
New 1,2,3-Triazole Scaffold Schiff Bases as Potential Anti-COVID-19: Design, Synthesis, DFT-Molecular Docking, and Cytotoxicity Aspects

 ,
, 
Abstract
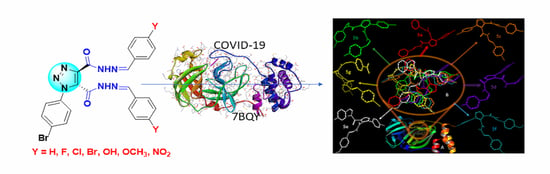
:
1. Introduction
2. Results and Discussion
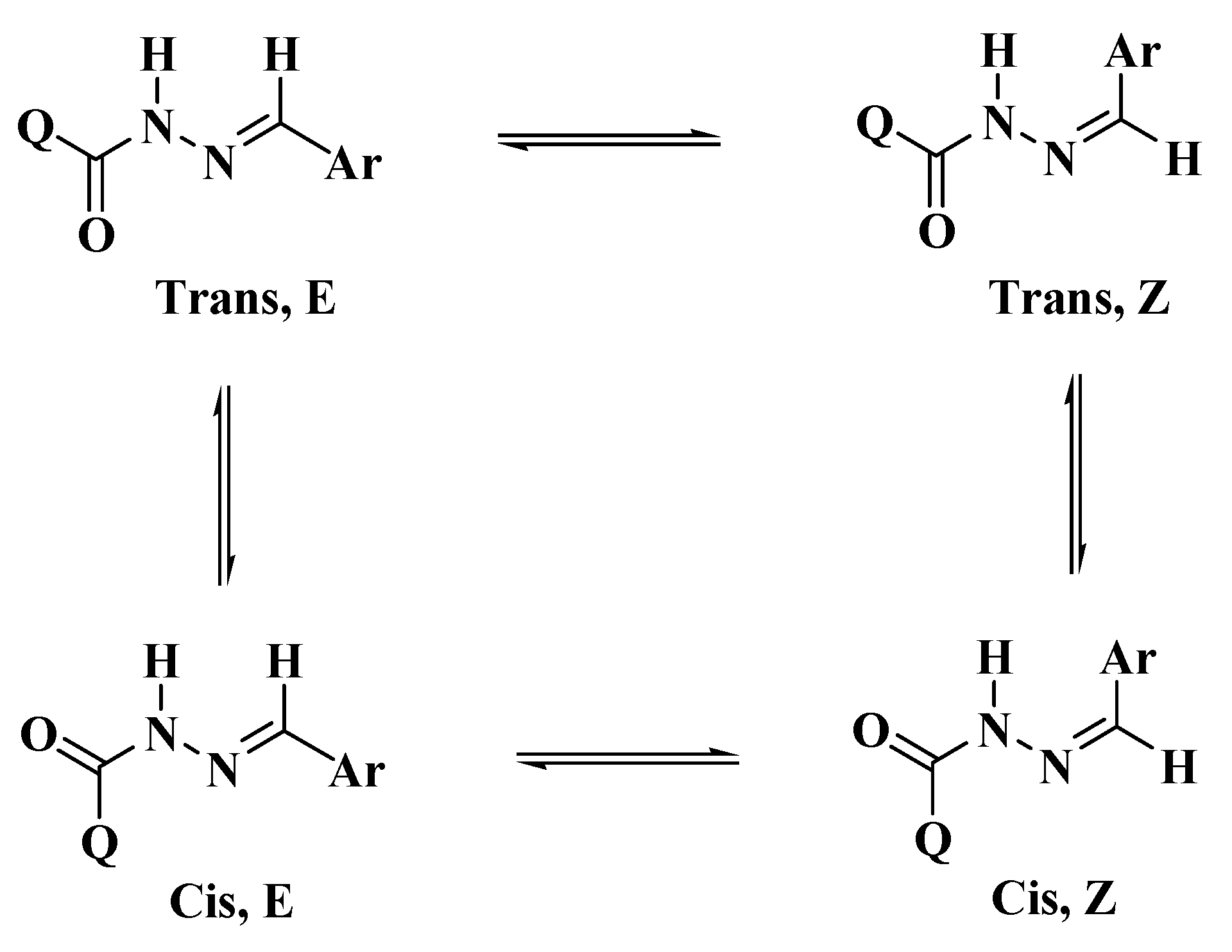
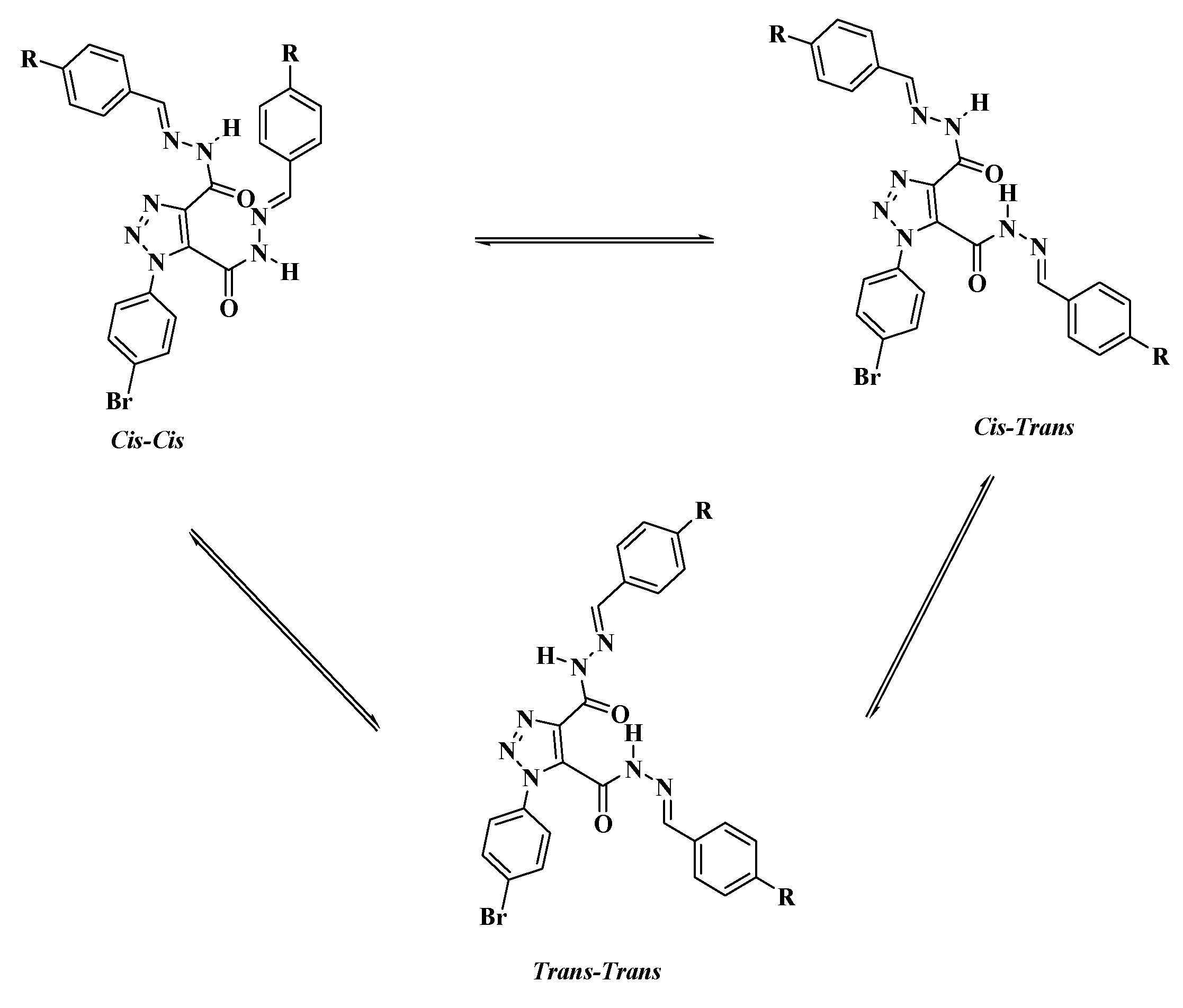
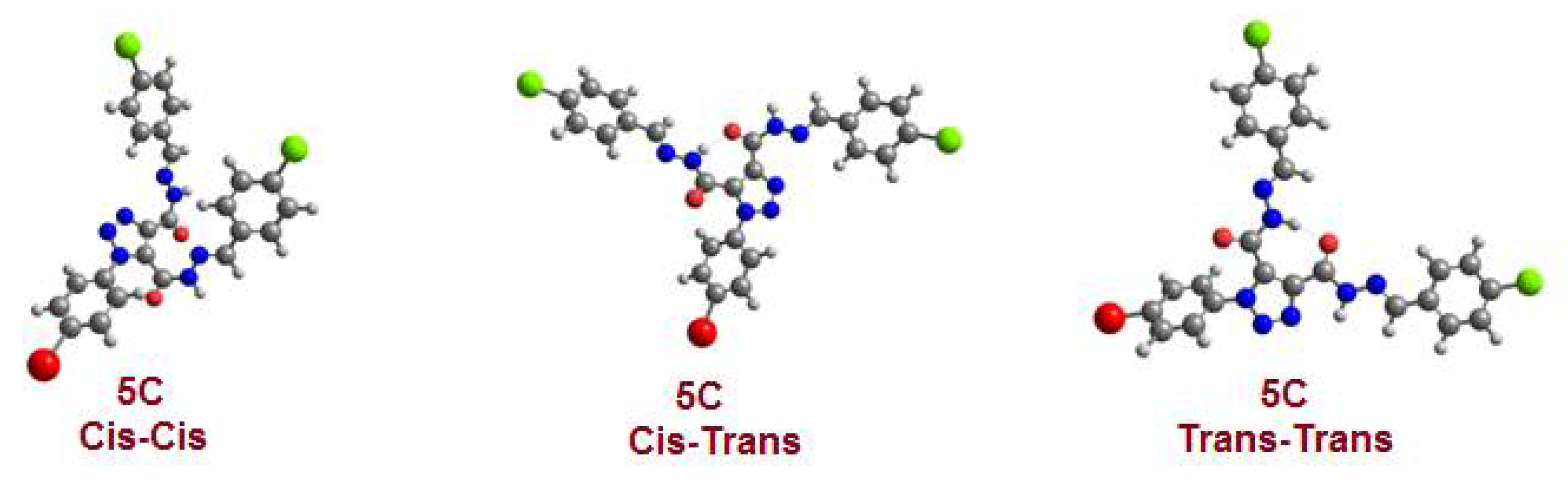
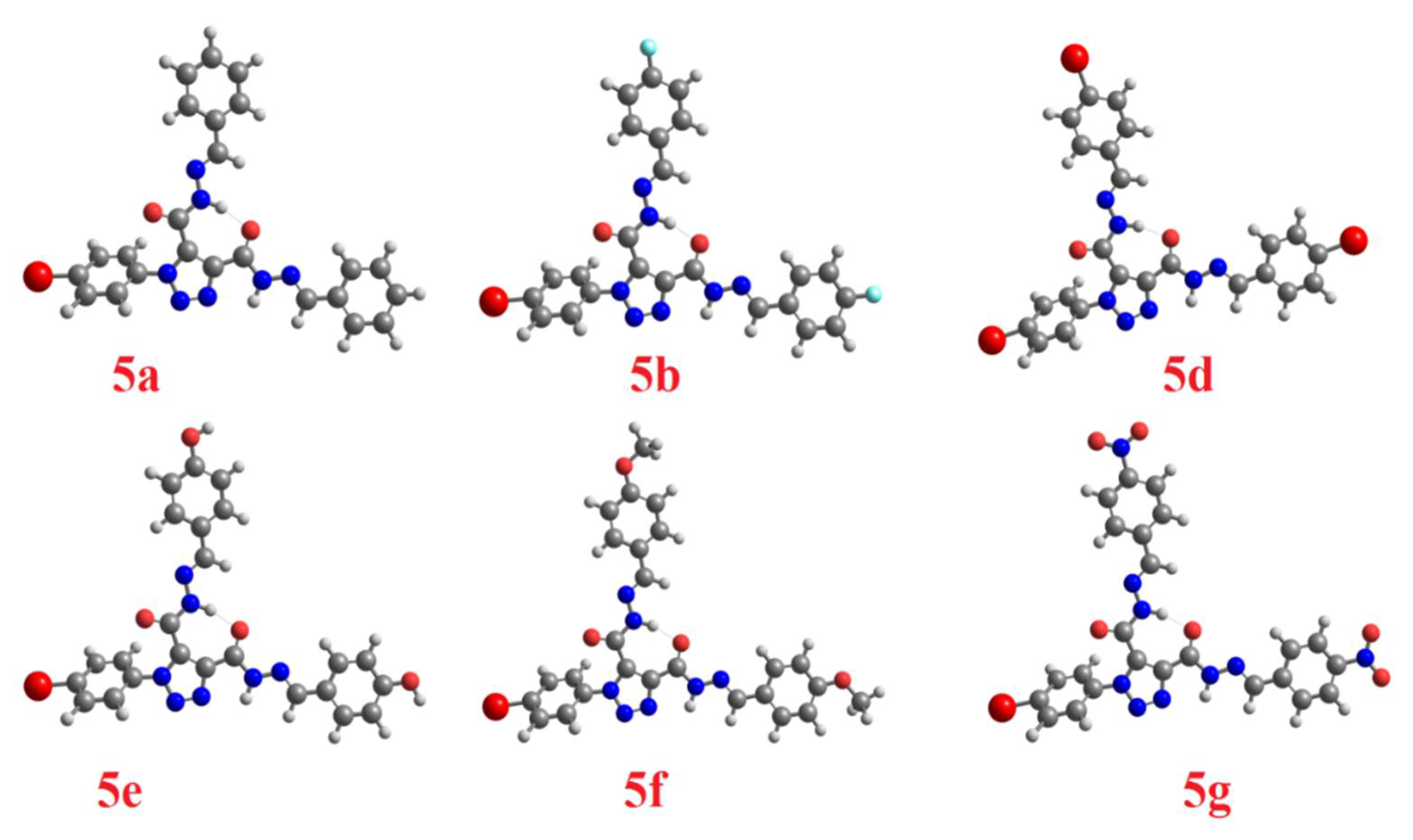
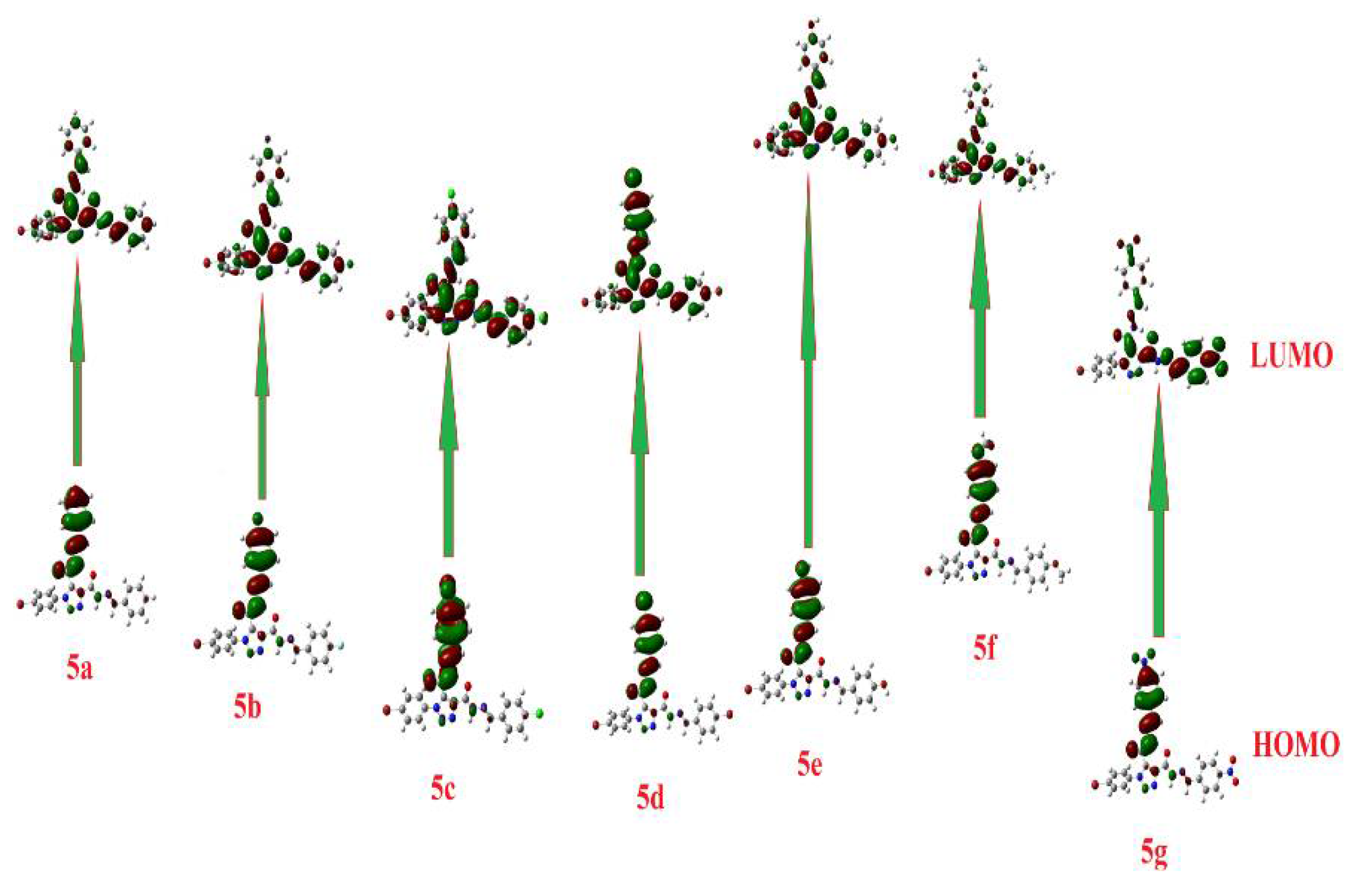
2.1. DFT Theoretical Calculations
Molecular Geometry
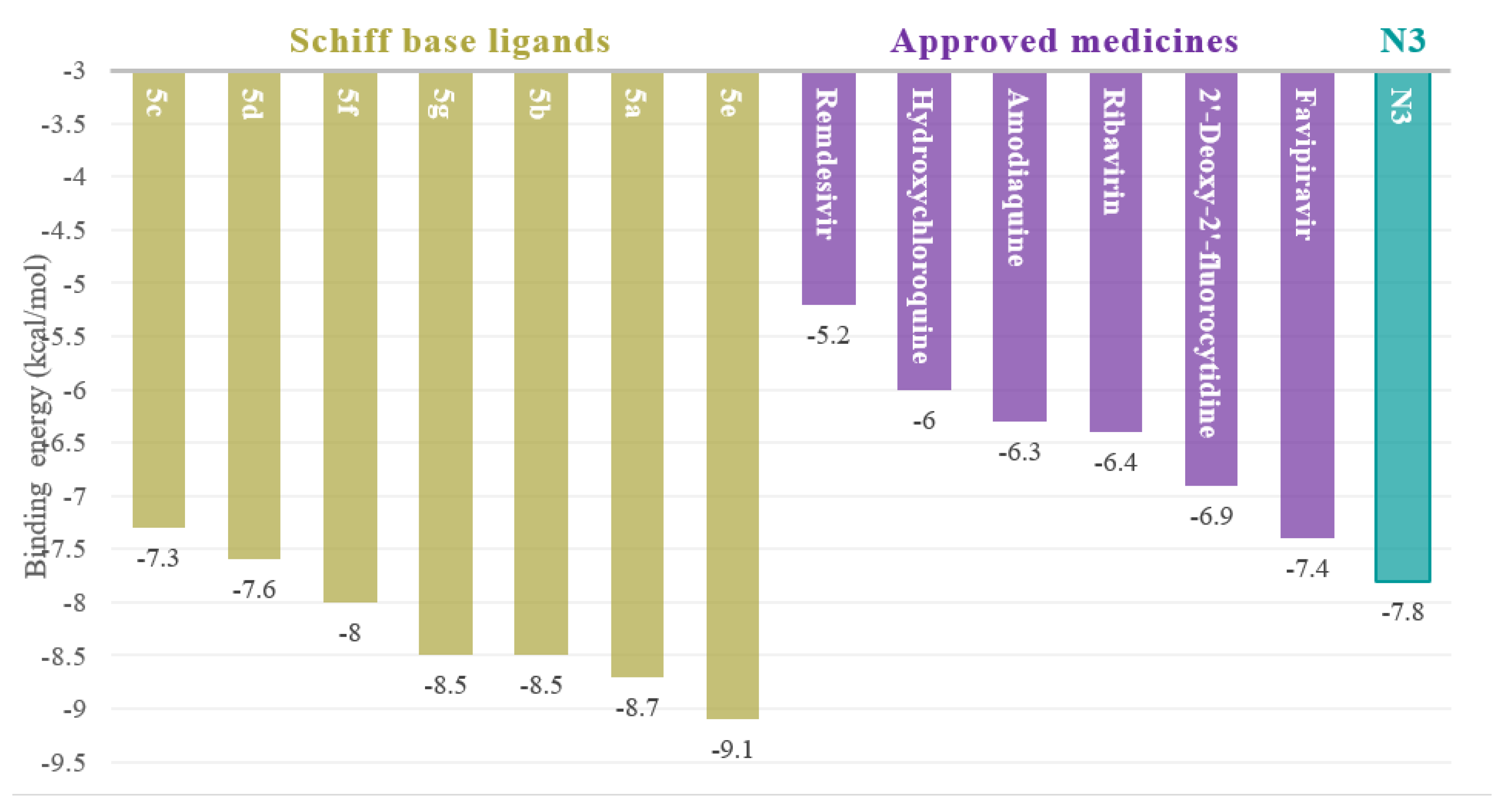
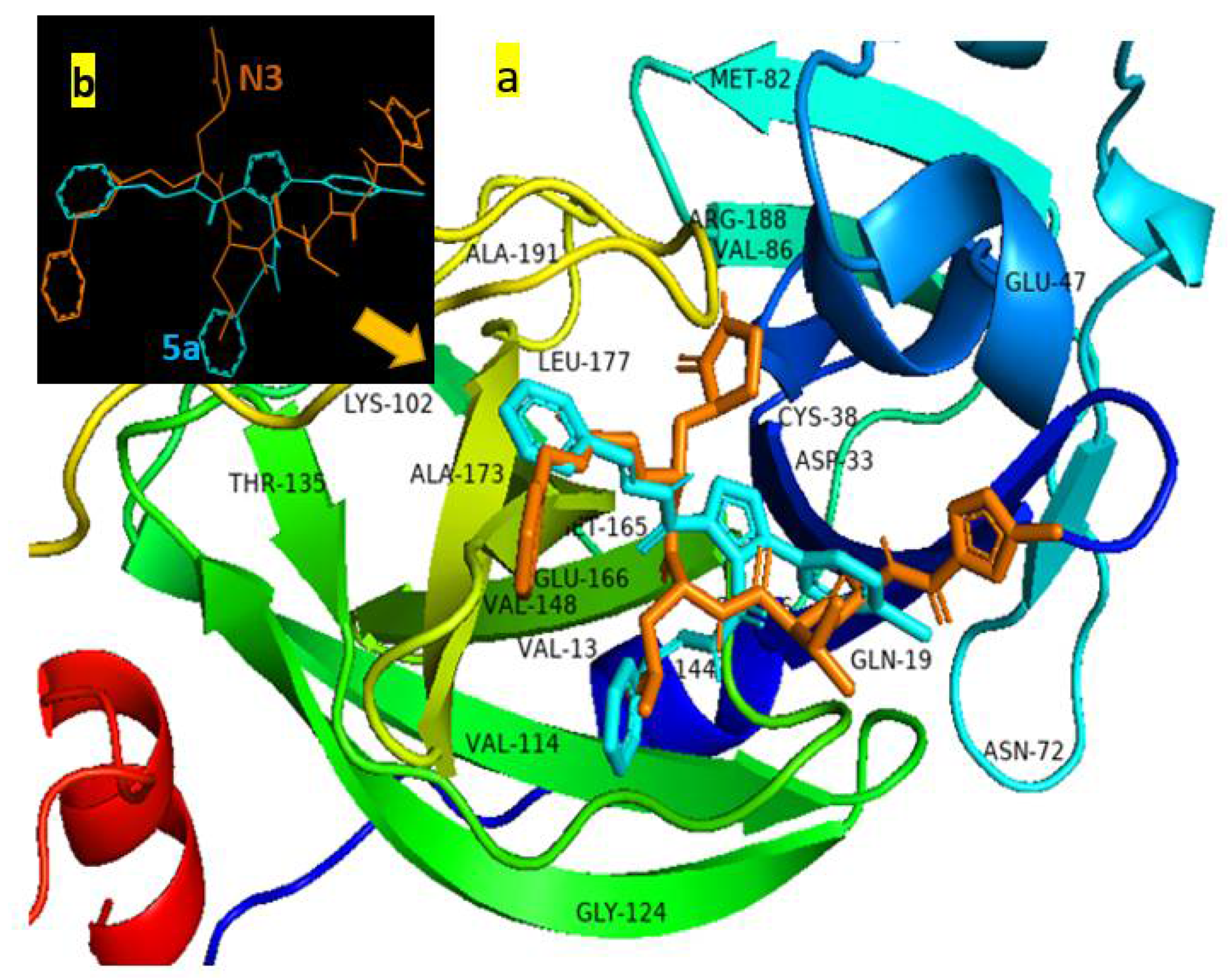
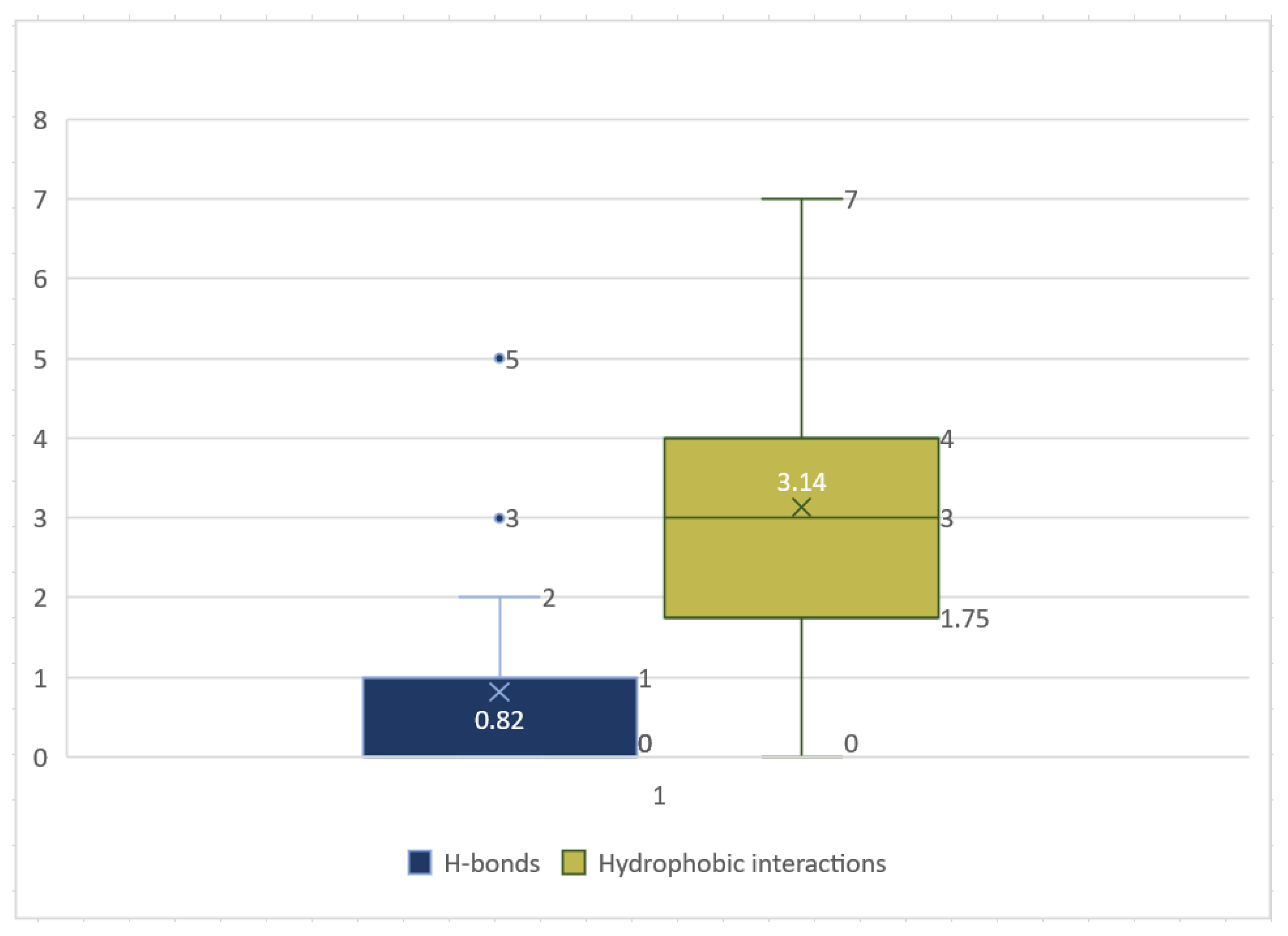
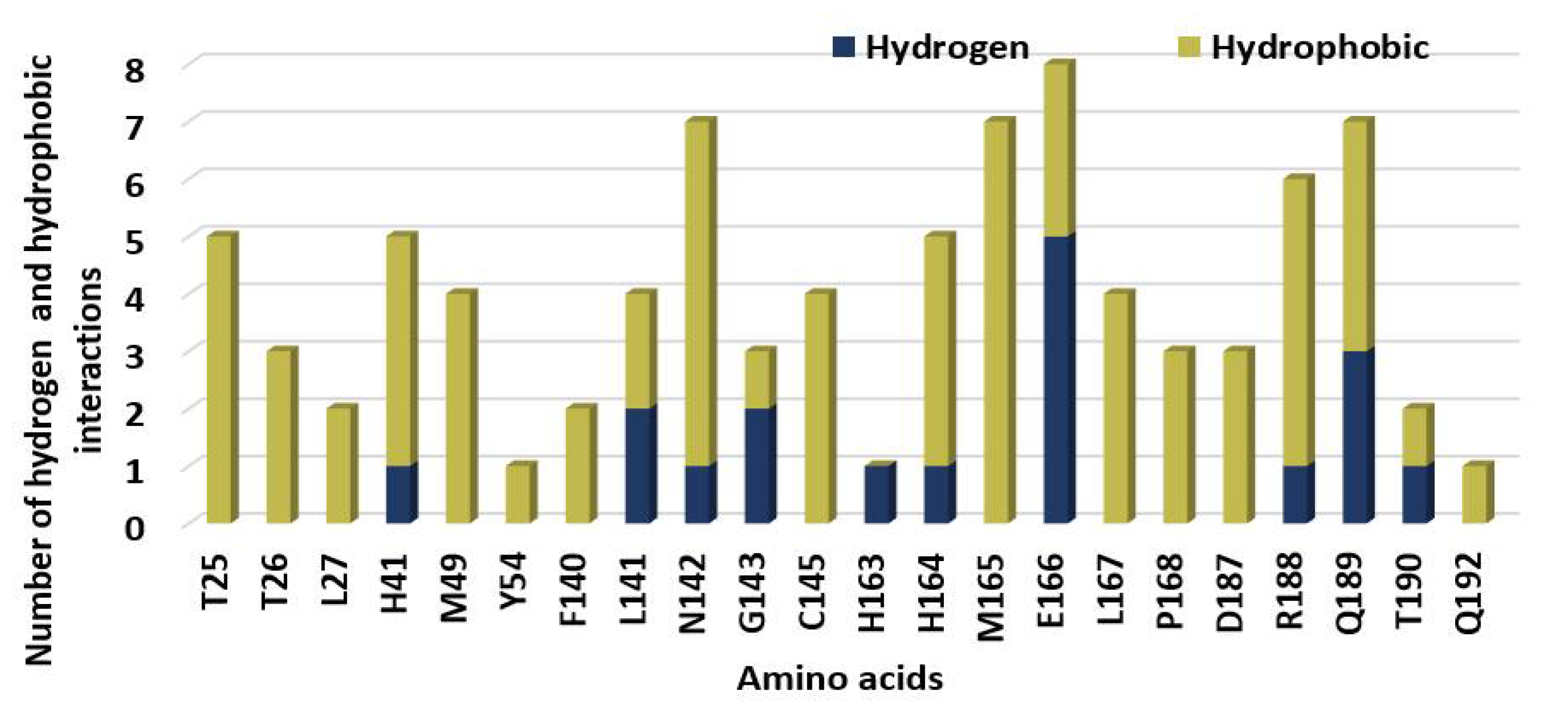
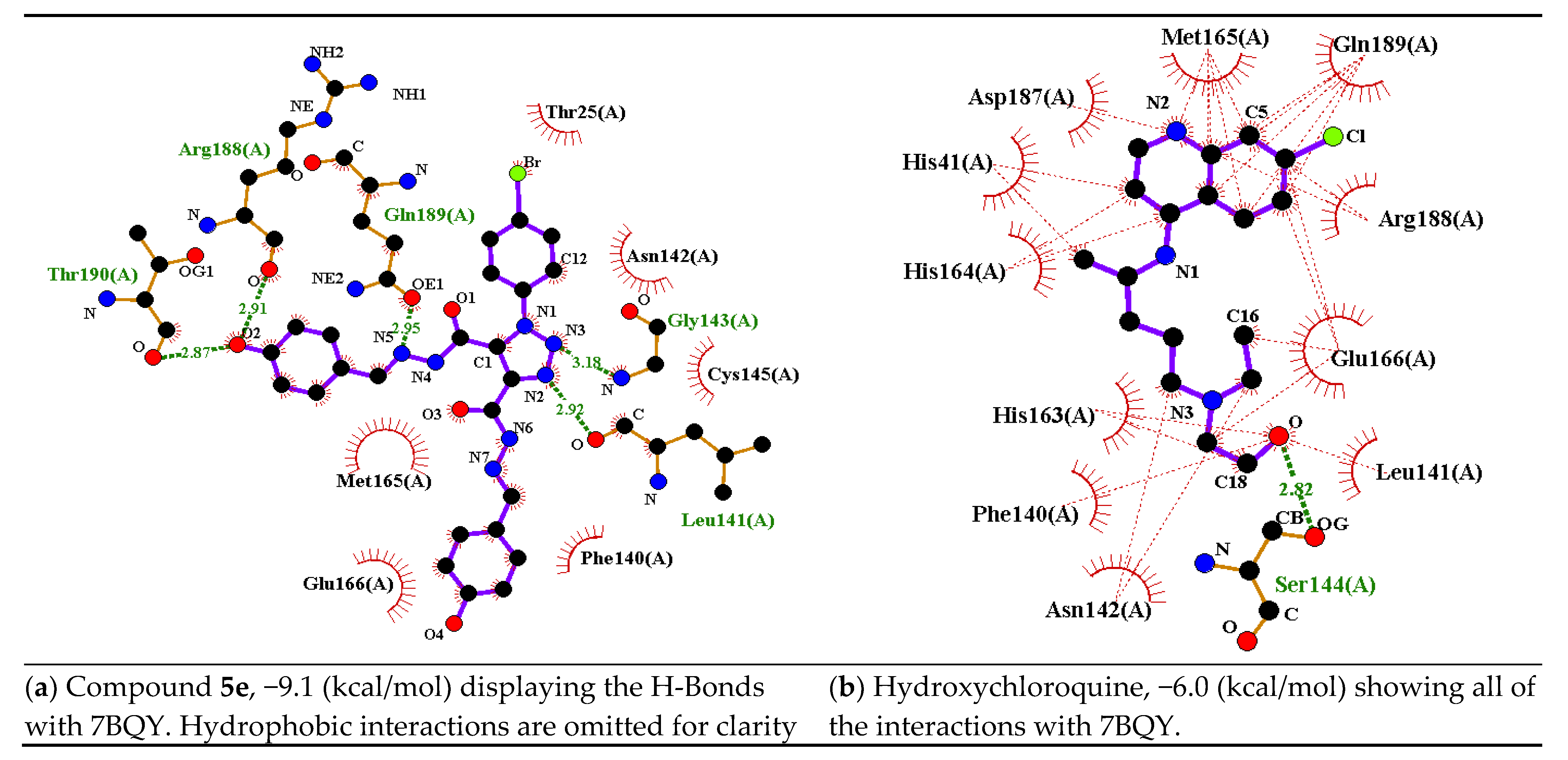
2.2. Docking Study
In Silico Cytotoxic Effect on Human Cancer Cell Lines
2.3. Quantitative Structure-Activity Relationships (QSAR)
3. Experimental
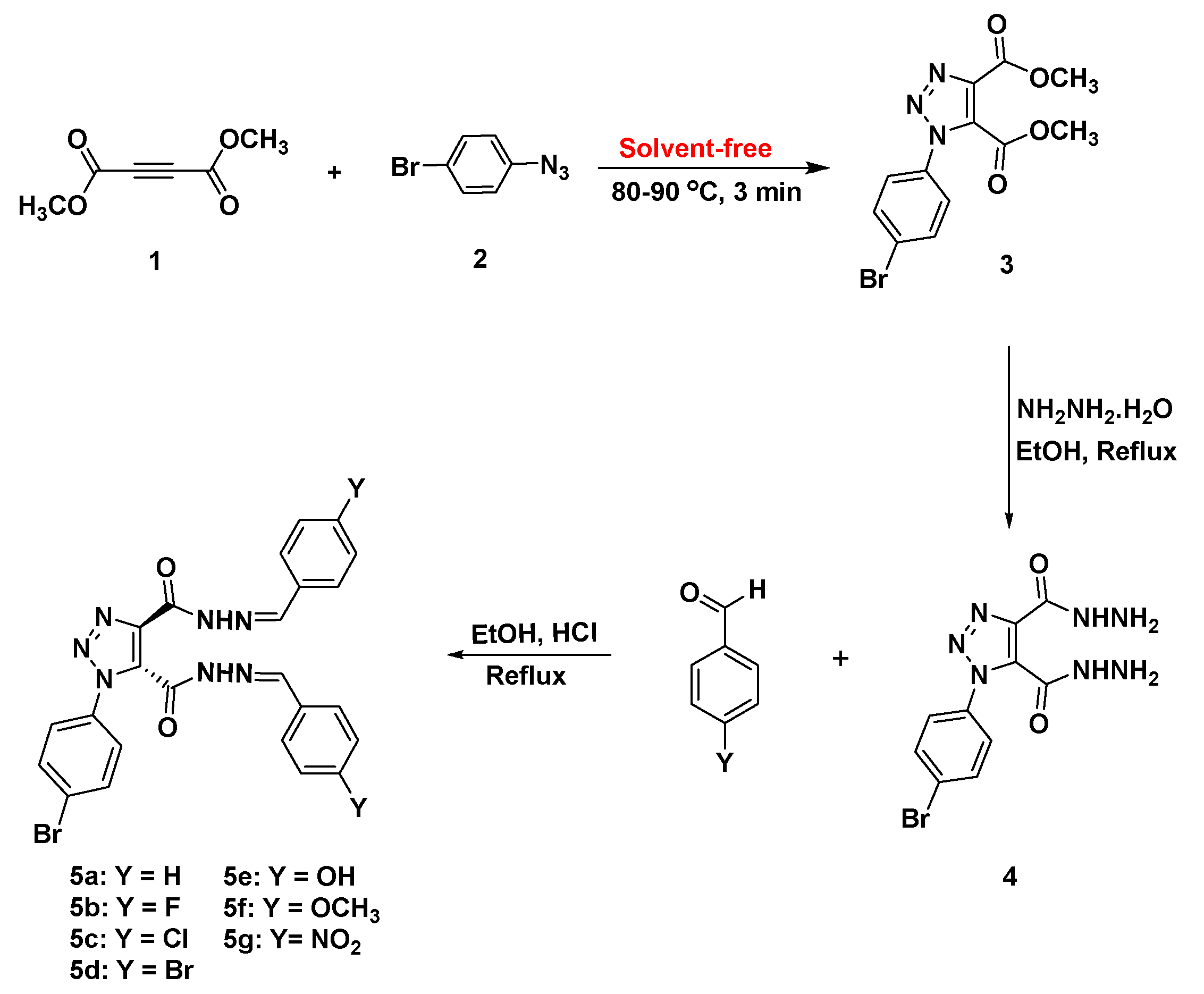
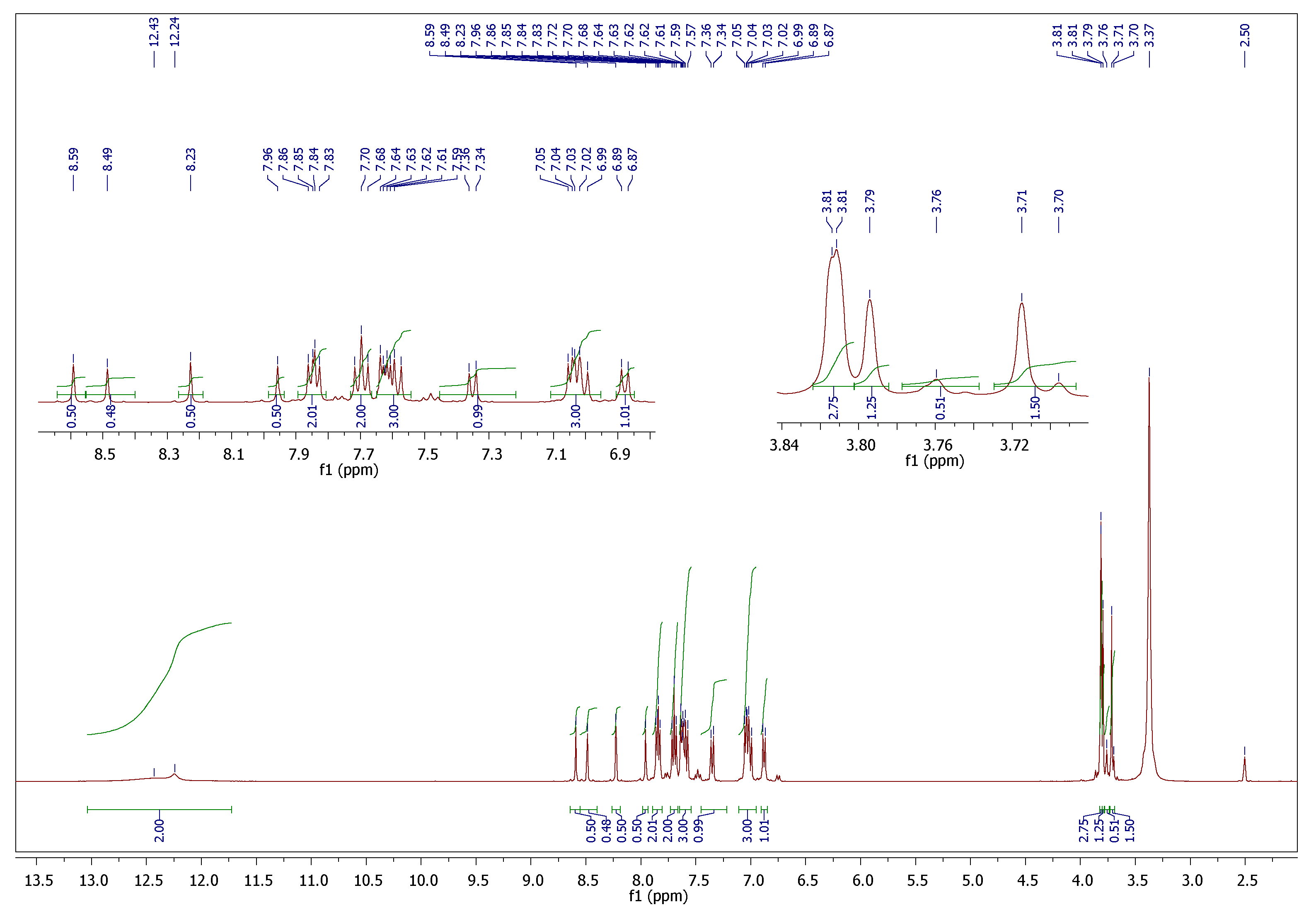
3.1. Synthesis
3.2. Docking in Silico Studies
3.3. In Silico Cytotoxic Effect on Human Cancer Cell Lines
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Narasimharao, K. Design, Spectroscopic Characterization, Electrical Conductivity and Molecular Modelling Studies of Biologically Puissant Co(II) and Ni(II) Complexes of N,N’-bis(furan-2-ylmethyl)benzene-1,2- dicarboxamide. Int. J. Electrochem. Sci. 2016, 11, 7282–7307. [Google Scholar] [CrossRef]
- Sharma, O.P.; Bhat, T.K. DPPH antioxidant assay revisited. Food Chem. 2009, 113, 1202–1205. [Google Scholar] [CrossRef]
- Florindo, H.F.; Kleiner, R.; Vaskovich-Koubi, D.; Acúrcio, R.C.; Carreira, B.; Yeini, E.; Tiram, G.; Liubomirski, Y.; Satchi-Fainaro, R. Immune-mediated approaches against COVID-19. Nat. Nanotechnol. 2020, 15, 630–645. [Google Scholar] [CrossRef]
- Asai, A.; Konno, M.; Ozaki, M.; Otsuka, C.; Vecchione, A.; Arai, T.; Kitagawa, T.; Ofusa, K.; Yabumoto, M.; Hirotsu, T.; et al. COVID-19 drug discovery using intensive approaches. Int. J. Mol. Sci. 2020, 21, 2839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. CoVID-19 Strategy Up Date; World Health Organization: Geneva, Switzerland, 2020; Volume 3, p. 18. [Google Scholar]
