
Membrane Domain Localization and Interaction of the Prion-Family Proteins, Prion and Shadoo with Calnexin

 , ,
, ,  , and
, and
Abstract
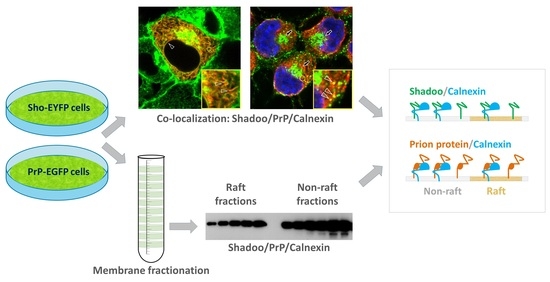
{kind=link}
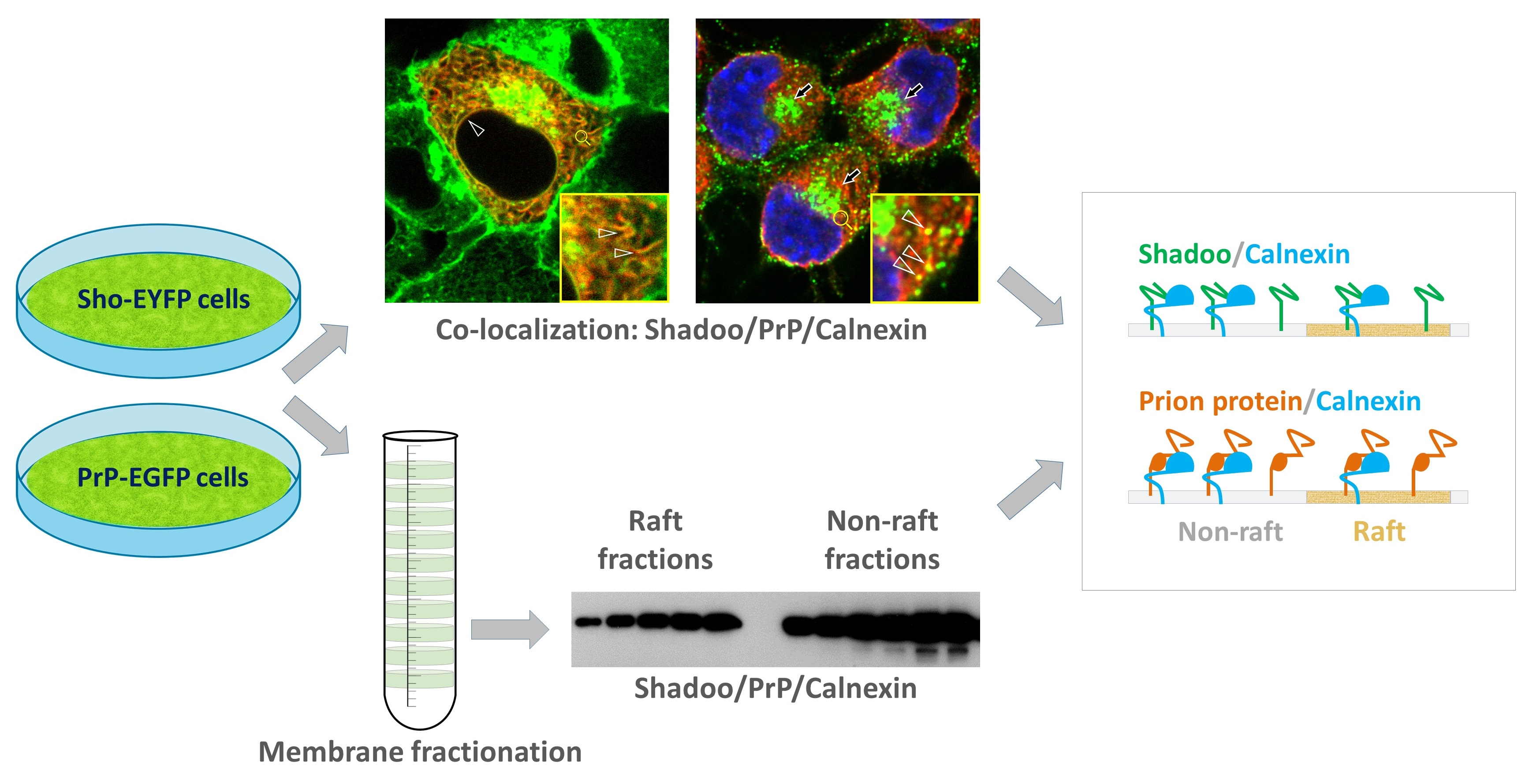
{kind=link}
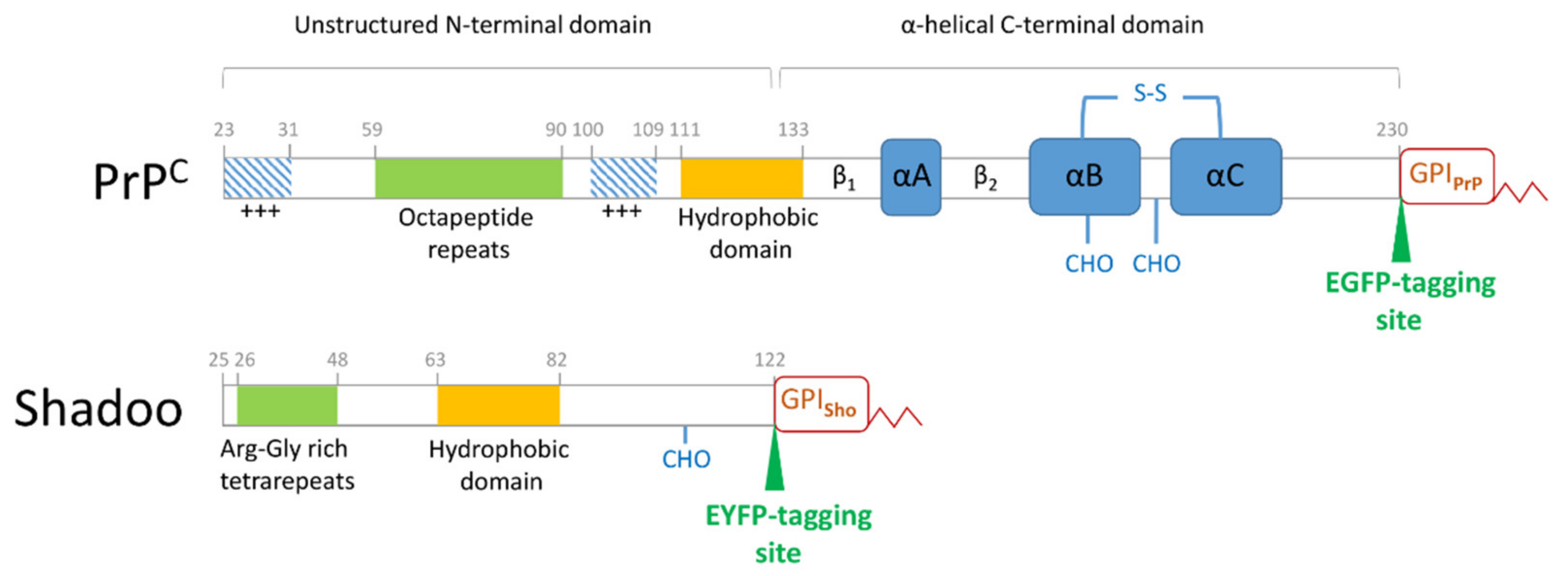
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Antibodies
2.3. Plasmid Constructs
2.4. Cell Culturing
2.5. Generation of Stable Transgenic Cells
2.5.1. N2a PrP(+EGFP) and N2a(EGFP) Cells
2.5.2. N2a PrP-EGFP, Sho-EYFP, Sho-FLAG-EYFP and Their Respective Control Cells (EGFP, EYFP and EYFP-FLAG) Cells
2.6. Transient Transfection of Cells
2.7. Golgi Complex Labelling and Confocal Microscopy
2.8. Extraction of Total Cell Lysates
2.9. Detergent-Free Separation of Lipid Rafts
2.10. PNGase F Treatment
2.11. Western Blotting
2.12. Cholesterol Determination
2.13. Immunocytochemistry
2.14. Live-Cell Analysis
2.15. In Vitro Pull-Down Assay
2.16. Co-Immunoprecipitation
3. Results
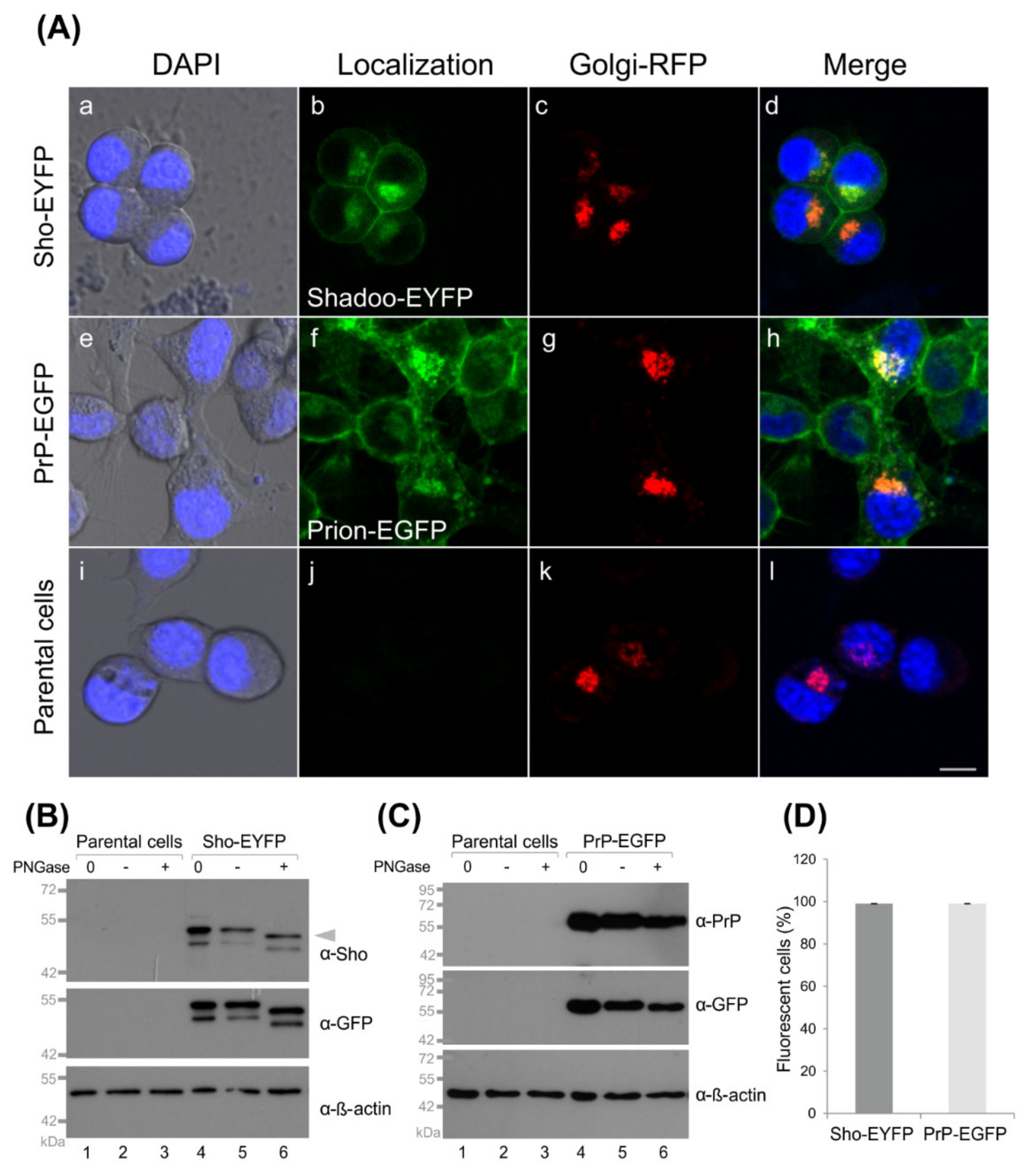
3.1. Fluorescent Protein-Tagged Shadoo and Prion Proteins Are Expressed and Localize as Expected in the Transgenic N2a Cells Developed
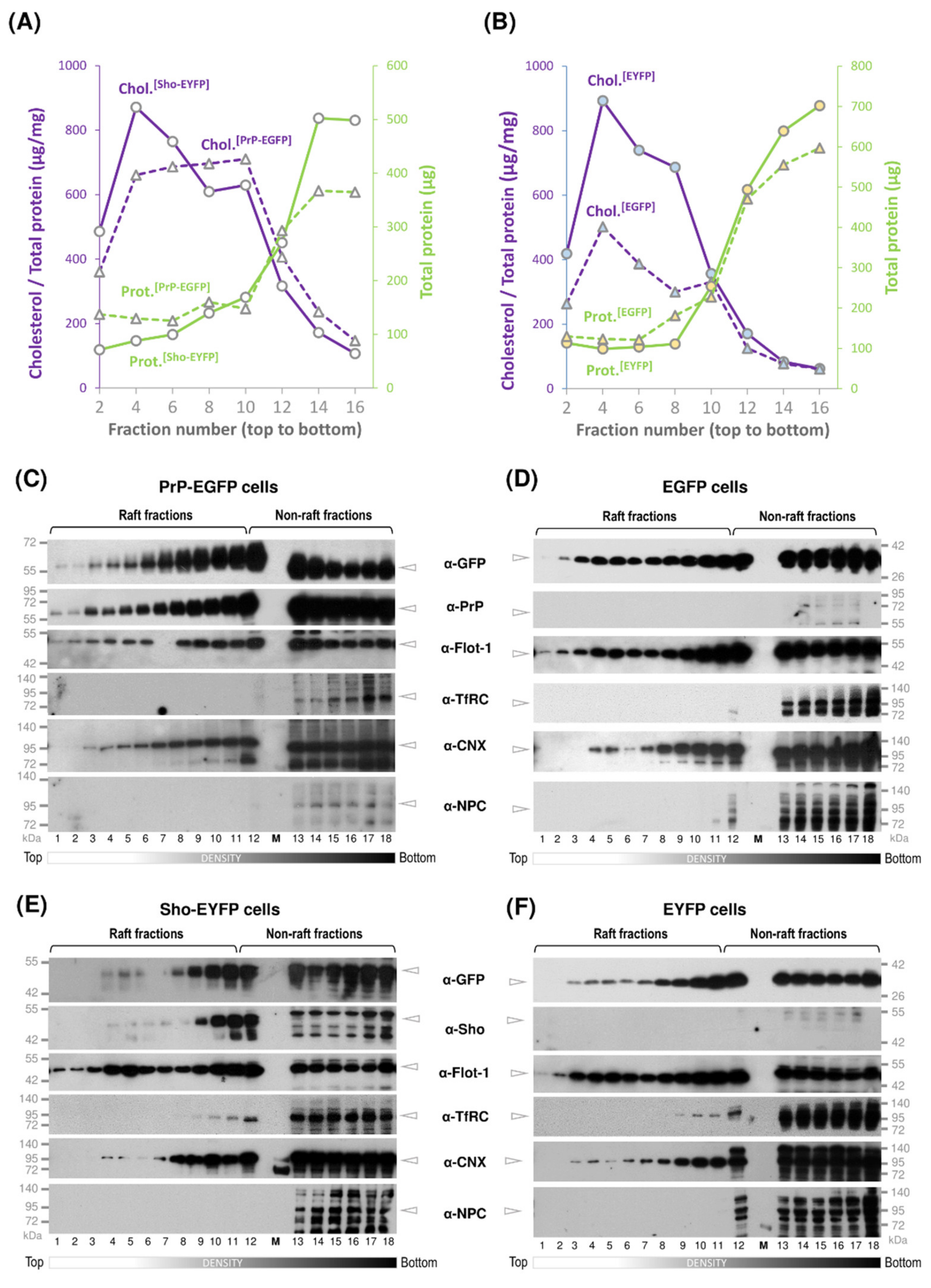
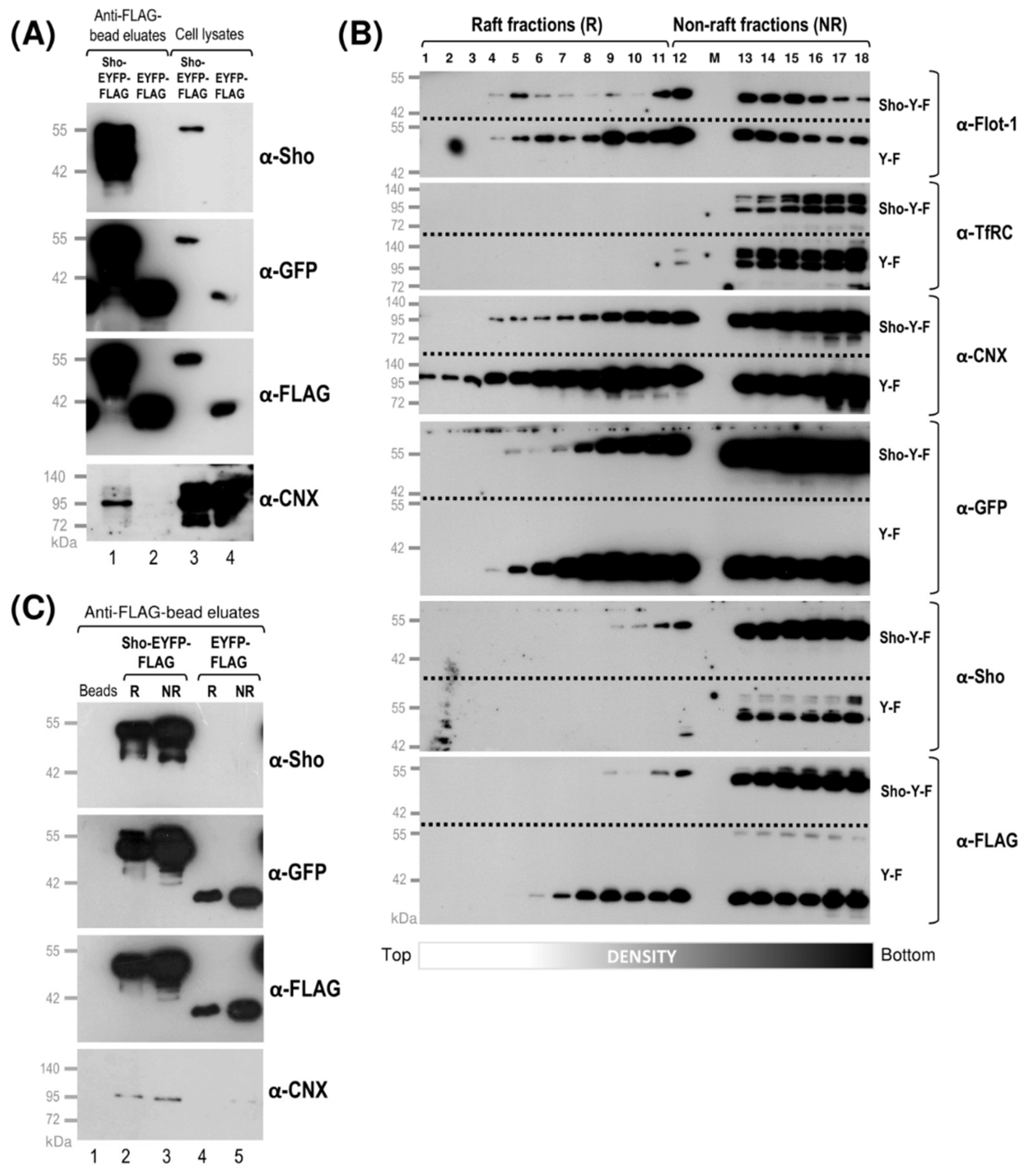
3.2. While Sho and PrP Are Membrane-Raft Localized, They Are Present in Non-Raft Membrane Fractions as Well
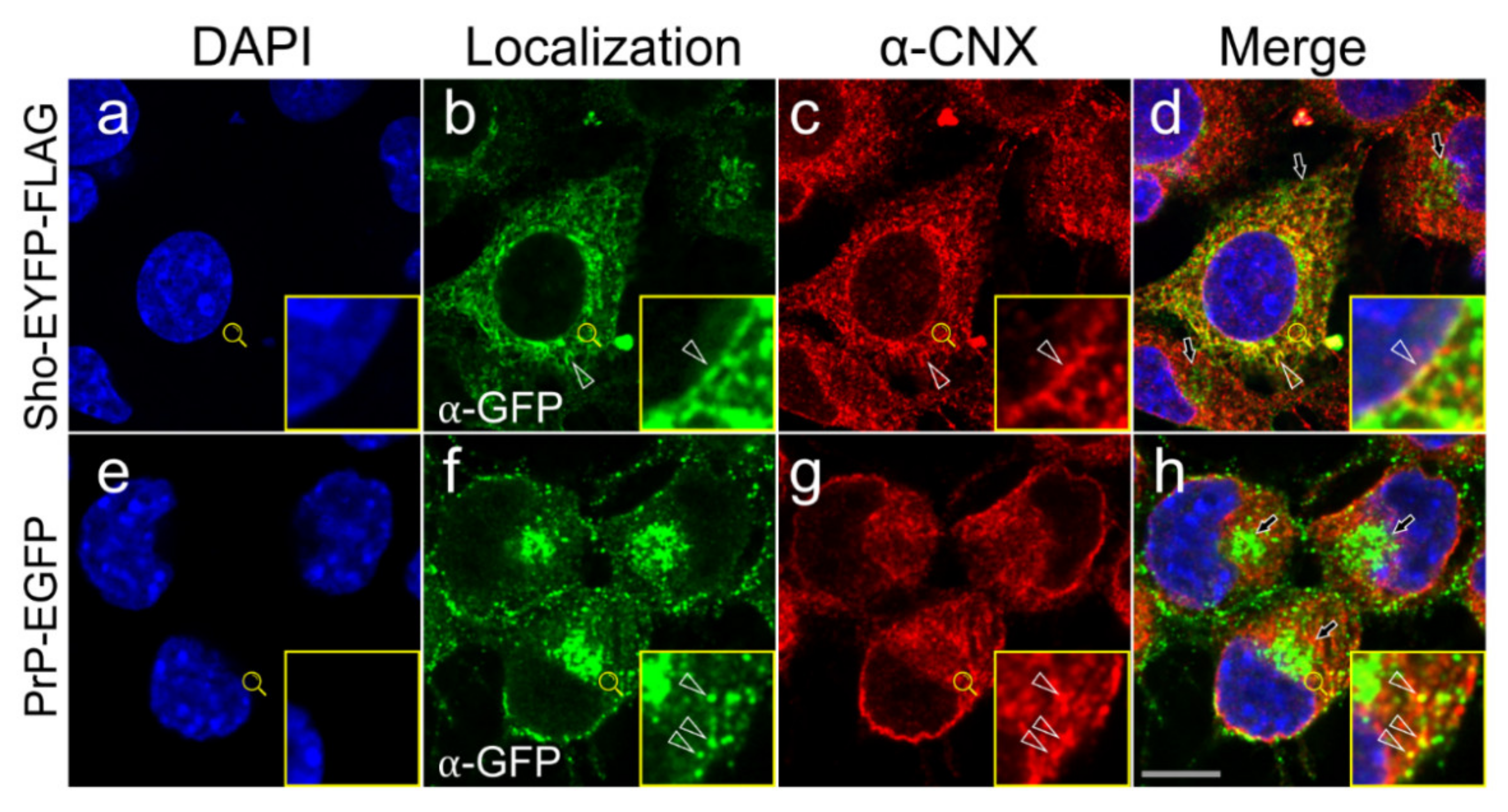
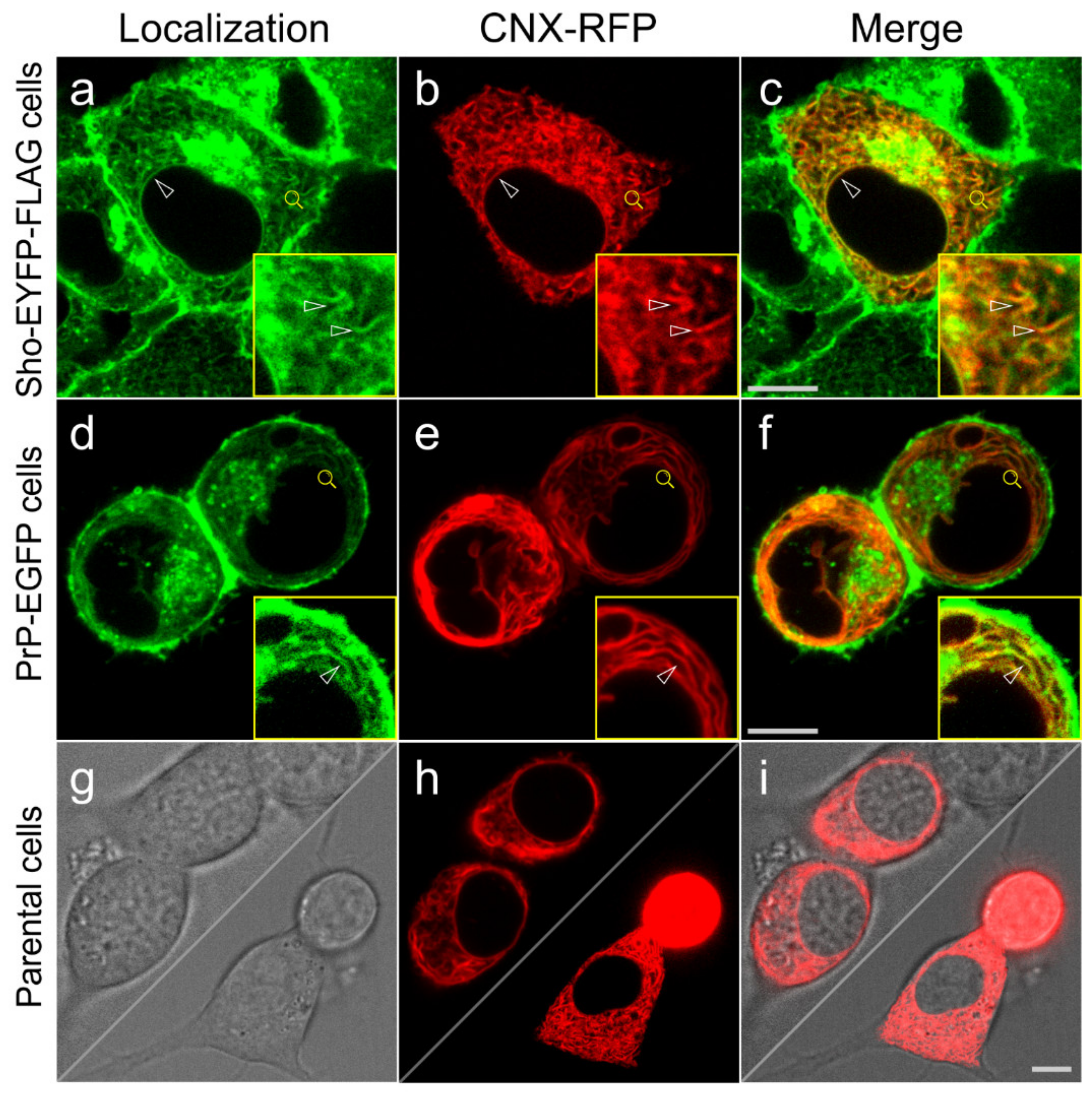
3.3. Shadoo and Prion Proteins Colocalize with Calnexin in the ER Compartments of N2a Cells
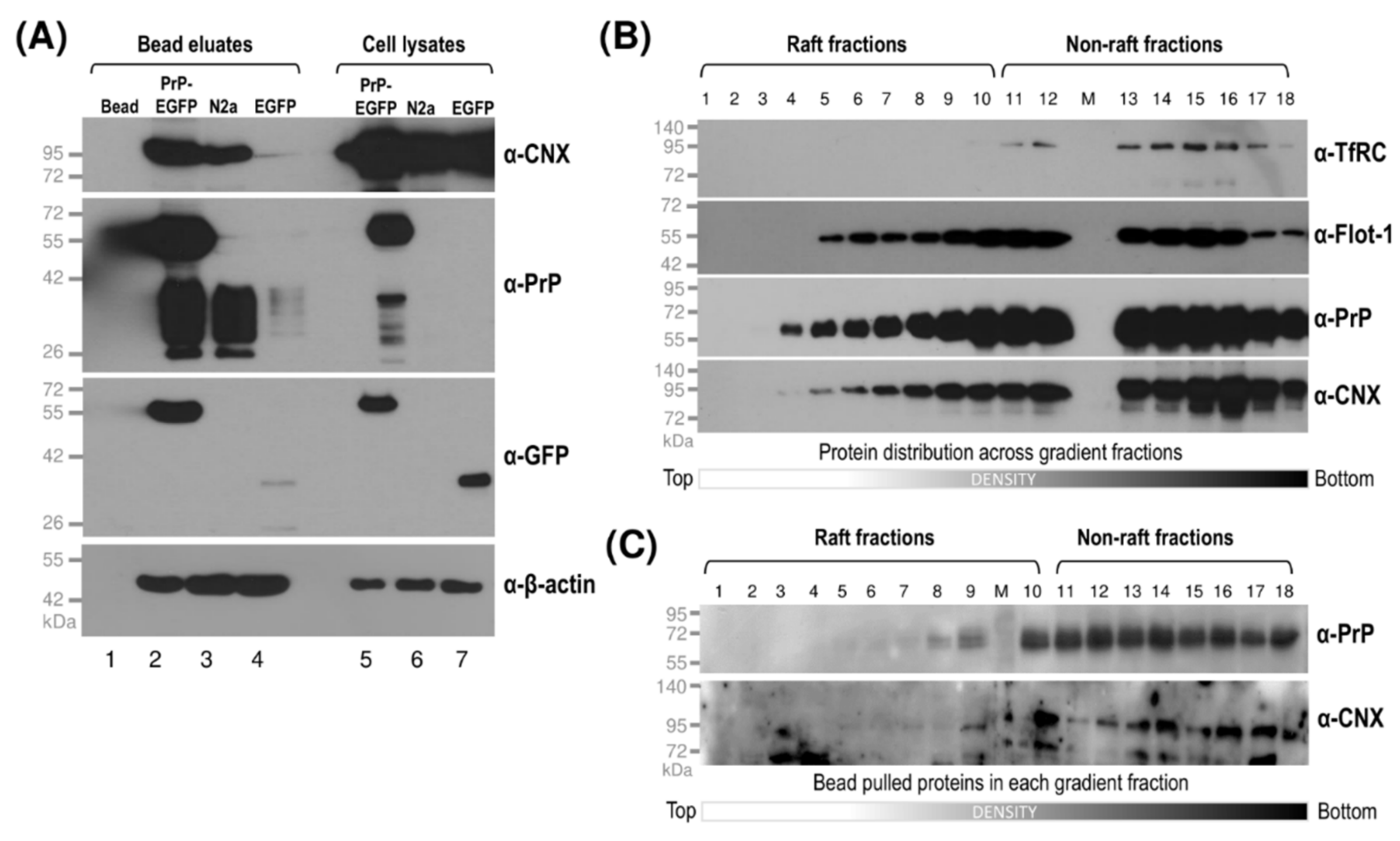
3.4. Interaction of Prion Protein and Shadoo with the ER Chaperon Calnexin in the Lipid-Raft and Non-Raft Membrane Domains
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prusiner, S.B. Novel Proteinaceous Infectious Particles Cause Scrapie. Source. Sci. New Ser. 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Silveira, J.R.; Caughey, B.; Baron, G.S. Prion Protein and the Molecular Features of Transmissible Spongiform Encephalopathy Agents. Curr. Top Microbiol. Immunol. 2004, 284, 1–50. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J. Prion disease of humans and animals: Their Causes and Molecular Basis. Annu. Rev. Neurosci. 2001, 24, 519–550. [Google Scholar] [CrossRef]
- Aguzzi, A. Prion diseases of humans and farm animals: Epidemiology, genetics, and pathogenesis. J. Neurochem. 2006, 97, 1726–1739. [Google Scholar] [CrossRef]
- Jeffrey, M.; González, L. Classical sheep transmissible spongiform encephalopathies: Pathogenesis, pathological phenotypes and clinical disease. Neuropathol. Appl. Neurobiol. 2007, 33, 373–394. [Google Scholar] [CrossRef] [PubMed]
- Wilesmith, J.W.; Wells, G.A.; Cranwell, M.P.; Ryan, J.B. Bovine spongiform encephalopathy: Epidemiological studies. Vet. Rec. 1988, 123, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.S.; Young, S. Chronic wasting disease of captive mule deer: A spongiform encephalopathy. J. Wildl. Dis. 1980, 16, 89–98. [Google Scholar] [CrossRef]
- Bernardi, L.; Bruni, A.C. Mutations in prion protein gene: Pathogenic mechanisms in c-terminal vs. n-terminal domain, a review. Int. J. Mol. Sci. 2019, 20, 3606. [Google Scholar] [CrossRef] [PubMed]
- Bonda, D.J.; Manjila, S.; Mehndiratta, P.; Khan, F.; Miller, B.R.; Onwuzulike, K.; Puoti, G.; Cohen, M.L.; Schonberger, L.B.; Cali, I. Human prion diseases: Surgical lessons learned from iatrogenic prion transmission. Neurosurg. Focus. 2016, 41, E10. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Lansbury, P.T. Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 2003, 26, 267–298. [Google Scholar] [CrossRef] [PubMed]
