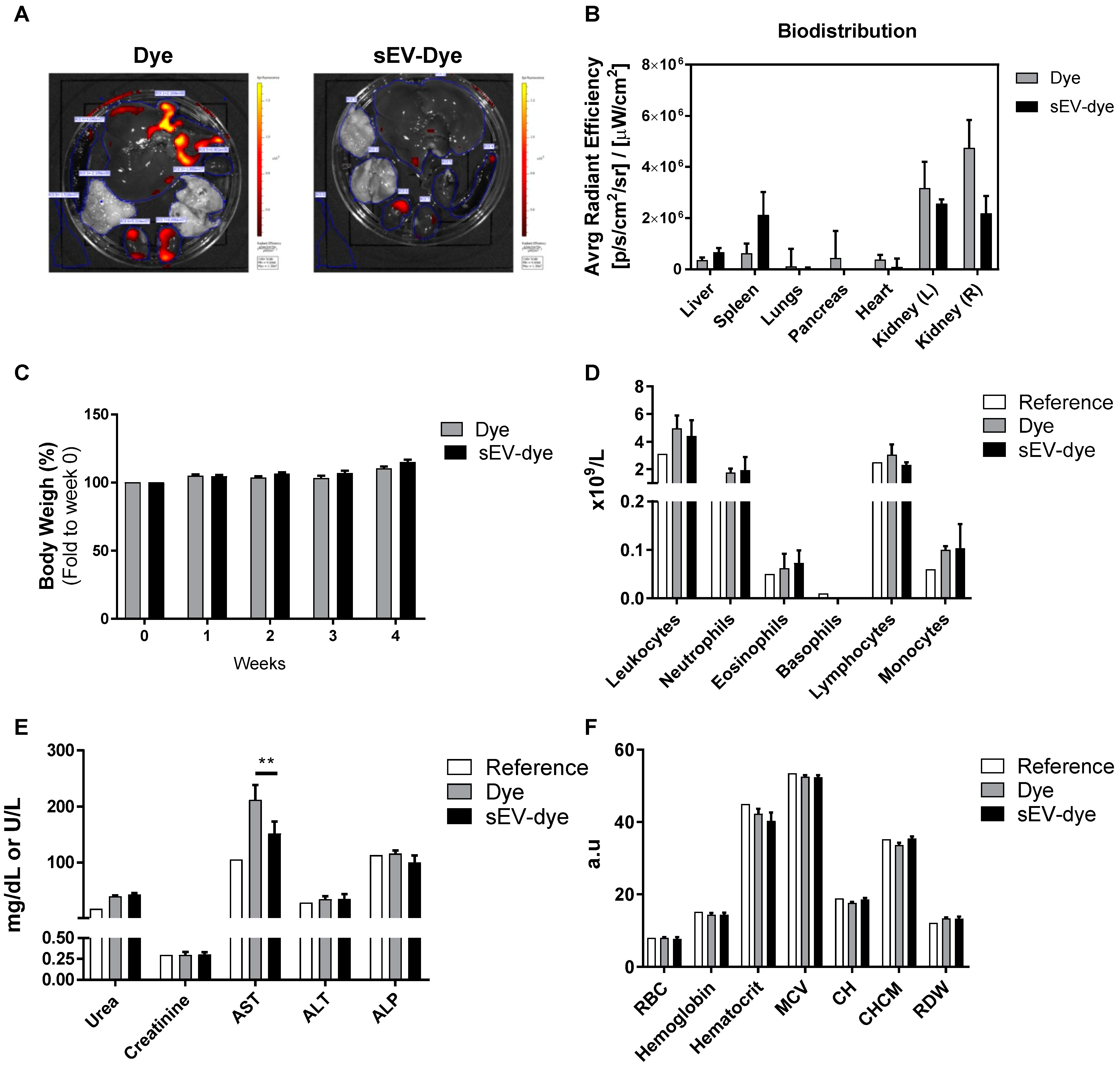
3.2. In Vivo Biodistribution and Toxicity: 4 Weeks
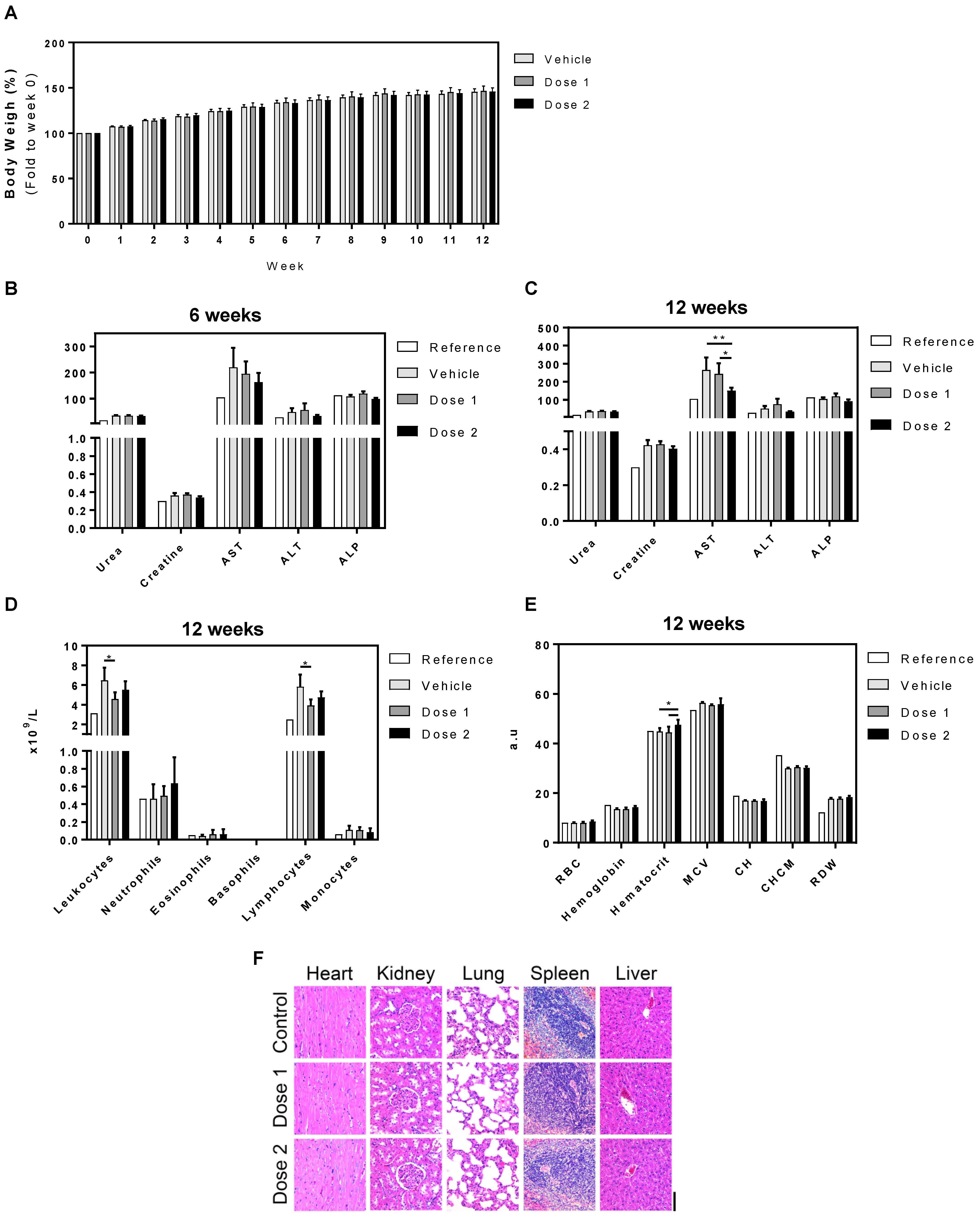
3.3. In Vivo Toxicity: 6 and 12 Weeks
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Skog, J.; Würdinger, T.; Van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Kritchevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- György, B.; Szabó, T.G.; Pásztói, M.; Pál, Z.; Misják, P.; Aradi, B.; Lásyló, V.; Pállinger, E.; Pap, E.; Kittel, A.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Ciferri, M.C.; Quarto, R.; Tasso, R. Extracellular Vesicles as Biomarkers and Therapeutic Tools: From Pre-Clinical to Clinical Applications. Biology 2021, 10, 359. [Google Scholar] [CrossRef]
- Herrmann, I.K.; Wood, M.J.A.; Fuhrmann, G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021, 16, 748–759. [Google Scholar] [CrossRef]
- Banks, W.A.; Sharma, P.; Bullock, K.M.; Hansen, K.M.; Ludwig, N.; Whiteside, T.L. Transport of Extracellular Vesicles across the Blood-Brain Barrier: Brain Pharmacokinetics and Effects of Inflammation. Int. J. Mol. Sci. 2020, 21, 4407. [Google Scholar] [CrossRef]
- Raimondo, S.; Giavaresi, G.; Lorico, A.; Alessandro, R. Extracellular Vesicles as Biological Shuttles for Targeted Therapies. Int. J. Mol. Sci. 2019, 20, 1848. [Google Scholar] [CrossRef] [Green Version]
- Lou, G.; Chen, Z.; Zheng, M.; Liu, Y. Mesenchymal stem cell-derived exosomes as a new therapeutic strategy for liver diseases. Exp. Mol. Med. 2017, 49, e346. [Google Scholar] [CrossRef]
- Lai, R.C.; Chen, T.S.; Lim, S.K. Mesenchymal stem cell exosome: A novel stem cell-based therapy for cardiovascular disease. Regen. Med. 2011, 6, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Xin, H.; Li, Y.; Cui, Y.; Yang, J.J.; Zhang, Z.G.; Chopp, M. Systemic administration of exosomes released from mesenchymal stromal cells promote functional recovery and neurovascular plasticity after stroke in rats. J. Cereb. Blood Flow Metab. 2013, 33, 1711–1715. [Google Scholar] [CrossRef] [Green Version]
