
Virus-Induced CD8+ T-Cell Immunity and Its Exploitation to Contain the SARS-CoV-2 Pandemic


Abstract
:1. Introduction
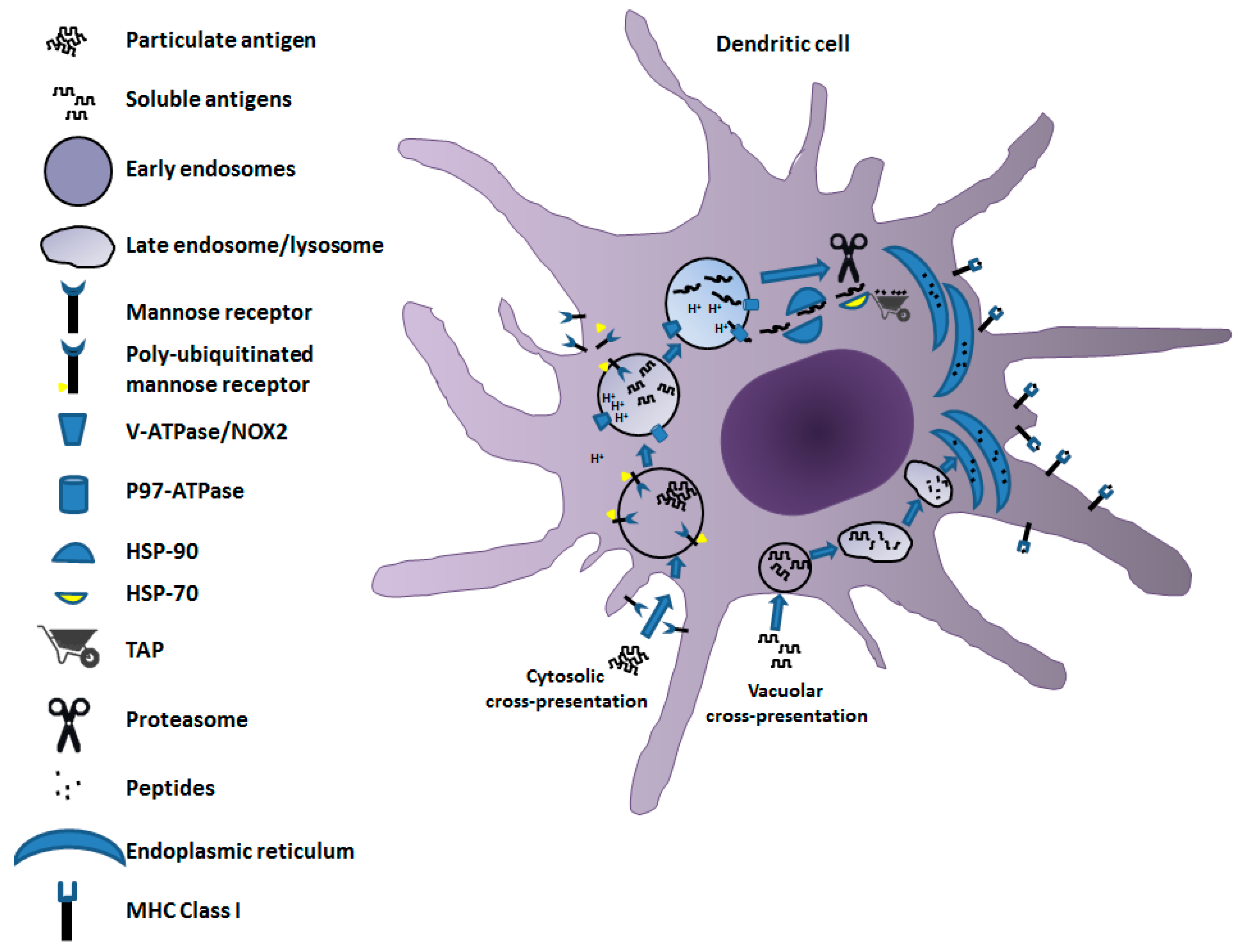
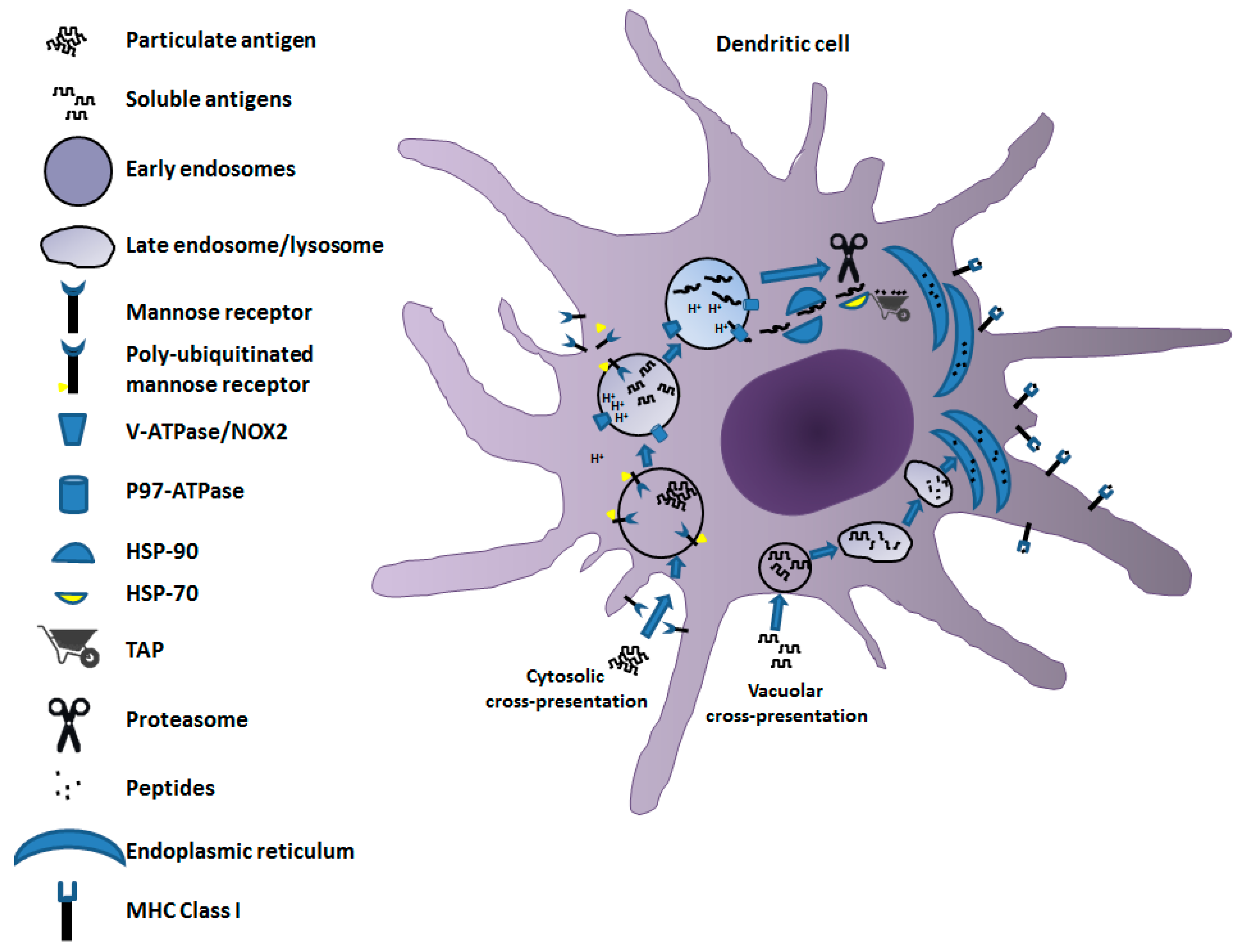
2. Mechanisms of Cross-Presentation
3. The Impact of CD8+ T-Cell Immunity in Viral Diseases
4. The CD8+ T-Cell Immune Response in SARS-CoV and SARS-CoV-2 Infections
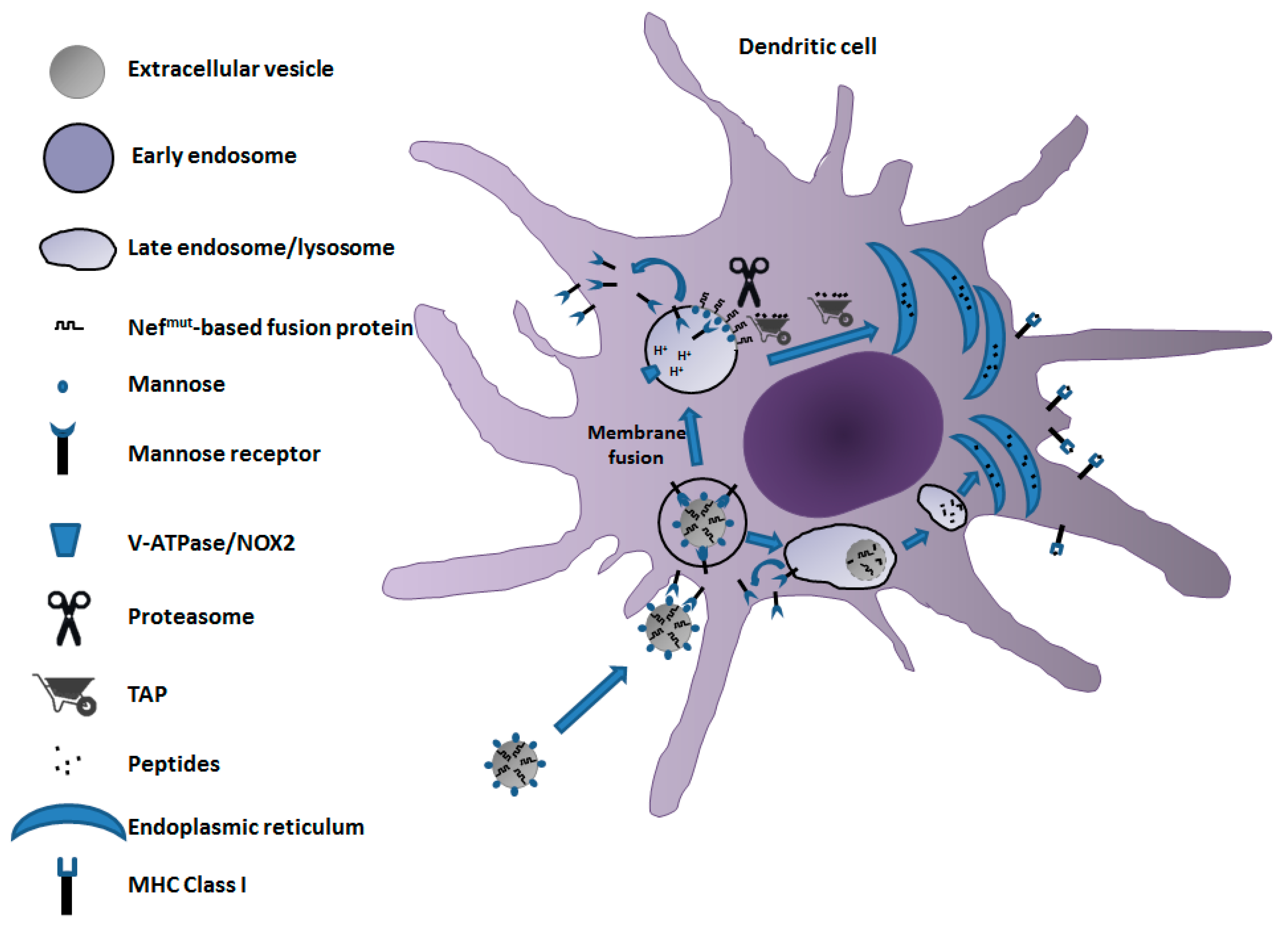
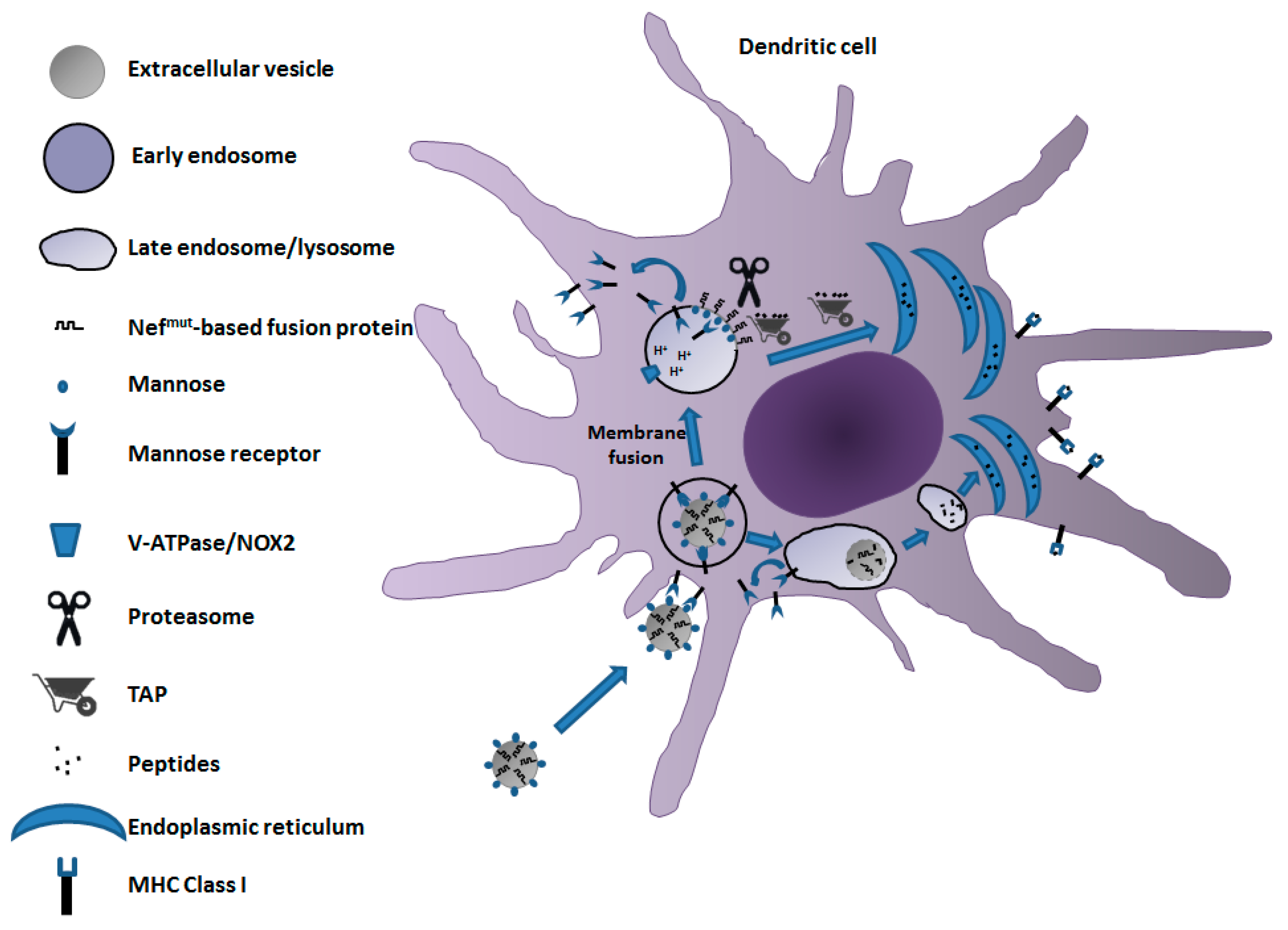
5. A Candidate CD8+ T-Cell-Based Vaccine to Fight COVID-19
6. Conclusions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Bevan, M.J. Cross-Priming for a Secondary Cytotoxic Response to Minor H Antigens with H-2 Congenic Cells Which Do Not Cross-React in the Cytotoxic Assay. J. Exp. Med. 1976, 143, 1283–1288. [Google Scholar] [CrossRef] [Green Version]
- Mallajosyula, V.; Ganjavi, C.; Chakraborty, S.; McSween, A.M.; Pavlovitch-Bedzyk, A.J.; Wilhelmy, J.; Nau, A.; Manohar, M.; Nadeau, K.C.; Davis, M.M. CD8+ T Cells Specific for Conserved Coronavirus Epitopes Correlate with Milder Disease in COVID-19 Patients. Sci. Immunol. 2021, 6, eabg5669. [Google Scholar] [CrossRef]
- Mackay, L.K.; Rahimpour, A.; Ma, J.Z.; Collins, N.; Stock, A.T.; Hafon, M.-L.; Vega-Ramos, J.; Lauzurica, P.; Mueller, S.N.; Stefanovic, T.; et al. The Developmental Pathway for CD103+CD8+ Tissue-Resident Memory T Cells of Skin. Nat. Immunol. 2013, 14, 1294–1301. [Google Scholar] [CrossRef]
- Slütter, B.; Van Braeckel-Budimir, N.; Abboud, G.; Varga, S.M.; Salek-Ardakani, S.; Harty, J.T. Dynamics of Influenza-Induced Lung-Resident Memory T Cells Underlie Waning Heterosubtypic Immunity. Sci. Immunol. 2017, 2, eaag2031. [Google Scholar] [CrossRef] [Green Version]
- Schenkel, J.M.; Fraser, K.A.; Vezys, V.; Masopust, D. Sensing and Alarm Function of Resident Memory CD8+ T Cells. Nat. Immunol. 2013, 14, 509–513. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, J.D.; Wick, M.J.; Roberts, R.L.; Findlay, K.; Normark, S.J.; Harding, C.V. Phagocytic Processing of Bacterial Antigens for Class I MHC Presentation to T Cells. Nature 1993, 361, 359–362. [Google Scholar] [CrossRef]
- Song, R.; Harding, C.V. Roles of Proteasomes, Transporter for Antigen Presentation (TAP), and Beta 2-Microglobulin in the Processing of Bacterial or Particulate Antigens via an Alternate Class I MHC Processing Pathway. J. Immunol. 1996, 156, 4182–4190. [Google Scholar]
- Shen, L.; Sigal, L.J.; Boes, M.; Rock, K.L. Important Role of Cathepsin S in Generating Peptides for TAP-Independent MHC Class I Crosspresentation in Vivo. Immunity 2004, 21, 155–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, C.V.; Song, R. Phagocytic Processing of Exogenous Particulate Antigens by Macrophages for Presentation by Class I MHC Molecules. J. Immunol. 1994, 153, 4925–4933. [Google Scholar] [PubMed]