- Bendheim, P.E.; Brown, H.R.; Rudelli, R.D.; Scala, L.J.; Goller, N.L.; Wen, G.Y.; Kascsak, R.J.; Cashman, N.R.; Bolton, D.C. Nearly ubiquitous tissue distribution of the scrapie agent precursor protein. Neurology 1992, 42, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, O.; Gavín, R.; del Río, J.A. New insights into cellular prion protein (PrPc) functions: The “ying and yang” of a relevant protein. Brain Res. Rev. 2009, 61, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Wopfner, F.; Weidenhöfer, G.; Schneider, R.; Von Brunn, A.; Gilch, S.; Schwarz, T.F.; Werner, T.; Schätzl, H.M. Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J. Mol. Biol. 1999, 289, 1163–1178. [Google Scholar] [CrossRef]
- Büeler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.P.; DeArmond, S.J.; Prusiner, S.B.; Aguet, M.W.C. Normal development and behaviour of mice lacking the neuronal cell surface PrP protein. Nature 1992, 356, 577–582. [Google Scholar] [CrossRef]
- Manson, J.C.; Clarke, A.R.; Hooper, M.L.; Aitchison, L.; McConnell, I.; Hope, J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol. Neurobiol. 1994, 8, 121–127. [Google Scholar] [CrossRef]
- Richt, J.A.; Kasinathan, P.; Hamir, A.N. Production of cattle lacking prion protein. Nat. Biotechnol. 2007, 25, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Chen, J.; Xu, Y.; Zhu, C.; Yu, H.; Liu, S.; Sha, H.; Chen, J.; Xu, X.; Wu, Y.; et al. Generation of goats lacking prion protein. Mol. Reprod. Dev. 2009, 76, 3. [Google Scholar] [CrossRef] [PubMed]
- Benestad, S.L.; Austbø, L.; Tranulis, M.A.; Espenes, A.; Olsaker, I. Healthy goats naturally devoid of prion protein. Vet. Res. 2012, 43, 87. [Google Scholar] [CrossRef]
- Büeler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar] [CrossRef]
- Watts, J.C.; Huo, H.; Bai, Y.; Ehsani, S.; Won Jeon, A.H.; Shi, T.; Daude, N.; Lau, A.; Young, R.; Xu, L.; et al. Interactome analyses identify ties of PrP and its mammalian paralogs to oligomannosidic N-glycans and endoplasmic reticulum-derived chaperones. PLoS Pathog. 2009, 5, e1000608. [Google Scholar] [CrossRef]
- Mahabadi, H.M.; Taghibiglou, C. Cellular prion protein (Prpc): Putative interacting partners and consequences of the interaction. Int. J. Mol. Sci. 2020, 21, 7058. [Google Scholar] [CrossRef]
- Castle, A.R.; Gill, A.C. Physiological functions of the cellular prion protein. Front. Mol. Biosci. 2017, 4, 19. [Google Scholar] [CrossRef]
- Martellucci, S.; Santacroce, C.; Santilli, F.; Manganelli, V.; Sorice, M.; Mattei, V. Prion protein in stem cells: A lipid raft component involved in the cellular differentiation process. Int. J. Mol. Sci. 2020, 21, 4168. [Google Scholar] [CrossRef]
- Gavín, R.; Lidón, L.; Ferrer, I.; Del Río, J.A. The Quest for Cellular Prion Protein Functions in the Aged and Neurodegenerating Brain. Cells 2020, 9, 591. [Google Scholar] [CrossRef]
- Taylor, D.R.; Hooper, N.M. The prion protein and lipid rafts (Review). Mol. Membr. Biol. 2006, 23, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Anilkumar, A.A.; Mayor, S. GPI-anchored protein organization and dynamics at the cell surface. J. Lipid Res. 2016, 57, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Linden, R. The biological function of the prion protein: A cell surface scaffold of signaling modules. Front. Mol. Neurosci. 2017, 10, 77. [Google Scholar] [CrossRef] [PubMed]
- Donne, D.G.; Viles, J.H.; Groth, D.; Mehlhorn, I.; James, T.L.; Cohen, F.E.; Prusiner, S.B.; Wright, P.E.; Dyson, H.J. Structure of the recombinant full-length hamster prion protein PrP(29-231): The N terminus is highly flexible. Proc. Natl. Acad. Sci. USA 1997, 94, 13452–13457. [Google Scholar] [CrossRef]
- Pastore, A.; Zagari, A. A structural overview of the vertebrate prion proteins. Prion 2007, 1, 185–197. [Google Scholar] [CrossRef][Green Version]