- Chaturvedi, D.; Kamboj, M. Role of Schiff Base in Drug Discovery Research. Chem. Sci. J. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Schiff, H. Mittheilungenaus dem Universitätslaboratorium in Pisa: Eine neueReiheorganischerBasen. Justus Liebigs Ann. Chem. 1864, 131, 118–119. [Google Scholar] [CrossRef] [Green Version]
- Bayrak, H.; Demirbas, A.; Karaoglu, S.A.; Demirbas, N. Synthesis of some new 1,2,4-triazoles, their Mannich and Schiff bases and evaluation of their antimicrobial activities. Eur. J. Med. Chem. 2009, 44, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- El-Naggar, M.; Abd El-All, A.S.; El-Naem, S.I.A.; Abdalla, M.M.; Rashdan, H.R.M. New Potent 5α- Reductase and Aromatase Inhibitors Derived from 1,2,3-Triazole Derivative. Molecules 2020, 25, 672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandile, N.G.; Mohamed, M.I.; Ismaeel, H.M. Synthesis of new Schiff bases bearing 1,2,4-triazole, thiazolidine and chloroazetidine moieties and their pharmacological evaluation. J. Enzyme Inhib. Med. Chem. 2017, 32, 119–129. [Google Scholar] [CrossRef]
- Khalaj, A.; Rastegi, H.R.; Jorjani, M. Synthesis and muscle relaxant activity of two analogues of dantrolene sodium in mice. Pharm. Pharmacol. Commun. 1998, 4, 477–479. [Google Scholar] [CrossRef]
- Sidorov, N.G.; Kravchenko, A.D.; Poddubikov, A.V.; Arzumanian, V.G. Synthesis and study of the antimicrobial activity of nifuroxazide derivatives. Microbiol. Indep. Res. J. 2019, 6, 10–17. [Google Scholar] [CrossRef]
- Coxon, G.D.; Craig, D.; Corrales, R.M.; Vialla, E.; Gannoun-Zaki, L.; Kremer, L. Synthesis, Antitubercular Activity and Mechanism of Resistance of Highly Effective Thiacetazone Analogues. PLoS ONE 2013, 8, e53162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alafeefy, A.M.; Bakht, M.A.; Ganaie, M.A.; Ansarie, M.N.; El-Sayed, N.N.; Awaad, A.S. Synthesis, analgesic, anti-inflammatory and anti-ulcerogenic activities of certain novel Schiff’s bases as fenamate isosteres. Bioorganic Med. Chem. Lett. 2015, 25, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Xu, F.Z.; Zhu, Y.Y.; Song, B.; Luo, D.; Yu, G.; Chen, S.; Xue, W.; Wu, J. Pyrazolo [3,4-d]pyrimidine derivatives containing a Schiff base moiety as potential antiviral agents. Bioorganic Med. Chem. Lett. 2018, 28, 2979–2984. [Google Scholar] [CrossRef]