- Savina, A.; Jancic, C.; Hugues, S.; Guermonprez, P.; Vargas, P.; Moura, I.C.; Lennon-Duménil, A.-M.; Seabra, M.C.; Raposo, G.; Amigorena, S. NOX2 Controls Phagosomal PH to Regulate Antigen Processing during Crosspresentation by Dendritic Cells. Cell 2006, 126, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jancic, C.; Savina, A.; Wasmeier, C.; Tolmachova, T.; El-Benna, J.; Dang, P.M.-C.; Pascolo, S.; Gougerot-Pocidalo, M.-A.; Raposo, G.; Seabra, M.C.; et al. Rab27a Regulates Phagosomal PH and NADPH Oxidase Recruitment to Dendritic Cell Phagosomes. Nat. Cell Biol. 2007, 9, 367–378. [Google Scholar] [CrossRef]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. Function of the P97-Ufd1-Npl4 Complex in Retrotranslocation from the ER to the Cytosol: Dual Recognition of Nonubiquitinated Polypeptide Segments and Polyubiquitin Chains. J. Cell Biol. 2003, 162, 71–84. [Google Scholar] [CrossRef]
- Ackerman, A.L.; Giodini, A.; Cresswell, P. A Role for the Endoplasmic Reticulum Protein Retrotranslocation Machinery during Crosspresentation by Dendritic Cells. Immunity 2006, 25, 607–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ménager, J.; Ebstein, F.; Oger, R.; Hulin, P.; Nedellec, S.; Duverger, E.; Lehmann, A.; Kloetzel, P.-M.; Jotereau, F.; Guilloux, Y. Cross-Presentation of Synthetic Long Peptides by Human Dendritic Cells: A Process Dependent on ERAD Component P97/VCP but Not Sec61 and/or Derlin-1. PLoS ONE 2014, 9, e89897. [Google Scholar] [CrossRef] [PubMed]
- Zehner, M.; Chasan, A.I.; Schuette, V.; Embgenbroich, M.; Quast, T.; Kolanus, W.; Burgdorf, S. Mannose Receptor Polyubiquitination Regulates Endosomal Recruitment of P97 and Cytosolic Antigen Translocation for Cross-Presentation. Proc. Natl. Acad. Sci. USA 2011, 108, 9933–9938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giodini, A.; Cresswell, P. Hsp90-Mediated Cytosolic Refolding of Exogenous Proteins Internalized by Dendritic Cells. EMBO J. 2008, 27, 201–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, T.; Kato, Y.; Kajiwara, C.; Mizukami, S.; Ishige, I.; Ichiyanagi, T.; Hikida, M.; Wang, J.-Y.; Udono, H. Heat Shock Protein 90 (HSP90) Contributes to Cytosolic Translocation of Extracellular Antigen for Cross-Presentation by Dendritic Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16363–16368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, Y.; Kajiwara, C.; Ishige, I.; Mizukami, S.; Yamazaki, C.; Eikawa, S.; Kakimi, K.; Udono, H. HSP70 and HSP90 Differentially Regulate Translocation of Extracellular Antigen to the Cytosol for Cross-Presentation. Autoimmune Dis. 2012, 2012, 745962. [Google Scholar] [CrossRef] [PubMed]