- Ciric, D.; Rezaei, H. Biochemical insight into the prion protein family. Front. Cell Dev. Biol. 2015, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Wille, H.; Stöhr, J.; Kendall, A.; Bian, W.; McDonald, M.; Tiggelaar, S.; Watts, J.C.; Prusiner, S.B.; Stubbs, G. Structural studies of truncated forms of the prion protein PrP. Biophys. J. 2015, 108, 1548–1554. [Google Scholar] [CrossRef] [PubMed]
- Riek, R.; Hornemann, S.; Wider, G.; Billeter, M.; Glockshuber, R.; Wuthrich, K. NMR structure of the mouse prion protein domain PrP(121-231). Nature 1996, 382, 180–182. [Google Scholar] [CrossRef]
- Wüthrich, K.; Riek, R. Three-dimensional structures of prion proteins. Adv. Protein Chem. 2001, 57, 55–82. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Sakaguchi, S. N-terminal regions of prion protein: Functions and roles in prion diseases. Int. J. Mol. Sci. 2020, 21, 6233. [Google Scholar] [CrossRef]
- Millhauser, G.L. Copper and the prion protein: Methods, structures, function, and disease. Annu. Rev. Phys. Chem. 2007, 58, 299–320. [Google Scholar] [CrossRef]
- Walter, E.D.; Stevens, D.J.; Visconte, M.P.; Millhauser, G.L. The prion protein is a combined zinc and copper binding protein: Zn 2+ alters the distribution of Cu2+ coordination modes. J. Am. Chem. Soc. 2007, 129, 15440–15441. [Google Scholar] [CrossRef] [PubMed]
- Watt, N.T.; Taylor, D.R.; Kerrigan, T.L.; Griffiths, H.H.; Rushworth, J.V.; Whitehouse, I.J.; Hooper, N.M. Prion protein facilitates uptake of zinc into neuronal cells. Nat. Commun. 2012, 3, 1134. [Google Scholar] [CrossRef]
- You, H.; Tsutsui, S.; Hameed, S.; Kannanayakal, T.J.; Chen, L.; Xia, P.; Engbers, J.D.T.; Lipton, S.A.; Stys, P.K.; Zamponi, G.W. Aβ neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 1737–1742. [Google Scholar] [CrossRef]
- Gasperini, L.; Meneghetti, E.; Pastore, B.; Benetti, F.; Legname, G. Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid. Redox Signal. 2015, 22, 772–784. [Google Scholar] [CrossRef]
- Evans, E.G.G.B.; Pushie, M.J.; Markham, K.A.A.; Lee, H.W.; Millhauser, G.L.L. Interaction between Prion Protein’s Copper-Bound Octarepeat Domain and a Charged C-Terminal Pocket Suggests a Mechanism for N-Terminal Regulation. Structure 2016, 24, 1057–1067. [Google Scholar] [CrossRef]
- Silva, J.L.; Lima, L.M.T.R.; Foguel, D.; Cordeiro, Y. Intriguing nucleic-acid-binding features of mammalian prion protein. Trends Biochem. Sci. 2008, 33, 132–140. [Google Scholar] [CrossRef]
- Lathe, R.; Darlix, J.L. Prion protein PrP nucleic acid binding and mobilization implicates retroelements as the replicative component of transmissible spongiform encephalopathy. Arch. Virol. 2020, 165, 535–556. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.C.; Lee, I.Y.; Silverman, G.L.; Harrison, P.M.; Strome, R.; Heinrich, C.; Karunaratne, A.; Pasternak, S.H.; Chishti, M.A.; Liang, Y.; et al. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J. Mol. Biol. 1999, 292, 797–817. [Google Scholar] [CrossRef]
- Premzl, M.; Sangiorgio, L.; Strumbo, B.; Marshall Graves, J.A.; Simonic, T.; Gready, J.E. Shadoo, a new protein highly conserved from fish to mammals and with similarity to prion protein. Gene 2003, 314, 89–102. [Google Scholar] [CrossRef]
- Watts, J.C.; Westaway, D. The prion protein family: Diversity, rivalry, and dysfunction. Biochim. Biophys. Acta Mol. Basis. Dis. 2007, 1772, 654–672. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Drisaldi, B.; Ng, V.; Yang, J.; Strome, B.; Horne, P.; Sy, M.S.; Yoong, L.; Young, R.; Mastrangelo, P.; et al. The CNS glycoprotein Shadoo has PrPC-like protective properties and displays reduced levels in prion infections. EMBO J. 2007, 26, 4038–4050. [Google Scholar] [CrossRef] [PubMed]
