
Dendritic Cell Tumor Vaccination via Fc Gamma Receptor Targeting: Lessons Learned from Pre-Clinical and Translational Studies


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Cancer Therapy and the Immune System
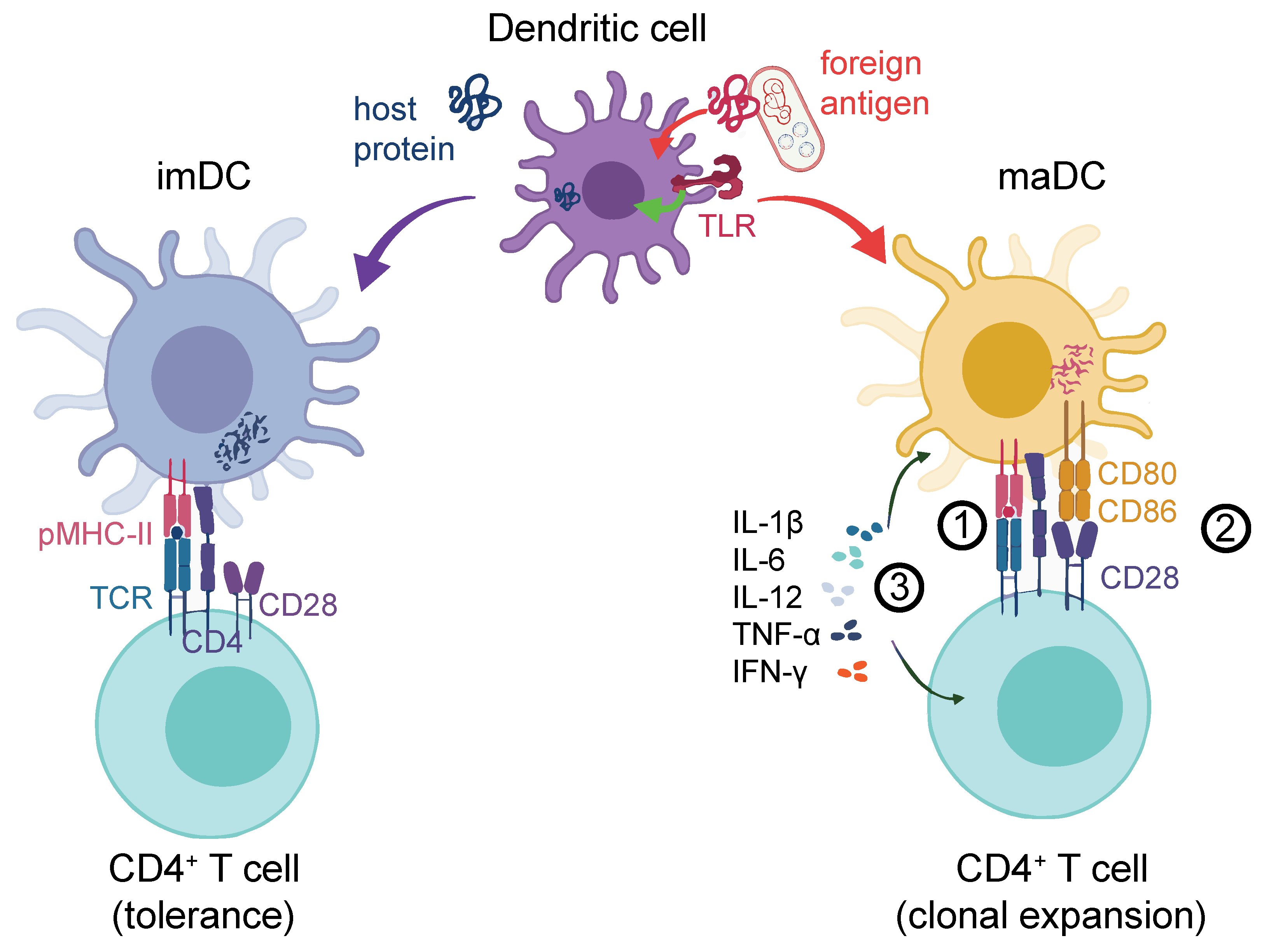
1.2. DCs Are Crucial for Effective Helper and Cytotoxic T-cell Activation
1.3. FcγR Crosslinking on DCs Leads to Effective T-Cell Activation and Proliferation
2. Targeting DCs for Cancer Vaccination via FcγRs: Mechanistic Principles
2.1. Allogenic Tumor IgG ICs Can Trigger Cancer Immunity via DC Activation
2.2. FcγR-Targeted Vaccination Strategies in Preclinical Tumor Models
3. The Long Way to the Clinic: Lessons Learned from Translational Models
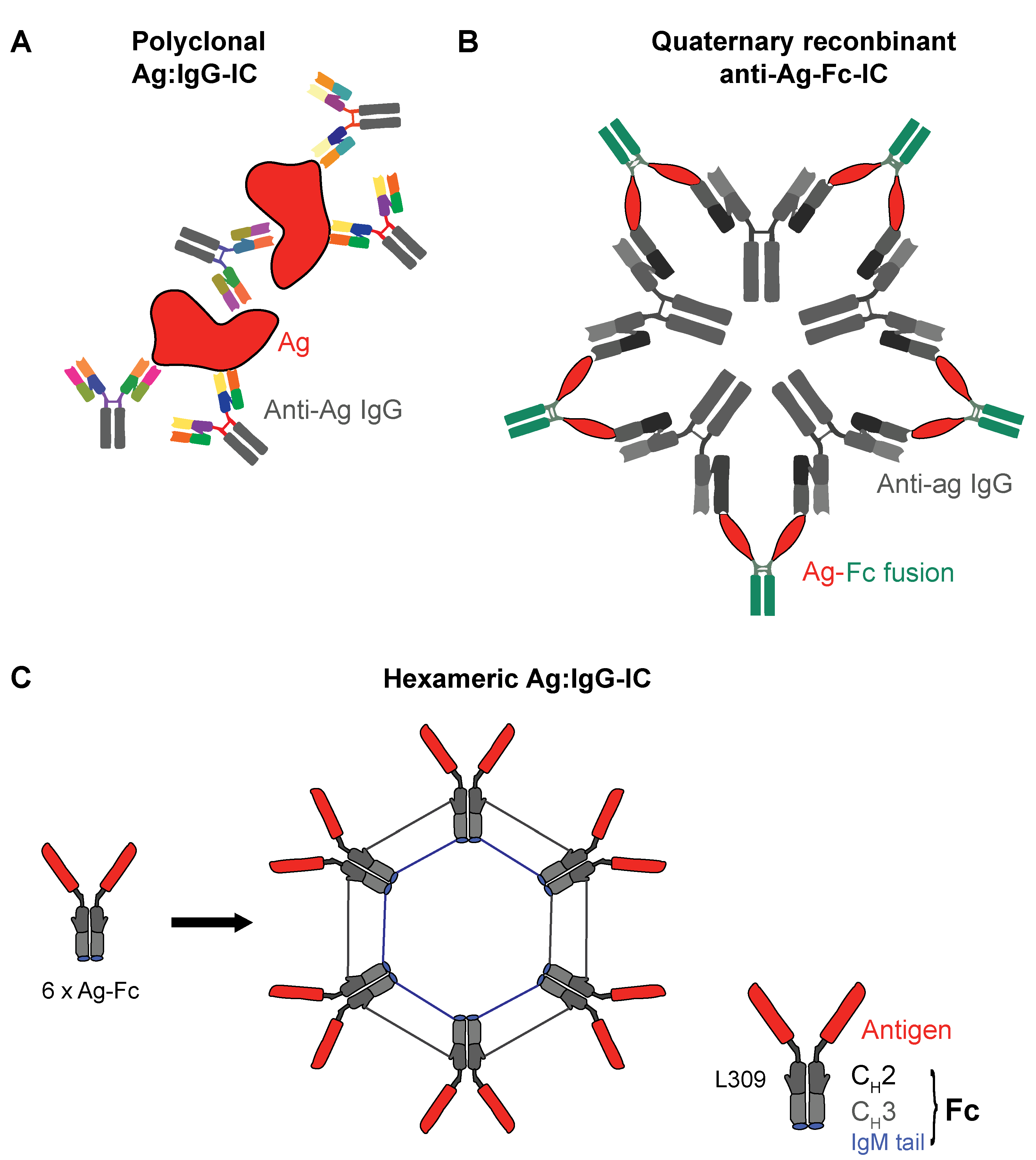
3.1. Ag:IgG IC or Ag plus Hapten?
3.2. Recombinant IgG ICs to Target Human FcγRs
3.3. How Translatable Are Preclinical IC Vaccination Models?