- Bonandi, E.; Christodoulou, M.S.; Fumagalli, G.; Perdicchia, D.; Rastelli, G.; Passarella, D. The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov. Today 2017, 22, 1572–1581. [Google Scholar] [CrossRef]
- Li, H.; Aneja, R.; Chaiken, I. Click Chemistry in Peptide-Based Drug Design. Molecules 2013, 18, 9797–9817. [Google Scholar] [CrossRef] [Green Version]
- Angell, Y.L.; Burgess, K. Peptidomimetics via copper-catalyzed azide–alkyne cycloadditions. Chem. Soc. Rev. 2007, 36, 1674–1689. [Google Scholar] [CrossRef] [PubMed]
- Agalave, S.G.; Maujan, S.R.; Pore, V.S. Click chemistry: 1,2,3-triazoles as pharmacophores. Chem. Asian J. 2011, 6, 2696–2718. [Google Scholar] [CrossRef]
- Muller, T.; Bräse, S. Click Chemistry Finds Its Way into Covalent Porous Organic Materials. Angew. Chemie Int. Ed. 2011, 50, 11844–11845. [Google Scholar] [CrossRef] [PubMed]
- Smit, F.J.; Seldon, R.; Aucamp, J.; Jordaan, A.; Warner, D.F.; N’Da, D.D. Synthesis and antimycobacterial activity of disubstituted benzyltriazoles. Med. Chem. Res. 2019, 28, 2279–2293. [Google Scholar] [CrossRef] [Green Version]
- Aziz Ali, A.; Gogoi, D.; Chaliha, A.K.; Buragohain, A.K.; Trivedi, P.; Saikia, P.J.; Gehlot, P.S.; Kumar, A.; Chaturvedi, V.; Sarma, D. Synthesis and biological evaluation of novel 1,2,3-triazole derivatives as anti-tubercular agents. Bioorganic Med. Chem. Lett. 2017, 27, 3698–3703. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xu, Z.; Gao, C.; Ren, Q.C.; Chang, L.; Lv, Z.S.; Feng, L.S. Triazole derivatives and their anti-tubercular activity. Eur. J. Med. Chem. 2017, 138, 501–513. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, P.; Xuan, L.-N.; Fu, X.-Y.; Jing, F.; Li, S.; Liu, Y.-M.; Chen, B.-Q. Synthesis and antitumor activities of novel hybrid molecules containing 1,3,4-oxadiazole and 1,3,4-thiadiazole bearing Schiff base moiety. Bioorganic Med. Chem. Lett. 2014, 24, 5154–5156. [Google Scholar] [CrossRef]
- Aouad, M. Synthesis, Characterization and Antimicrobial Evaluation of Some New Schiff, Mannich and Acetylenic Mannich Bases Incorporating a 1,2,4-Triazole Nucleus. Molecules 2014, 19, 18897–18910. [Google Scholar] [CrossRef] [Green Version]
- Aouad, M.R. Efficient eco-friendly solvent-free click synthesis and antimicrobial evaluation of new fluorinated 1,2,3-triazoles and their conversion into schiff bases. J. Braz. Chem. Soc. 2015, 26, 2105–2115. [Google Scholar] [CrossRef]