- Young, R.; Passet, B.; Vilotte, M.; Cribiu, E.P.; Béringue, V.; Le Provost, F.; Laude, H.; Vilotte, J.L. The prion or the related Shadoo protein is required for early mouse embryogenesis. FEBS Lett. 2009, 583, 3296–3300. [Google Scholar] [CrossRef]
- Young, R.; Bouet, S.; Polyte, J.; Le Guillou, S.; Passet, B.; Vilotte, M.; Castille, J.; Beringue, V.; Le Provost, F.; Laude, H.; et al. Expression of the prion-like protein Shadoo in the developing mouse embryo. Biochem. Biophys. Res. Commun. 2011, 416, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Passet, B.; Castille, J.; Makhzami, S.; Truchet, S.; Vaiman, A.; Floriot, S.; Moazami-Goudarzi, K.; Vilotte, M.; Gaillard, A.L.; Helary, L.; et al. The Prion-like protein Shadoo is involved in mouse embryonic and mammary development and differentiation. Sci. Rep. 2020, 10, 6765. [Google Scholar] [CrossRef]
- Passet, B.; Young, R.; Makhzami, S.; Vilotte, M.; Jaffrezic, F.; Halliez, S.; Bouet, S.; Marthey, S.; Khalifé, M.; Kanellopoulos-Langevin, C.; et al. Prion protein and shadoo are involved in overlapping embryonic pathways and trophoblastic development. PLoS ONE 2012, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Daude, N.; Wohlgemuth, S.; Brown, R.; Pitstick, R.; Gapeshina, H.; Yang, J.; Carlson, G.A.; Westaway, D. Knockout of the prion protein (PrP)-like Sprn gene does not produce embryonic lethality in combination with PrPC-deficiency. Proc. Natl. Acad. Sci. USA 2012, 109, 9035–9040. [Google Scholar] [CrossRef]
- Daude, N.; Westaway, D. Shadoo/PrP (Sprn0/0/Prnp0/0) double knockout mice: More than zeroes. Prion 2012, 6, 420–424. [Google Scholar] [CrossRef]
- Jiayu, W.; Zhu, H.; Ming, X.; Xiong, W.; Songbo, W.; Bocui, S.; Wensen, L.; Jiping, L.; Keying, M.; Zhongyi, L. Mapping the interaction site of prion protein and Sho. Mol. Biol. Rep. 2010, 37, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Ciric, D.; Richard, C.-A.; Moudjou, M.; Chapuis, J.; Sibille, P.; Daude, N.; Westaway, D.; Adrover, M.; Béringue, V.; Martin, D.; et al. Interaction between Shadoo and PrP Affects the PrP-Folding Pathway. J. Virol. 2015, 89, 6287–6293. [Google Scholar] [CrossRef] [PubMed]
- Sangeetham, S.B.; Huszár, K.; Bencsura, P.; Nyeste, A.; Hunyadi-Gulyás, É.; Fodor, E.; Welker, E. Interrogating the Dimerization Interface of the Prion Protein Via Site-Specific Mutations to p-Benzoyl-L-Phenylalanine. J. Mol. Biol. 2018, 430, 2784–2801. [Google Scholar] [CrossRef]
- Corley, S.M.; Gready, J.E. Identification of the RGG Box Motif in Shadoo: RNA-Binding and Signaling Roles? Bioinform. Biol. Insights. 2008, 2, 383–400. [Google Scholar] [CrossRef]
- Lau, A.; Mays, C.E.; Genovesi, S.; Westaway, D. RGG repeats of PrP-like shadoo protein bind nucleic acids. Biochemistry 2012, 51, 9029–9031. [Google Scholar] [CrossRef] [PubMed]
- Tóth, E.; Kulcsár, P.I.; Fodor, E.; Ayaydin, F.; Kalmár, L.; Borsy, A.É.; László, L.; Welker, E. The highly conserved, N-terminal (RXXX) 8 motif of mouse Shadoo mediates nuclear accumulation. BBA-Mol. Cell Res. 2013, 1833, 1199–1211. [Google Scholar] [CrossRef]
- Kang, S.; Mays, C.E.; Daude, N.; Yang, J.; Kar, S.; Westaway, D. Proteasomal Inhibition Redirects the PrP-Like Shadoo Protein to the Nucleus. Mol. Neurobiol. 2019, 56, 7888–7904. [Google Scholar] [CrossRef] [PubMed]
- Shmerling, D.; Hegyi, I.; Fischer, M.; Blättler, T.; Brandner, S.; Götz, J.; Rülicke, T.; Flechsig, E.; Cozzio, A.; Von Mering, C.; et al. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 1998, 93, 203–214. [Google Scholar] [CrossRef]