- Olinger, G.G.; Bailey, M.A.; Dye, J.M.; Bakken, R.; Kuehne, A.; Kondig, J.; Wilson, J.; Hogan, R.J.; Hart, M.K. Protective Cytotoxic T-Cell Responses Induced by Venezuelan Equine Encephalitis Virus Replicons Expressing Ebola Virus Proteins. J. Virol. 2005, 79, 14189–14196. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.A.; Hart, M.K. Protection from Ebola Virus Mediated by Cytotoxic T Lymphocytes Specific for the Viral Nucleoprotein. J. Virol. 2001, 75, 2660–2664. [Google Scholar] [CrossRef] [Green Version]
- Sakabe, S.; Sullivan, B.M.; Hartnett, J.N.; Robles-Sikisaka, R.; Gangavarapu, K.; Cubitt, B.; Ware, B.C.; Kotliar, D.; Branco, L.M.; Goba, A.; et al. Analysis of CD8+ T Cell Response during the 2013-2016 Ebola Epidemic in West Africa. Proc. Natl. Acad. Sci. USA 2018, 115, E7578–E7586. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, B.; Diamond, M.S. Role of CD8+ T Cells in Control of West Nile Virus Infection. J. Virol. 2004, 78, 8312–8321. [Google Scholar] [CrossRef] [Green Version]
- Netland, J.; Bevan, M.J. CD8 and CD4 T Cells in West Nile Virus Immunity and Pathogenesis. Viruses 2013, 5, 2573–2584. [Google Scholar] [CrossRef] [PubMed]
- McMichael, A.J.; Gotch, F.M.; Noble, G.R.; Beare, P.A. Cytotoxic T-Cell Immunity to Influenza. N. Engl. J. Med. 1983, 309, 13–17. [Google Scholar] [CrossRef]
- Valkenburg, S.A.; Josephs, T.M.; Clemens, E.B.; Grant, E.J.; Nguyen, T.H.O.; Wang, G.C.; Price, D.A.; Miller, A.; Tong, S.Y.C.; Thomas, P.G.; et al. Molecular Basis for Universal HLA-A*0201-Restricted CD8+ T-Cell Immunity against Influenza Viruses. Proc. Natl. Acad. Sci. USA 2016, 113, 4440–4445. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Hu, Y.; Lee, Y.-T.; Bouchard, K.R.; Benechet, A.; Khanna, K.; Cauley, L.S. Lung-Resident Memory CD8 T Cells (TRM) Are Indispensable for Optimal Cross-Protection against Pulmonary Virus Infection. J. Leukoc. Biol 2014, 95, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Elong Ngono, A.; Regla-Nava, J.A.; Kim, K.; Gorman, M.J.; Diamond, M.S.; Shresta, S. Dengue Virus-Reactive CD8+ T Cells Mediate Cross-Protection against Subsequent Zika Virus Challenge. Nat. Commun. 2017, 8, 1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peiris, J.S.M.; Guan, Y.; Yuen, K.Y. Severe Acute Respiratory Syndrome. Nat. Med. 2004, 10 (Suppl. 12), S88–S97. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhao, J.; Perlman, S. T Cell Responses Are Required for Protection from Clinical Disease and for Virus Clearance in Severe Acute Respiratory Syndrome Coronavirus-Infected Mice. J. Virol. 2010, 84, 9318–9325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Channappanavar, R.; Fett, C.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Virus-Specific Memory CD8 T Cells Provide Substantial Protection from Lethal Severe Acute Respiratory Syndrome Coronavirus Infection. J. Virol. 2014, 88, 11034–11044. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.S.M.; Wu, A.; To, K.F.; Lee, N.; Lam, C.W.K.; Wong, C.K.; Chan, P.K.S.; Ng, M.H.L.; Yu, L.M.; Hui, D.S.; et al. Haematological Manifestations in Patients with Severe Acute Respiratory Syndrome: Retrospective Analysis. BMJ 2003, 326, 1358–1362. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Qiu, Z.; Zhang, L.; Han, Y.; He, W.; Liu, Z.; Ma, X.; Fan, H.; Lu, W.; Xie, J.; et al. Significant Changes of Peripheral T Lymphocyte Subsets in Patients with Severe Acute Respiratory Syndrome. J. Infect. Dis. 2004, 189, 648–651. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, T.; Hill, T.; Li, K.; Peters, C.J.; Tseng, C.-T.K. Severe Acute Respiratory Syndrome (SARS) Coronavirus-Induced Lung Epithelial Cytokines Exacerbate SARS Pathogenesis by Modulating Intrinsic Functions of Monocyte-Derived Macrophages and Dendritic Cells. J. Virol. 2009, 83, 3039–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Zhao, J.; Van Rooijen, N.; Perlman, S. Evasion by Stealth: Inefficient Immune Activation Underlies Poor T Cell Response and Severe Disease in SARS-CoV-Infected Mice. PLoS Pathog. 2009, 5, e1000636. [Google Scholar] [CrossRef]
- Wu, L.-P.; Wang, N.-C.; Chang, Y.-H.; Tian, X.-Y.; Na, D.-Y.; Zhang, L.-Y.; Zheng, L.; Lan, T.; Wang, L.-F.; Liang, G.-D. Duration of Antibody Responses after Severe Acute Respiratory Syndrome. Emerg. Infect. Dis. 2007, 13, 1562–1564. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Quan, Y.; Xin, Z.-T.; Wrammert, J.; Ma, M.-J.; Lv, H.; Wang, T.-B.; Yang, H.; Richardus, J.H.; Liu, W.; et al. Lack of Peripheral Memory B Cell Responses in Recovered Patients with Severe Acute Respiratory Syndrome: A Six-Year Follow-up Study. J. Immunol. 2011, 186, 7264–7268. [Google Scholar] [CrossRef] [Green Version]
- Le Bert, N.; Tan, A.T.; Kunasegaran, K.; Tham, C.Y.L.; Hafezi, M.; Chia, A.; Chng, M.H.Y.; Lin, M.; Tan, N.; Linster, M.; et al. SARS-CoV-2-Specific T Cell Immunity in Cases of COVID-19 and SARS, and Uninfected Controls. Nature 2020, 584, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Lai, X.; Sun, J.; Chen, Z.; Zhang, Z.; Dai, J.; Liu, D.; Li, Y.; Li, F.; Wang, Y.; et al. Mapping and Role of T Cell Response in SARS-CoV-2-Infected Mice. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.E.; Brasel, T.; Massey, C.; Herst, C.V.; Burkholz, S.; Lloyd, P.; Blankenberg, T.; Bey, T.M.; Carback, R.; Hodge, T.; et al. A Synthetic Peptide CTL Vaccine Targeting Nucleocapsid Confers Protection from SARS-CoV-2 Challenge in Rhesus Macaques. Vaccines 2021, 9, 520. [Google Scholar] [CrossRef]
- McMahan, K.; Yu, J.; Mercado, N.B.; Loos, C.; Tostanoski, L.H.; Chandrashekar, A.; Liu, J.; Peter, L.; Atyeo, C.; Zhu, A.; et al. Correlates of Protection against SARS-CoV-2 in Rhesus Macaques. Nature 2021, 590, 630–634. [Google Scholar] [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e15. [Google Scholar] [CrossRef]