- Rezki, N.; Al-Yahyawi, A.; Bardaweel, S.; Al-Blewi, F.; Aouad, M. Synthesis of Novel 2,5-Disubstituted-1,3,4-thiadiazoles Clubbed 1,2,4-Triazole, 1,3,4-Thiadiazole, 1,3,4-Oxadiazole and/or Schiff Base as Potential Antimicrobial and Antiproliferative Agents. Molecules 2015, 20, 16048–16067. [Google Scholar] [CrossRef] [Green Version]
- Almehmadi, M.A.; Aljuhani, A.; Alraqa, S.Y.; Ali, I.; Rezki, N.; Aouad, M.R.; Hagar, M. Design, synthesis, DNA binding, modeling, anticancer studies and DFT calculations of Schiff bases tethering benzothiazole-1,2,3-triazole conjugates. J. Mol. Struct. 2021, 1225, 129148. [Google Scholar] [CrossRef]
- Tanoli, S.T.; Ramzan, M.; Hassan, A.; Sadiq, A.; Jan, M.S.; Khan, F.A.; Ullah, F.; Ahmad, H.; Bibi, M.; Mahmood, T.; et al. Design, synthesis and bioevaluation of tricyclic fused ring system as dual binding site acetylcholinesterase inhibitors. Int. J. Mol. Sci. 2020, 83, 1–25. [Google Scholar] [CrossRef]
- Rezki, N.; Al-Sodies, S.A.; Aouad, M.R.; Bardaweel, S.; Messali, M.; El Ashry, E.; Ashry, E.S.H.E. An eco-friendly ultrasound-assisted synthesis of novel fluorinated pyridinium salts-based hydrazones and antimicrobial and antitumor screening. Int. J. Mol. Sci. 2016, 17, 766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aouad, M.R.; Messali, M.; Rezki, N.; Ali, A.A.S.; Lesimple, A. Synthesis and characterization of some novel 1,2,4-triazoles, 1,3,4-thiadiazoles and Schiff bases incorporating imidazole moiety as potential antimicrobial agents. Acta Pharm. 2015, 65, 117–132. [Google Scholar] [CrossRef] [Green Version]
- D’yakonov, V.A.; Finkelshtein, E.S.; Ibragimov, A.G. Dzhemilev reaction for the synthesis of spiro[3.3]heptane and spiro[3.4]octanes. Tetrahedron Lett. 2007, 48, 8583–8586. [Google Scholar] [CrossRef]
- Dheer, D.; Singh, V.; Shankar, R. Medicinal attributes of 1,2,3-triazoles: Current developments. Bioorganic Chem. 2017, 71, 30–54. [Google Scholar] [CrossRef]
- Wasilenko, W.J.; Palad, A.J.; Somers, K.D.; Blackmore, P.F.; Kohn, E.C.; Rhim, J.S.; Wright, G.L.; Schellhammer, P.F. Effects of the calcium influx inhibitor carboxyamido-triazole on the proliferation and invasiveness of human prostate tumor cell lines. Int. J. Cancer 1996, 68, 259–264. [Google Scholar] [CrossRef]