- Sakthivelu, V.; Seidel, R.P.; Winklhofer, K.F.; Tatzelt, J. Conserved stress-protective activity between prion protein and Shadoo. J. Biol. Chem. 2011, 286, 8901–8908. [Google Scholar] [CrossRef] [PubMed]
- Nyeste, A.; Bencsura, P.; Fodor, E.; Welker, E. Expression of the Prion Protein Family Member Shadoo Causes Drug Hypersensitivity That Is Diminished by the Coexpression of the Wild Type Prion Protein. J. Biol. Chem. 2016, 291, 4473–4486. [Google Scholar] [CrossRef]
- Nyeste, A.; Stincardini, C.; Bencsura, P.; Cerovic, M.; Biasini, E.; Welker, E. The prion protein family member Shadoo induces spontaneous ionic currents in cultured cells. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Stöhr, J.; Bhardwaj, S.; Wille, H.; Oehler, A.; DeArmond, S.J.; Giles, K.; Prusiner, S.B. Protease-resistant prions selectively decrease Shadoo protein. PLoS Pathog. 2011, 7, e1002382. [Google Scholar] [CrossRef]
- Westaway, D.; Genovesi, S.; Daude, N.; Brown, R.; Lau, A.; Lee, I.; Mays, C.E.; Coomaraswamy, J.; Canine, B.; Pitstick, R.; et al. Down-regulation of shadoo in prion infections traces a pre-clinical event inversely related to PrP sc accumulation. PLoS Pathog. 2011, 7, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Mays, C.E.; Kim, C.; Haldiman, T.; Van Der Merwe, J.; Lau, A.; Yang, J.; Grams, J.; Di Bari, M.A.; Nonno, R.; Telling, G.C.; et al. Prion disease tempo determined by host-dependent substrate reduction. J. Clin. Invest. 2014, 124, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Manuelidis, L. Agent-specific shadoo responses in transmissible encephalopathies. J. Neuroimmune Pharmacol. 2010, 5, 155–163. [Google Scholar] [CrossRef]
- Wang, H.; Wan, J.; Wang, W.; Wang, D.; Li, S.; Liao, P.; Hao, Z.; Wu, S.; Xu, J.; Li, N.; et al. Overexpression of Shadoo protein in transgenic mice does not impact the pathogenesis of scrapie. Neurosci. Lett. 2011, 496, 1–4. [Google Scholar] [CrossRef]
- Graner, E.; Mercadante, A.F.; Zanata, S.M.; Forlenza, O.V.; Cabral, A.L.B.; Veiga, S.S.; Juliano, M.A.; Roesler, R.; Walz, R.; Minetti, A.; et al. Cellular prion protein binds laminin and mediates neuritogenesis. Mol. Brain Res. 2000, 76, 85–92. [Google Scholar] [CrossRef]
- Santuccione, A.; Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 2005, 169, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Zanata, S.M.; Lopes, M.H.; Mercadante, A.F.; Hajj, G.N.M.; Chiarini, L.B.; Nomizo, R.; Freitas, A.R.O.; Cabral, A.L.B.; Lee, K.S.; Juliano, M.A.; et al. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002, 21, 3307–3316. [Google Scholar] [CrossRef]
- Lopes, M.H.; Hajj, G.N.M.; Muras, A.G.; Mancini, G.L.; Castro, R.M.P.S.; Ribeiro, K.C.B.; Brentani, R.R.; Linden, R.; Martins, V.R. Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J. Neurosci. 2005, 25, 11330–11339. [Google Scholar] [CrossRef] [PubMed]
- Baron, G.S.; Wehrly, K.; Dorward, D.W.; Chesebro, B.; Caughey, B. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrP(Sc)) into contiguous membranes. EMBO J. 2002, 21, 1031–1040. [Google Scholar] [CrossRef]
- Sarnataro, D.; Campana, V.; Paladino, S.; Stornaiuolo, M.; Nitsch, L.; Zurzolo, C. PrPC association with lipid rafts in the early secretory pathway stabilizes its cellular conformation. Mol. Biol. Cell. 2004, 15, 4031–4042. [Google Scholar] [CrossRef] [PubMed]
- Pepe, A.; Avolio, R.; Matassa, D.S.; Esposito, F.; Nitsch, L.; Zurzolo, C.; Paladino, S.; Sarnataro, D. Regulation of sub-compartmental targeting and folding properties of the Prion-like protein Shadoo. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Wang, W.; Chen, R.; Luo, K.; Wu, D.; Huang, L.; Huang, T.; Xiao, G. Calnexin inhibits thermal aggregation and neurotoxicity of prion protein. J. Cell Biochem. 2010, 111, 343–349. [Google Scholar] [CrossRef]