- Schulien, I.; Kemming, J.; Oberhardt, V.; Wild, K.; Seidel, L.M.; Killmer, S.; Sagar, N.; Daul, F.; Salvat Lago, M.; Decker, A.; et al. Characterization of Pre-Existing and Induced SARS-CoV-2-Specific CD8+ T Cells. Nat. Med. 2021, 27, 78–85. [Google Scholar] [CrossRef]
- Rydyznski Moderbacher, C.; Ramirez, S.I.; Dan, J.M.; Grifoni, A.; Hastie, K.M.; Weiskopf, D.; Belanger, S.; Abbott, R.K.; Kim, C.; Choi, J.; et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell 2020, 183, 996–1012.e19. [Google Scholar] [CrossRef]
- Peng, Y.; Mentzer, A.J.; Liu, G.; Yao, X.; Yin, Z.; Dong, D.; Dejnirattisai, W.; Rostron, T.; Supasa, P.; Liu, C.; et al. Broad and Strong Memory CD4+ and CD8+ T Cells Induced by SARS-CoV-2 in UK Convalescent Individuals Following COVID-19. Nat. Immunol. 2020, 21, 1336–1345. [Google Scholar] [CrossRef]
- Nelde, A.; Bilich, T.; Heitmann, J.S.; Maringer, Y.; Salih, H.R.; Roerden, M.; Lübke, M.; Bauer, J.; Rieth, J.; Wacker, M.; et al. SARS-CoV-2-Derived Peptides Define Heterologous and COVID-19-Induced T Cell Recognition. Nat. Immunol. 2021, 22, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.T.; Linster, M.; Tan, C.W.; Le Bert, N.; Chia, W.N.; Kunasegaran, K.; Zhuang, Y.; Tham, C.Y.L.; Chia, A.; Smith, G.J.D.; et al. Early Induction of Functional SARS-CoV-2-Specific T Cells Associates with Rapid Viral Clearance and Mild Disease in COVID-19 Patients. Cell Rep. 2021, 34, 108728. [Google Scholar] [CrossRef] [PubMed]
- Westmeier, J.; Paniskaki, K.; Karaköse, Z.; Werner, T.; Sutter, K.; Dolff, S.; Overbeck, M.; Limmer, A.; Liu, J.; Zheng, X.; et al. Impaired Cytotoxic CD8+ T Cell Response in Elderly COVID-19 Patients. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Bange, E.M.; Han, N.A.; Wileyto, P.; Kim, J.Y.; Gouma, S.; Robinson, J.; Greenplate, A.R.; Hwee, M.A.; Porterfield, F.; Owoyemi, O.; et al. CD8+ T Cells Contribute to Survival in Patients with COVID-19 and Hematologic Cancer. Nat. Med. 2021. [Google Scholar] [CrossRef]
- Redd, A.D.; Nardin, A.; Kared, H.; Bloch, E.M.; Pekosz, A.; Laeyendecker, O.; Abel, B.; Fehlings, M.; Quinn, T.C.; Tobian, A.A. CD8+ T Cell Responses in COVID-19 Convalescent Individuals Target Conserved Epitopes from Multiple Prominent SARS-CoV-2 Circulating Variants. medRxiv 2021. [Google Scholar] [CrossRef]
- Tarke, A.; Sidney, J.; Methot, N.; Zhang, Y.; Dan, J.M.; Goodwin, B.; Rubiro, P.; Sutherland, A.; da Silva Antunes, R.; Frazier, A.; et al. Negligible Impact of SARS-CoV-2 Variants on CD4 + and CD8 + T Cell Reactivity in COVID-19 Exposed Donors and Vaccinees. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ferretti, A.P.; Kula, T.; Wang, Y.; Nguyen, D.M.V.; Weinheimer, A.; Dunlap, G.S.; Xu, Q.; Nabilsi, N.; Perullo, C.R.; Cristofaro, A.W.; et al. Unbiased Screens Show CD8+ T Cells of COVID-19 Patients Recognize Shared Epitopes in SARS-CoV-2 That Largely Reside Outside the Spike Protein. Immunity 2020, 53, 1095–1107.e3. [Google Scholar] [CrossRef]