- Xin, H.; Katakowski, M.; Wang, F.; Qian, J.-Y.; Liu, X.S.; Ali, M.M.; Buller, B.; Zhang, Z.G.; Chopp, M. MicroRNA cluster miR-17-92 Cluster in Exosomes Enhance Neuroplasticity and Functional Recovery after Stroke in Rats. Stroke 2017, 48, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Mathiyalagan, P.; Liang, Y.; Kim, D.; Misener, S.; Thorne, T.; Kamide, C.E.; Klyachko, E.; Losordo, D.; Hajjar, R.J.; Sahoo, S. Angiogenic Mechanisms of Human CD34+ Stem Cell Exosomes in the Repair of Ischemic Hindlimb. Circ. Res. 2017, 120, 1466–1476. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, S.; Klychko, E.; Thorne, T.; Misener, S.; Schultz, K.M.; Millay, M.; Ito, A.; Liu, T.; Kamide, C.; Agrawal, H.; et al. Exosomes from human CD34+ stem cells mediate their proangiogenic paracrine activity. Circ. Res. 2011, 109, 724–728. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Rao, S.-S.; Wang, Z.-X.; Cao, J.; Tan, Y.-J.; Luo, J.; Li, H.-M.; Zhang, W.-S.; Chen, C.-Y.; Xie, H. Exosomes from human umbilical cord blood accelerate cutaneous wound healing through miR-21-3p-mediated promotion of angiogenesis and fibroblast function. Theranostics 2018, 8, 169–184. [Google Scholar] [CrossRef]
- Henriques-Antunes, H.; Cardoso, R.; Zonari, A.; Correia, J.S.; Leal, E.; Jiménez-Balsa, A.; Lino, M.M.; Barradas, A.; Kostic, I.; Gomes, C.; et al. The Kinetics of Small Extracellular Vesicle Delivery Impacts Skin Tissue Regeneration. ACS Nano 2019, 13, 8694–8707. [Google Scholar] [CrossRef]
- Lener, T.; Gimona, M.; Aigner, L.; Börger, V.; Buzas, E.; Camussi, G.; Chaput, N.; Chatterjee, D.; Court, F.A.; Del Portillo, H.A.; et al. Applying extracellular vesicles based therapeutics in clinical trials—an ISEV position paper. J. Extracell. Vesicles 2015, 4, 1–31. [Google Scholar] [CrossRef]
- Furlani, D.; Ugurlucan, M.; Ong, L.; Bieback, K.; Pittermann, E.; Westien, I.; Wang, W.; Yerebakan, C.; Li, W.; Gaebel, R.; et al. Is the intravascular administration of mesenchymal stem cells safe? Microvasc. Res. 2009, 77, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Pak, H.-N.; Qayyum, M.; Kim, D.T.; Hamabe, A.; Miyauchi, Y.; Lill, M.C.; Frantzen, M.; Takizawa, K.; Chen, L.S.; Fishbein, M.C.; et al. Mesenchymal stem cell injection induces cardiac nerve sprouting and increased tenascin expression in a swine model of myocardial infarction. J. Cardiovasc. Electrophysiol. 2003, 14, 841–848. [Google Scholar] [CrossRef]
- Rani, S.; Ryan, A.E.; Griffin, M.D.; Ritter, T. Mesenchymal stem cell-derived extracellular vesicles: Toward cell-free therapeutic applications. Mol. Ther. 2015, 23, 812–823. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Badawi, M.; Pomeroy, S.; Sutaria, D.; Xie, Z.; Baek, A.; Jiang, J.; Elgamal, O.A.; Mo, X.; La Perle, K.; et al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J. Extracell. Vesicles 2017, 6, 1324730. [Google Scholar] [CrossRef]