- Allouche, A. Software News and Updates Gabedit—A Graphical User Interface for Computational Chemistry Softwares. J. Comput. Chem. 2012, 32, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kadyan, K.; Duhan, M.; Sindhu, J.; Singh, V.; Saharan, B.S. Design, synthesis, conformational and molecular docking study of some novel acyl hydrazone based molecular hybrids as antimalarial and antimicrobial agents. Chem. Cent. J. 2017, 11, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patorski, P.; Wyrzykiewicz, E.; Bartkowiak, G. Synthesis and conformational assignment of N-(E)- stilbenyloxymethylenecarbonyl-substituted hydrazones of acetone and o-(m-and p-) chloro- (nitro-) benzaldehydes by means of 1H and 13C NMR spectroscopy. J. Spectrosc. 2013, 1, 197475. [Google Scholar] [CrossRef] [Green Version]
- Wyrzykiewicz, E.; Prukała, D. New isomeric N -substituted hydrazones of 2-, 3- and 4-pyridinecarboxaldehydes. J. Heterocycl. Chem. 1998, 35, 381–387. [Google Scholar] [CrossRef]
- Palla, G.; Predieri, G.; Domiano, P.; Vignali, C.; Turner, W. Conformational behaviour and E/Z isomerization of N-acyl and N-aroylhydrazones. Tetrahedron 1986, 42, 3649–3654. [Google Scholar] [CrossRef]
- Chauhan, N. Possible Drug Candidates for COVID-19 Possible Drug Candidates for COVID-19. ChemRxiv 2020, 1, 1–16. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. PyMOL Reference Guide; Delano Science: San Carlos, CA, USA, 2004. [Google Scholar]
- Shawon, J.; Khan, A.M.; Rahman, A.; Hoque, M.M.; Khan, M.A.K.; Sarwar, M.G.; Halim, M.A. Molecular Recognition of Azelaic Acid and Related Molecules with DNA Polymerase I Investigated by Molecular Modeling Calculations. Interdiscip. Sci. Comput. Life Sci. 2018, 10, 525–537. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. Ligplot: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Alsafi, M.A.; Hughes, D.L.; Said, M.A. First COVID-19 molecular docking with a chalcone-based compound: Synthesis, single-crystal structure and Hirshfeld surface analysis study. Acta Crystallogr. Sect. C Struct. Chem. 2020, 76, 1043–1050. [Google Scholar] [CrossRef]