- Kared, H.; Redd, A.D.; Bloch, E.M.; Bonny, T.S.; Sumatoh, H.; Kairi, F.; Carbajo, D.; Abel, B.; Newell, E.W.; Bettinotti, M.P.; et al. SARS-CoV-2-Specific CD8+ T Cell Responses in Convalescent COVID-19 Individuals. J. Clin. Investig. 2021, 131, 145476. [Google Scholar] [CrossRef]
- Gonzalez-Galarza, F.F.; McCabe, A.; Santos, E.J.M.D.; Jones, J.; Takeshita, L.; Ortega-Rivera, N.D.; Cid-Pavon, G.M.D.; Ramsbottom, K.; Ghattaoraya, G.; Alfirevic, A.; et al. Allele Frequency Net Database (AFND) 2020 Update: Gold-Standard Data Classification, Open Access Genotype Data and New Query Tools. Nucleic Acids Res. 2020, 48, D783–D788. [Google Scholar] [CrossRef]
- Borges da Silva, H. Navigating in Deep Waters: How Tissue Damage and Inflammation Shape Effector and Memory CD8+ T Cell Responses. Immunohorizons 2021, 5, 338–348. [Google Scholar] [CrossRef]
- Zhu, F.-C.; Guan, X.-H.; Li, Y.-H.; Huang, J.-Y.; Jiang, T.; Hou, L.-H.; Li, J.-X.; Yang, B.-F.; Wang, L.; Wang, W.-J.; et al. Immunogenicity and Safety of a Recombinant Adenovirus Type-5-Vectored COVID-19 Vaccine in Healthy Adults Aged 18 Years or Older: A Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet 2020, 396, 479–488. [Google Scholar] [CrossRef]
- Ewer, K.J.; Barrett, J.R.; Belij-Rammerstorfer, S.; Sharpe, H.; Makinson, R.; Morter, R.; Flaxman, A.; Wright, D.; Bellamy, D.; Bittaye, M.; et al. T Cell and Antibody Responses Induced by a Single Dose of ChAdOx1 NCoV-19 (AZD1222) Vaccine in a Phase 1/2 Clinical Trial. Nat. Med. 2021, 27, 270–278. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding Light on the Cell Biology of Extracellular Vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The Biology, Function, and Biomedical Applications of Exosomes. Science 2020, 367. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, L.; Federico, M. A Strategy of Antigen Incorporation into Exosomes: Comparing Cross-Presentation Levels of Antigens Delivered by Engineered Exosomes and by Lentiviral Virus-like Particles. Vaccine 2012, 30, 7229–7237. [Google Scholar] [CrossRef]
- Chiozzini, C.; Manfredi, F.; Arenaccio, C.; Ferrantelli, F.; Leone, P.; Federico, M. N-Terminal Fatty Acids of NEFMUT Are Required for the CD8+ T-Cell Immunogenicity of In Vivo Engineered Extracellular Vesicles. Vaccines 2020, 8, 243. [Google Scholar] [CrossRef]
- Di Bonito, P.; Chiozzini, C.; Arenaccio, C.; Anticoli, S.; Manfredi, F.; Olivetta, E.; Ferrantelli, F.; Falcone, E.; Ruggieri, A.; Federico, M. Antitumor HPV E7-Specific CTL Activity Elicited by in Vivo Engineered Exosomes Produced through DNA Inoculation. Int. J. Nanomed. 2017, 12, 4579–4591. [Google Scholar] [CrossRef] [Green Version]
- Chiozzini, C.; Manfredi, F.; Ferrantelli, F.; Leone, P.; Giovannelli, A.; Olivetta, E.; Federico, M. The C-Terminal Domain of Nefmut Is Dispensable for the CD8+ T Cell Immunogenicity of In Vivo Engineered Extracellular Vesicles. Vaccines 2021, 9, 373. [Google Scholar] [CrossRef]