- Macdonald, J.L.; Pike, L.J. A simplified method for the preparation of detergent-free lipid rafts. J. Lipid Res. 2005, 46, 1061–1067. [Google Scholar] [CrossRef]
- Race, R.E.; Fadness, L.H.; Chesebro, B. Characterization of scrapie infection in mouse neuroblastoma cells. J. Gen. Virol. 1987, 68, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, N.V.; Dirndorfer, D.; Lang, S.; Resenberger, U.K.; Restelli, L.M.; Hemion, C.; Miesbauer, M.; Frank, S.; Neutzner, A.; Zimmermann, R.; et al. Structural features within the nascent chain regulate alternative targeting of secretory proteins to mitochondria. EMBO J. 2013, 32, 1036–1051. [Google Scholar] [CrossRef] [PubMed]
- Engelke, A.D.; Gonsberg, A.; Thapa, S.; Jung, S.; Ulbrich, S.; Seidel, R.; Basu, S.; Multhaup, G.; Baier, M.; Engelhard, M.; et al. Dimerization of the cellular prion protein inhibits propagation of scrapie prions. J. Biol. Chem. 2018, 293, 8020–8031. [Google Scholar] [CrossRef] [PubMed]
- Tóth, E.; Welker, E.; Construction, A.P. Comparison of Anti-Shadoo Antibodies–Where is the endogenous Shadoo protein? Int. J. Biotechnol. Bioeng. 2012, 6, 259–261. [Google Scholar]
- Zanusso, G.; Liu, D.; Ferrari, S.; Hegyi, I.; Yin, X.; Aguzzi, A.; Hornemann, S.; Liemann, S.; Glockshuber, R.; Manson, J.C.; et al. Prion protein expression in different species: Analysis with a panel of new mAbs. Proc. Natl. Acad. Sci. USA 1998, 95, 8812–8816. [Google Scholar] [CrossRef]
- Barmada, S.; Piccardo, P.; Yamaguchi, K.; Ghetti, B.; Harris, D.A. GFP-tagged prion protein is correctly localized and functionally active in the brains of transgenic mice. Neurobiol. Dis. 2004, 16, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Varma, R.; Mayor, S. GPI-anchored proteins are organized in submicron domains at the cell surface. Nature 1998, 394, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Madore, N.; Smith, K.L.; Graham, C.H.; Jen, A.; Brady, K.; Hall, S.; Morris, R. Functionally different GPI proteins are organized in different domains on the neuronal surface. EMBO J. 1999, 18, 6917–6926. [Google Scholar] [CrossRef]
- Brügger, B.; Graham, C.; Leibrecht, I.; Mombelli, E.; Jen, A.; Wieland, F.; Morris, R. The Membrane Domains Occupied by Glycosylphosphatidylinositol-anchored Prion Protein and Thy-1 Differ in Lipid Composition. J. Biol. Chem. 2004, 279, 7530–7536. [Google Scholar] [CrossRef]
- Hancock, J.F. Lipid rafts: Contentious only from simplistic standpoints. Nat. Rev. Mol. Cell Biol. 2006, 7, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Persaud-Sawin, D.A.; Lightcap, S.; Harry, G.J. Isolation of rafts from mouse brain tissue by a detergent-free method. J. Lipid Res. 2009, 50, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, H.; Helms, J.B.; Ohno-Iwashita, Y.; Shimada, Y.; Horikoshi, Y.; Yamaguchi, H. Ultrastructural localization of flotillin-1 to cholesterol-rich membrane microdomains, rafts, in rat brain tissue. Brain Res. 2003, 965, 83–90. [Google Scholar] [CrossRef]
- Otto, G.P.; Nichols, B.J. The roles of flotillin microdomains-endocytosis and beyond. J. Cell Sci. 2011, 124, 3933–3940. [Google Scholar] [CrossRef]
- Chamberlain, L.H.; Burgoyne, R.D.; Gould, G.W. SNARE proteins are highly enriched in lipid rafts in PC12 cells: Implications for the spatial control of exocytosis. Proc. Natl. Acad. Sci. USA 2001, 98, 5619–5624. [Google Scholar] [CrossRef] [PubMed]
- Hering, H.; Lin, C.C.; Sheng, M. Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J. Neurosci. 2003, 23, 3262–3271. [Google Scholar] [CrossRef]
- Eckert, G.P.; Igbavboa, U.; Müller, W.E.; Wood, W.G. Lipid rafts of purified mouse brain synaptosomes prepared with or without detergent reveal different lipid and protein domains. Brain Res. 2003, 962, 144–150. [Google Scholar] [CrossRef]