- Wang, F.; Chen, C.; Tan, W.; Yang, K.; Yang, H. Structure of Main Protease from Human Coronavirus NL63: Insights for Wide Spectrum Anti-Coronavirus Drug Design. Sci. Rep. 2016, 6, 22677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liming, Y.; Yang, C.; Rao, Z.; Ren, Z.; Yan, L.; Zhang, N.; Guo, Y.; Yang, C.; Lou, Z.; Rao, Z. The newly emerged SARS-Like coronavirus HCoV-EMC also has an “Achilles’’ heel: Current effective inhibitor targeting a 3C-like protease. Protein Cell 2013, 4, 248–250. [Google Scholar] [CrossRef]
- Xue, X.; Yu, H.; Yang, H.; Xue, F.; Wu, Z.; Shen, W.; Li, J.; Zhou, Z.; Ding, Y.; Zhao, Q.; et al. Structures of Two Coronavirus Main Proteases: Implications for Substrate Binding and Antiviral Drug Design Downloaded from. J. Virol. 2008, 82, 2515–2527. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yang, M.; Ding, Y.; Liu, Y.; Lou, Z.; Zhou, Z.; Sun, L.; Mo, L.; Ye, S.; Pang, H.; et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [Google Scholar] [CrossRef] [Green Version]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef] [PubMed]
- Bursulaya, B.D.; Totrov, M.; Abagyan, R.; Brooks, C.L. Comparative study of several algorithms for flexible ligand docking. J. Comput. Aided. Mol. Des. 2003, 17, 755–763. [Google Scholar] [CrossRef] [PubMed]
- De Magalhães, C.S.; Barbosa, H.J.C.C.; Dardenne, L.E. A Genetic Algorithm for the Ligand-Protein Docking Problem. Genet. Mol. Biol. 2004, 27, 605–610. [Google Scholar] [CrossRef] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15-Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A.; Sterling, T.; Irwin, J.J. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020, 39, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, D.C.; Ji, H.F. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Med. Infect. Dis. 2020, 35, 101646. [Google Scholar] [CrossRef]
- Wang, Q.; Zhou, Y.; Rychahou, P.; Fan, T.W.M.; Lane, A.N.; Weiss, H.L.; Evers, B.M. Ketogenesis contributes to intestinal cell differentiation. Cell Death Differ. 2017, 24, 458–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Schneider, T.D.; Stephens, R.M. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 1990, 18, 6097–6100. [Google Scholar] [CrossRef]
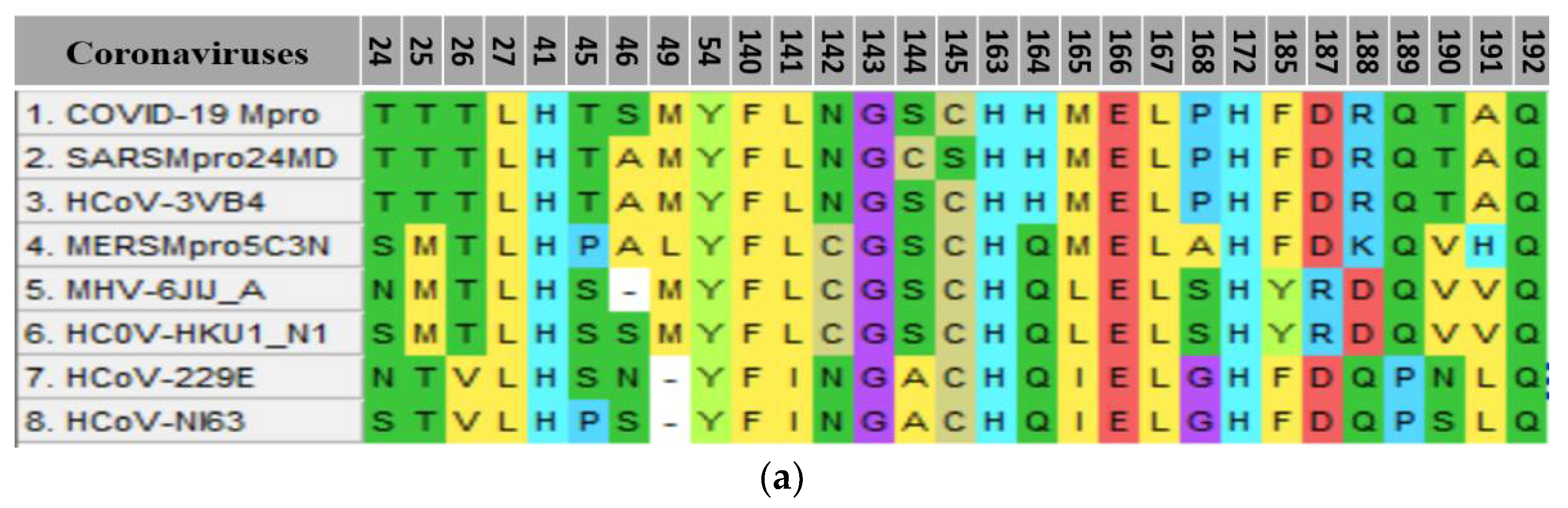