- Anticoli, S.; Manfredi, F.; Chiozzini, C.; Arenaccio, C.; Olivetta, E.; Ferrantelli, F.; Capocefalo, A.; Falcone, E.; Ruggieri, A.; Federico, M. An Exosome-Based Vaccine Platform Imparts Cytotoxic T Lymphocyte Immunity Against Viral Antigens. Biotechnol. J. 2018, 13, e1700443. [Google Scholar] [CrossRef]
- Anticoli, S.; Aricò, E.; Arenaccio, C.; Manfredi, F.; Chiozzini, C.; Olivetta, E.; Ferrantelli, F.; Lattanzi, L.; D’Urso, M.T.; Proietti, E.; et al. Engineered Exosomes Emerging from Muscle Cells Break Immune Tolerance to HER2 in Transgenic Mice and Induce Antigen-Specific CTLs upon Challenge by Human Dendritic Cells. J. Mol. Med. 2018, 96, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.; Royo, F.; Aizpurua-Olaizola, O.; Pazos, R.; Boons, G.-J.; Reichardt, N.-C.; Falcon-Perez, J.M. Glycosylation of Extracellular Vesicles: Current Knowledge, Tools and Clinical Perspectives. J. Extracell Vesicles 2018, 7, 1442985. [Google Scholar] [CrossRef]
- Shimoda, A.; Sawada, S.-I.; Sasaki, Y.; Akiyoshi, K. Exosome Surface Glycans Reflect Osteogenic Differentiation of Mesenchymal Stem Cells: Profiling by an Evanescent Field Fluorescence-Assisted Lectin Array System. Sci. Rep. 2019, 9, 11497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, E.S.; Song, J.; Kang, Y.Y.; Mok, H. Mannose-Modified Serum Exosomes for the Elevated Uptake to Murine Dendritic Cells and Lymphatic Accumulation. Macromol. Biosci. 2019, 19, e1900042. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.E.; Drickamer, K. Mammalian Sugar-Binding Receptors: Known Functions and Unexplored Roles. FEBS J. 2019, 286, 1800–1814. [Google Scholar] [CrossRef]
- Martinez-Pomares, L. The Mannose Receptor. J. Leukoc. Biol. 2012, 92, 1177–1186. [Google Scholar] [CrossRef]
- Hu, Z.; Shi, X.; Yu, B.; Li, N.; Huang, Y.; He, Y. Structural Insights into the PH-Dependent Conformational Change and Collagen Recognition of the Human Mannose Receptor. Structure 2018, 26, 60–71.e3. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of Secretion and Uptake of Exosomes and Other Extracellular Vesicles for Cell-to-Cell Communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Joshi, B.S.; de Beer, M.A.; Giepmans, B.N.G.; Zuhorn, I.S. Endocytosis of Extracellular Vesicles and Release of Their Cargo from Endosomes. ACS Nano. 2020, 14, 4444–4455. [Google Scholar] [CrossRef] [Green Version]
- Ferrantelli, F.; Manfredi, F.; Chiozzini, C.; Leone, P.; Giovannelli, A.; Olivetta, E.; Federico, M. Long-Term Antitumor CD8+ T Cell Immunity Induced by Endogenously Engineered Extracellular Vesicles. Cancers 2021, 13, 2263. [Google Scholar] [CrossRef]