- Saleh, A.F.; Lázaro-Ibáñez, E.; Forsgard, M.A.-M.; Shatnyeva, O.; Osteikoetxea, X.; Karlsson, F.; Heath, N.; Ingelsten, M.; Rose, J.; Harris, J.; et al. Extracellular vesicles induce minimal hepatotoxicity and immunogenicity. Nanoscale 2019, 11, 6990–7001. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, R.M.S.; Rodrigues, S.C.; Gomes, C.F.; Duarte, F.V.; Romao, M.; Leal, E.C.; Freire, P.C.; Neves, R.; Simões-Correia, J. Development of an optimized and scalable method for isolation of umbilical cord blood-derived small extracellular vesicles for future clinical use. Stem Cells Transl. Med. 2021, 10, 910–921. [Google Scholar] [CrossRef]
- Böing, A.N.; van der Pol, E.; Grootemaat, A.E.; Coumans, F.A.W.; Sturk, A.; Nieuwland, R. Single-step isolation of extracellular vesicles by size-exclusion chromatography. J. Extracell. Vesicles 2014, 3, 23430. [Google Scholar] [CrossRef]
- Gámez-Valero, A.; Monguió-Tortajada, M.; Carreras-Planella, L.; Franquesa, M.; Beyer, K.; Borràs, F.E. Size-Exclusion Chromatography-based isolation minimally alters Extracellular Vesicles’ characteristics compared to precipitating agents. Sci. Rep. 2016, 6, 33641. [Google Scholar] [CrossRef] [Green Version]
- Ha, D.; Yang, N.; Nadithe, V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: Current perspectives and future challenges. Acta Pharm. Sin. B 2016, 6, 287–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.; Gu, H.; Wu, M. Mesenchymal Stem Cells, Exosomes and Cutaneous Wound Healing. J. Stem Cells Res. Dev. Ther. 2020, 6, 1–5. [Google Scholar] [CrossRef]
- Jiang, T.; Wang, Z.; Sun, J. Human bone marrow mesenchymal stem cell-derived exosomes stimulate cutaneous wound healing mediates through TGF-β/Smad signaling pathway. Stem Cell Res. Ther. 2020, 11, 198. [Google Scholar] [CrossRef]
- Casado-Díaz, A.; Quesada-Gómez, J.M.; Dorado, G. Extracellular Vesicles Derived From Mesenchymal Stem Cells (MSC) in Regenerative Medicine: Applications in Skin Wound Healing. Front. Bioeng. Biotechnol. 2020, 8, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiklander, O.P.B.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mäger, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles. 2015, 4, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, T.; Kullberg, M.; Malik, N.; Smith-Jones, P.; Graner, M.W.; Anchordoquy, T.J. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J. Control. Release 2015, 199, 145–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, S.-I.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically injected exosomes targeted to EGFR deliver antitumor microrna to breast cancer cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, Y.W.; Lee, J.H.; Kim, S.-Y.; Pack, C.-G.; Ha, D.H.; Park, S.R.; Youn, J.; Cho, B.S. Advances in Analysis of Biodistribution of Exosomes by Molecular Imaging. Int. J. Mol. Sci. 2020, 21, 665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Kuang, H.; Zhang, W.; Aguilar, Z.P.; Wei, H.; Xu, H. Comparisons of the biodistribution and toxicological examinations after repeated intravenous administration of silver and gold nanoparticles in mice. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Driedonks, T.; Carlson, B.; Queen, S.; Gololobova, O.; Han, Z.; Liu, G.; Nyberg, L.; Lima, G.; Schonvisky, K.; Castell, N.; et al. CC7.5. Pharmacokinetics and biodistribution of EV administered intravenously versus intranasally in mice and macaque models. ISEV2021 Abstract Book. J. Extracell. Vesicles 2021, 10 (Suppl. 1), 28. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, S.C.; Cardoso, R.M.S.; Gomes, C.F.; Duarte, F.V.; Freire, P.C.; Neves, R.; Simoes-Correia, J. Toxicological Profile of Umbilical Cord Blood-Derived Small Extracellular Vesicles. Membranes 2021, 11, 647. https://doi.org/10.3390/membranes11090647
Rodrigues SC, Cardoso RMS, Gomes CF, Duarte FV, Freire PC, Neves R, Simoes-Correia J. Toxicological Profile of Umbilical Cord Blood-Derived Small Extracellular Vesicles. Membranes. 2021; 11(9):647. https://doi.org/10.3390/membranes11090647
Chicago/Turabian StyleRodrigues, Silvia C., Renato M. S. Cardoso, Claudia F. Gomes, Filipe V. Duarte, Patricia C. Freire, Ricardo Neves, and Joana Simoes-Correia. 2021. "Toxicological Profile of Umbilical Cord Blood-Derived Small Extracellular Vesicles" Membranes 11, no. 9: 647. https://doi.org/10.3390/membranes11090647
APA StyleRodrigues, S. C., Cardoso, R. M. S., Gomes, C. F., Duarte, F. V., Freire, P. C., Neves, R., & Simoes-Correia, J. (2021). Toxicological Profile of Umbilical Cord Blood-Derived Small Extracellular Vesicles. Membranes, 11(9), 647. https://doi.org/10.3390/membranes11090647