- Ortega, J.T.; Serrano, M.L.; Pujol, F.H.; Rangel, H.R. Unrevealing sequence and structural features of novel coronavirus using in silico approaches: The main protease as molecular target. EXCLI J. 2020, 19, 400–409. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 5c Cis-Cis | 5c Cis-Trans | 5c Trans-Trans |
|---|---|---|---|
| Ecorr | 0.366677 | 0.367067 | 0.367365 |
| ZPVE | −4952.400554 | −4952.406195 | −4952.414964 |
| Etot | −4952.368837 | −4952.374717 | −4952.383537 |
| H | −4952.367893 | −4952.373773 | −4952.382593 |
| G | −4952.472308 | −4952.477581 | −4952.486093 |
| ΔE in kcal/mol | 9.26 | 5.56 | 0.00 |
| Parameter | 5a Trans-Trans | 5b Trans-Trans | 5c Trans-Trans | 5d Trans-Trans | 5e Trans-Trans | 5f Trans-Trans | 5g Trans-Trans |
|---|---|---|---|---|---|---|---|
| H-bond length (Ǻ) | 1.73097 | 1.72869 | 1.72583 | 1.72985 | 1.72905 | 1.72551 | 1.72658 |
| Comp. No. | Dihedral Angles, ϕ | No. of Flexible Bonds | Binding Affinities kcal/mol | 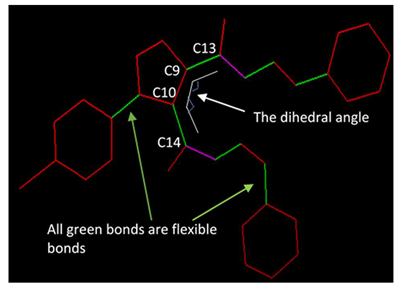 5a is a representative example to show the dihedral angles. Bonds in green are flexible bonds. |
| 5a | 2.4 | 7 | −8.7 | |
| 5b | 0.3 | 8 | −8.5 | |
| 5c | 7.0 | 7 | −7.3 | |
| 5d | 4.1 | 7 | −7.6 | |
| 5e | 7.2 | 7 | −9.1 | |
| 5f | 5.7 | 10 | −8.0 | |
| 5g | 6.3 | 10 | −8.5 |
| Biological Activities on Tumor Cell Line. | 5a | 5b | 5c | 5d | 5e | 5f | 5g | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | |
| HMGCS2 expression enhancer | 0.89 | 0.003 | 0.888 | 0.003 | 0.88 | 0.003 | 0.892 | 0.003 | 0.864 | 0.003 | 0.856 | 0.004 | 0.892 | 0.003 |
| Antimycobacterial | 0.79 | 0.004 | 0.787 | 0.004 | 0.783 | 0.004 | 0.789 | 0.004 | 0.798 | 0.004 | 0.791 | 0.004 | 0.859 | 0.003 |
| PfA-M1 aminopeptidase inhibitor | 0.73 | 0.003 | 0.729 | 0.003 | 0.71 | 0.003 | 0.736 | 0.003 | 0.692 | 0.004 | 0.666 | 0.004 | 0.721 | 0.003 |
| Antituberculosic | 0.7 | 0.004 | 0.703 | 0.004 | 0.681 | 0.004 | 0.702 | 0.004 | 0.725 | 0.004 | 0.682 | 0.004 | 0.799 | 0.003 |
| Age-related macular degeneration treatment | 0.61 | 0.003 | 0.605 | 0.003 | 0.611 | 0.003 | 0.608 | 0.003 | 0.593 | 0.004 | 0.646 | 0.003 | 0.633 | 0.003 |
| Orexin receptor 1 antagonist | 0.54 | 0.003 | 0.54 | 0.003 | 0.503 | 0.004 | 0.532 | 0.004 | 0 | 0 | 0.57 | 0.003 | 0.457 | 0.004 |