- Ferrantelli, F.; Chiozzini, C.; Manfredi, F.; Giovannelli, A.; Leone, P.; Federico, M. Simultaneous CD8+ T-Cell Immune Response against SARS-Cov-2 S, M, and N Induced by Endogenously Engineered Extracellular Vesicles in Both Spleen and Lungs. Vaccines 2021, 9, 240. [Google Scholar] [CrossRef]
- Takamura, S.; Kato, S.; Motozono, C.; Shimaoka, T.; Ueha, S.; Matsuo, K.; Miyauchi, K.; Masumoto, T.; Katsushima, A.; Nakayama, T.; et al. Interstitial-Resident Memory CD8+ T Cells Sustain Frontline Epithelial Memory in the Lung. J. Exp. Med. 2019, 216, 2736–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| MHC Class I Allele | Viral Protein | Epitope Sequence | % of Positivity and [Reference] |
|---|---|---|---|
| A*01:01 | ORF1ab | PTDNYITTY | 60% [51] |
| A*01:01 | ORF1ab | TTDPSFLGRY | 83% [45]; 80% [51] |
| A*01:01 | ORF1ab | NTCDGTTFTY | 60% [51] |
| A*01:01 | ORF1ab | CTDDNALAYY | 100% [51] |
| A*02:01 | ORF1ab | KLWAQCVQL | 88.9% [51] |
| A*02:01 | ORF1ab | ALWEIQQVV | 88.9% [51] |
| A*11:01 | ORF1ab | VTDTPKGPK | 60% [51] |
| A*24:02 | ORF1ab | VYIGDPAQL | 70% [45] |
| B*07:02 | ORF1ab | RPDTRYVL | 80% [51] |
| B*40:01 | ORF1ab | IEYPIIGDEL | 58% [45] |
| A*01:01 | S | LTDEMIAQY | 50% [45] |
| A*02:01 | S | YLQPRTFLL | 77.8% [51] |
| A*03:01 | S | KCYGVSPTK | 100% [51] |
| A*24:02 | S | QYIKWPWYI | 60% [45]; 60% [51] |
| A*01:01 | ORF3a | FTSDYYQLY | 100% [51] |
| A*02:01 | ORF3a | LLYDANYFL | 88.9% [51] |
| A*02:01 | ORF3a | ALSKGVHFV | 55% [45] |
| A*24:02 | ORF3a | VYFLQSINF | 70% [45]; 80% [51] |
| A*01:01 | M | ATSRTLSYY | 60% [51] |
| A*11:01 | M | ATSRTLSYYK | 60% [51] |
| B*40:01 | M | SELVIGAVIL | 50% [45] |
| C*07:01 | M | NRFLYIIKL | 50% [45] |
| A*01:01 | NP | FTSDYYQLY | 100% [52] |
| A*01:01 | NP | PTDNYITTY | 70% [52] |
| A*01:01 | NP | HTTDPSFLGRY | 100% [52] |
| A*02:01 | NP | YLQPRTFLL | 65% [52] |
| A*03:01 | NP | KTFPPTEPK | 64% [45]; 100% [51]; 75% [52] |
| A*11:01 | NP | KTFPPTEPK | 100% [51] |
| A*11:01 | NP | ATEGALNTPK | 82% [45] |
| A*24:02 | NP | NYNYLYRLF | 65% [52] |
| A*24:02 | NP | QYIKWPWYI | 75% [52] |
| B*07:02 | NP | RARSVSPKL | 75% [52] |
| B*07:02 | NP | SPRWYFYYL | 80% [51]; 100% [52] |
| B*40:01 | NP | MEVTPSGTWL | 75% [45] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Federico, M. Virus-Induced CD8+ T-Cell Immunity and Its Exploitation to Contain the SARS-CoV-2 Pandemic. Vaccines 2021, 9, 922. https://doi.org/10.3390/vaccines9080922
Federico M. Virus-Induced CD8+ T-Cell Immunity and Its Exploitation to Contain the SARS-CoV-2 Pandemic. Vaccines. 2021; 9(8):922. https://doi.org/10.3390/vaccines9080922
Chicago/Turabian StyleFederico, Maurizio. 2021. "Virus-Induced CD8+ T-Cell Immunity and Its Exploitation to Contain the SARS-CoV-2 Pandemic" Vaccines 9, no. 8: 922. https://doi.org/10.3390/vaccines9080922
APA StyleFederico, M. (2021). Virus-Induced CD8+ T-Cell Immunity and Its Exploitation to Contain the SARS-CoV-2 Pandemic. Vaccines, 9(8), 922. https://doi.org/10.3390/vaccines9080922