- Kinnun, J.J.; Bolmatov, D.; Lavrentovich, M.O.; Katsaras, J. Lateral heterogeneity and domain formation in cellular membranes. Chem. Phys. Lipids. 2020, 232, 104976. [Google Scholar] [CrossRef]
- Levental, I.; Levental, K.R.; Heberle, F.A. Lipid Rafts: Controversies Resolved, Mysteries Remain. Trends Cell Biol. 2020, 30, 341–353. [Google Scholar] [CrossRef]
- Kalappurakkal, J.M.; Sil, P.; Mayor, S. Toward a new picture of the living plasma membrane. Protein Sci. 2020, 29, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.J. Rafts defined: A report on the Keystone symposium on lipid rafts and cell function. J. Lipid Res. 2006, 47, 1597–1598. [Google Scholar] [CrossRef] [PubMed]
- Nickels, J.D.; Chatterjee, S.; Stanley, C.B.; Qian, S.; Cheng, X.; Myles, D.A.A.; Standaert, R.F.; Elkins, J.G.; Katsaras, J. The in vivo structure of biological membranes and evidence for lipid domains. PLoS Biol. 2017, 15, e2002214. [Google Scholar] [CrossRef] [PubMed]
- Schuck, S.; Honsho, M.; Ekroos, K.; Shevchenko, A.; Simons, K. Resistance of cell membranes to different detergents. Proc. Natl. Acad. Sci. USA 2003, 100, 5795–5800. [Google Scholar] [CrossRef] [PubMed]
- Shogomori, H.; Brown, D.A. Use of detergents to study membrane rafts: The good, the bad, and the ugly. Biol. Chem. 2003, 384, 1259–1263. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.R.; Watt, N.T.; Perera, W.S.S.; Hooper, N.M. Assigning functions to distinct regions of the N-terminus of the prion protein that are involved in its copper-stimulated, clathrin-dependent endocytosis. J. Cell Sci. 2005, 118, 5141–5153. [Google Scholar] [CrossRef] [PubMed]
- Lichtenberg, D.; Goñi, F.M.; Heerklotz, H. Detergent-resistant membranes should not be identified with membrane rafts. Trends Biochem. Sci. 2005, 30, 430–436. [Google Scholar] [CrossRef]
- Wang, J.; Yu, R.K. Association of glycolipids and growth factor receptors in lipid rafts. Methods Mol. Biol. 2021, 2187, 131–145. [Google Scholar] [CrossRef]
- Shah, M.B.; Sehgal, P.B. Nondetergent isolation of rafts. Methods Mol. Biol. 2007, 398, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Paladino, S.; Lebreton, S.; Tivodar, S.; Campana, V.; Tempre, R.; Zurzolo, C. Different GPI-attachment signals affect the oligomerisation of GPI-anchored proteins and their apical sorting. J. Cell Sci. 2008, 121, 4001–4007. [Google Scholar] [CrossRef] [PubMed]
- Puig, B.; Altmeppen, H.C.; Thurm, D.; Geissen, M.; Conrad, C.; Braulke, T.; Glatzel, M. N-glycans and glycosylphosphatidylinositol-anchor act on polarized sorting of mouse PrP c in Madin-Darby canine kidney cells. PLoS ONE 2011, 6, e24624. [Google Scholar] [CrossRef] [PubMed]
- Sunyach, C.; Jen, A.; Deng, J.; Fitzgerald, K.T.; Frobert, Y.; Grassi, J.; McCaffrey, M.W.; Morris, R. The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J. 2003, 22, 3591–3601. [Google Scholar] [CrossRef]
- Lucero, H.A.; Robbins, P.W. Lipid rafts-protein association and the regulation of protein activity. Arch. Biochem. Biophys. 2004, 426, 208–224. [Google Scholar] [CrossRef] [PubMed]
- Orberger, G.; Geyer, R.; Stirm, S.; Tauber, R. Structure of the N-linked oligosaccharides of the human transferrin receptor. Eur. J. Biochem. 1992, 205, 257–267. [Google Scholar] [CrossRef]
- Ohe, Y.; Ohnishi, H.; Okazawa, H.; Tomizawa, K.; Kobayashi, H.; Okawa, K.; Matozaki, T. Characterization of nucleotide pyrophosphatase-5 as an oligomannosidic glycoprotein in rat brain. Biochem. Biophys. Res. Commun. 2003, 308, 719–725. [Google Scholar] [CrossRef]
- Kucherer, A.; Faissner, A.; Schachner, M. The novel carbohydrate epitope L3 is shared by some neural cell adhesion molecules. J. Cell Biol. 1987, 104, 1597–1602. [Google Scholar] [CrossRef] [PubMed]