| Mcl-1 antagonist | 0.54 | 0.007 | 0.541 | 0.007 | 0.484 | 0.009 | 0.558 | 0.006 | 0.499 | 0.008 | 0.465 | 0.009 | 0.602 | 0.005 |
| Biological Activities on Non-Tumor Cell Line. | 5a | 5b | 5c | 5d | 5e | 5f | 5g | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | |
| Neutrophilic dermatosis (Sweet’s syndrome) | 0.564 | 0.101 | 0.564 | 0.101 | 0.676 | 0.06 | 0.575 | 0.097 | 0.306 | 0.265 | 0 | 0 | 0.559 | 0.103 |
| Nail discoloration | 0.355 | 0.211 | 0.355 | 0.211 | 0 | 0 | 0.314 | 0.105 | 0 | 0 | 0 | 0 | 0 | 0 |
| Adrenal cortex hypoplasia | 0 | 0 | 0 | 0 | 0.316 | 0.129 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Multiple organ failure | 0 | 0 | 0 | 0 | 0.342 | 0.194 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Acneiform eruption | 0 | 0 | 0 | 0 | 0 | 0 | 0.314 | 0.105 | 0 | 0 | 0 | 0 | 0 | 0 |
| Gastrointestinal hemorrhage | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.385 | 0.16 | 0 | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Said, M.A.; Khan, D.J.O.; Al-blewi, F.F.; Al-Kaff, N.S.; Ali, A.A.; Rezki, N.; Aouad, M.R.; Hagar, M. New 1,2,3-Triazole Scaffold Schiff Bases as Potential Anti-COVID-19: Design, Synthesis, DFT-Molecular Docking, and Cytotoxicity Aspects. Vaccines 2021, 9, 1012. https://doi.org/10.3390/vaccines9091012
Said MA, Khan DJO, Al-blewi FF, Al-Kaff NS, Ali AA, Rezki N, Aouad MR, Hagar M. New 1,2,3-Triazole Scaffold Schiff Bases as Potential Anti-COVID-19: Design, Synthesis, DFT-Molecular Docking, and Cytotoxicity Aspects. Vaccines. 2021; 9(9):1012. https://doi.org/10.3390/vaccines9091012
Chicago/Turabian StyleSaid, Musa A., Daoud J. O. Khan, Fawzia F. Al-blewi, Nadia S. Al-Kaff, Adeeb A. Ali, Nadjet Rezki, Mohamed Reda Aouad, and Mohamed Hagar. 2021. "New 1,2,3-Triazole Scaffold Schiff Bases as Potential Anti-COVID-19: Design, Synthesis, DFT-Molecular Docking, and Cytotoxicity Aspects" Vaccines 9, no. 9: 1012. https://doi.org/10.3390/vaccines9091012
APA StyleSaid, M. A., Khan, D. J. O., Al-blewi, F. F., Al-Kaff, N. S., Ali, A. A., Rezki, N., Aouad, M. R., & Hagar, M. (2021). New 1,2,3-Triazole Scaffold Schiff Bases as Potential Anti-COVID-19: Design, Synthesis, DFT-Molecular Docking, and Cytotoxicity Aspects. Vaccines, 9(9), 1012. https://doi.org/10.3390/vaccines9091012