- Pesheva, P.; Horwitz, A.F.; Schachner, M. Integrin, the cell surface receptor for fibronectin and laminin, expresses the L2/HNK-1 and L3 carbohydrate structures shared by adhesion molecules. Neurosci. Lett. 1987, 83, 303–306. [Google Scholar] [CrossRef]
- Horstkorte, R.; Schachner, M.; Magyar, J.P.; Vorherr, T.; Schmitz, B. The fourth immunoglobulin-like domain of NCAM contains a carbohydrate recognition domain for oligomannosidic glycans implicated in association with L1 and neurite outgrowth. J. Cell Biol. 1993, 121, 1409–1422. [Google Scholar] [CrossRef] [PubMed]
- Heller, M.; Von der Ohe, M.; Kleene, R.; Mohajeri, M.H.; Schachner, M. The immunoglobulin-superfamily molecule basigin is a binding protein for oligomannosidic carbohydrates: An anti-idiotypic approach. J. Neurochem. 2003, 84, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Baumann, O.; Walz, B. Endoplasmic reticulum of animal cells and its organization into structural and functional domains. Int. Rev. Cytol. 2001, 205, 149–214. [Google Scholar] [CrossRef] [PubMed]
- Voeltz, G.K.; Prinz, W.A. Sheets, ribbons and tubules-How organelles get their shape. Nat. Rev. Mol. Cell Biol. 2007, 8, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Nixon-Abell, J.; Obara, C.J.; Weigel, A.V.; Li, D.; Legant, W.R.; Xu, C.S.; Pasolli, H.A.; Harvey, K.; Hess, H.F.; Betzig, E.; et al. Increased spatiotemporal resolution reveals highly dynamic dense tubular matrices in the peripheral ER. Science 2016, 354, aaf3928. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; Shemesh, T.; Prinz, W.A.; Palazzo, A.F.; Kozlov, M.M.; Rapoport, T.A. Mechanisms determining the morphology of the peripheral ER. Cell 2010, 143, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; Voeltz, G.K.; Rapoport, T.A. Rough Sheets and Smooth Tubules. Cell 2006, 126, 435–439. [Google Scholar] [CrossRef]
- Johnson, S.; Michalak, M.; Opas, M.; Eggleton, P. The ins and outs of calreticulin: From the ER lumen to the extracellular space. Trends Cell Biol. 2001, 11, 122–129. [Google Scholar] [CrossRef]
- Okazaki, Y.; Ohno, H.; Takase, K.; Ochiai, T.; Saito, T. Cell surface expression of calnexin, a molecular chaperone in the endoplasmic reticulum. J. Biol. Chem. 2000, 275, 35751–35758. [Google Scholar] [CrossRef]
- Zavodszky, E.; Hegde, R.S. Misfolded GPI-anchored proteins are escorted through the secretory pathway by ER-derived factors. Elife 2019, 8, e46740. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dondapati, D.T.; Cingaram, P.R.; Ayaydin, F.; Nyeste, A.; Kanyó, A.; Welker, E.; Fodor, E. Membrane Domain Localization and Interaction of the Prion-Family Proteins, Prion and Shadoo with Calnexin. Membranes 2021, 11, 978. https://doi.org/10.3390/membranes11120978
Dondapati DT, Cingaram PR, Ayaydin F, Nyeste A, Kanyó A, Welker E, Fodor E. Membrane Domain Localization and Interaction of the Prion-Family Proteins, Prion and Shadoo with Calnexin. Membranes. 2021; 11(12):978. https://doi.org/10.3390/membranes11120978
Chicago/Turabian StyleDondapati, Divya Teja, Pradeep Reddy Cingaram, Ferhan Ayaydin, Antal Nyeste, Andor Kanyó, Ervin Welker, and Elfrieda Fodor. 2021. "Membrane Domain Localization and Interaction of the Prion-Family Proteins, Prion and Shadoo with Calnexin" Membranes 11, no. 12: 978. https://doi.org/10.3390/membranes11120978
APA StyleDondapati, D. T., Cingaram, P. R., Ayaydin, F., Nyeste, A., Kanyó, A., Welker, E., & Fodor, E. (2021). Membrane Domain Localization and Interaction of the Prion-Family Proteins, Prion and Shadoo with Calnexin. Membranes, 11(12), 978. https://doi.org/10.3390/membranes11120978