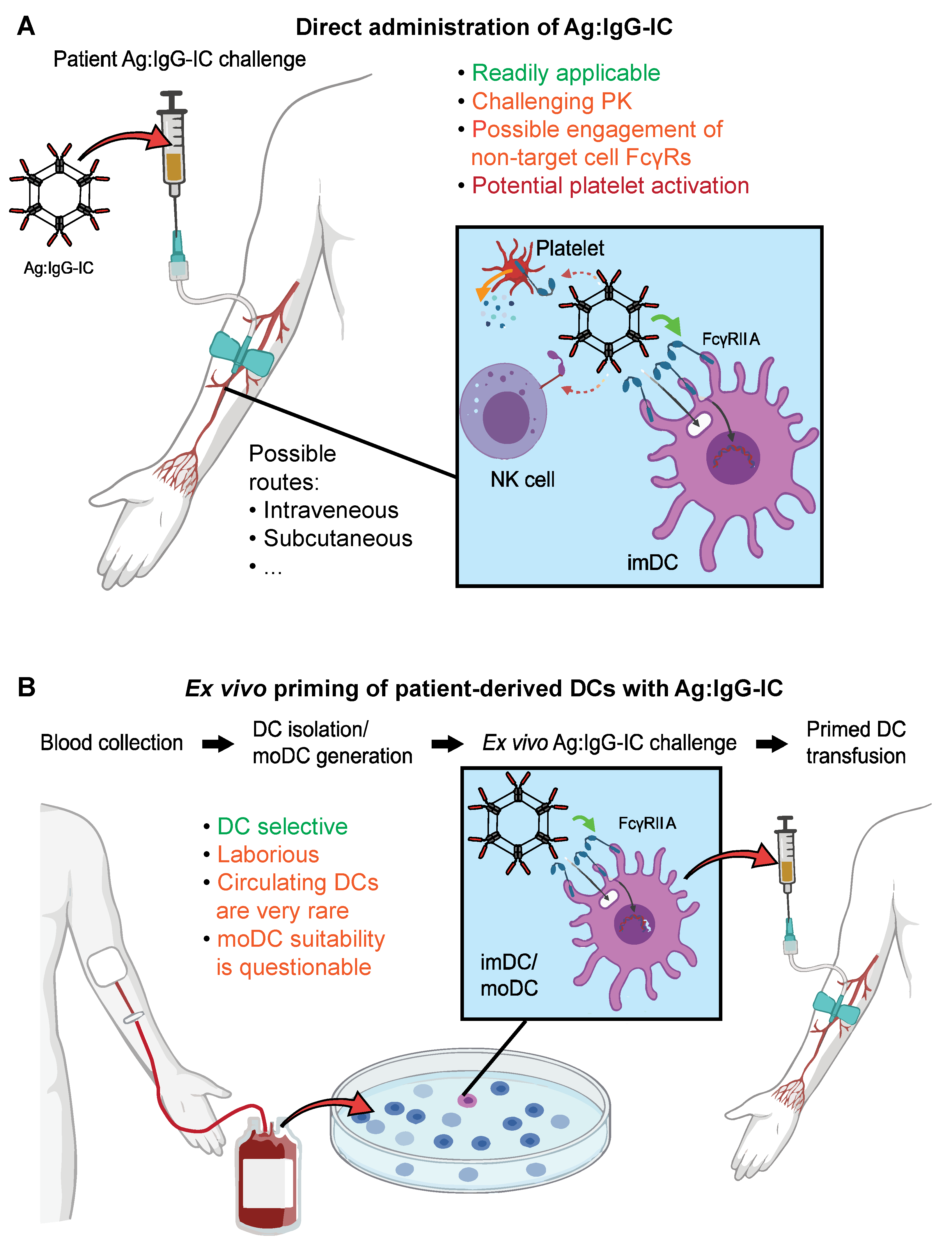
3.4. Advantages and Challenges of Recombinant ICs as DC Targeted Vaccines
3.5. FcγRs in Clinical Trials: More Than a Biomarker?
4. FcγRs as DC Targets for Tumor Vaccination: Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADCC | Antibody-dependent Cell-mediated Cytotoxicity |
| Ag | Antigen |
| alum | Aluminum hydroxide gel |
| APC | Antigen-presenting cell |
| BALB/c | Bagg and Albino mouse strain |
| BMDC | Bone marrow-derived dendritic cell |
| CD | Cluster of differentiation |
| cDC | Conventional dendritic cell |
| cEDIII | Consensus domain III sequence |
| CLR | C-type lectin receptors |
| CTL | Cytotoxic T lymphocyte |
| DC | Dendritic cell |
| DNA | Deoxyribonucleic acid |
| EpCAM | Epithelial cell adhesion molecule |
| Fc | Fragment, crystallizable |
| FcγR | Fc-gamma receptors |
| HLA | Human Leukocyte Antigen |
| IC | Immune complex |
| IFN-γ | Interferon-γ |
| Ig | Immunoglobulin |
| IL | Interleukin |
| imDC | Immature dendritic cell |
| IS | Immune system |
| ITAM | Immunoreceptor tyrosine-based activation motif |
| ITAMi | ITAM-mediated inhibitory signaling |
| IVIg | Intravenous immunoglobulin |
| KO | Knockout |
| LN | Lymph node |
| maDC | Mature dendritic cell |
| mAb | Monoclonal antibody |
| MAPK | Mitogen-activated protein kinase |
| MHC | Major histocompatibility complex |
| MoA | Mode of action |
| moDC | Monocyte-derived dendritic cell |
| MSP1-19 | Merozoite surface protein 1, C-terminal 19 kDa region |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK | Natural killer cell |
| OVA | Ovalbumin |
| PAMP | Pathogen-associated molecular patterns |
| PBMC | Peripheral blood mononuclear cell |
| pDC | Plasmacytoid dendritic cell |
| PIGS | Polymeric immunoglobulin G scaffold |
| PK | Pharmacokinetics |
| PRR | Pattern recognition receptor |
| SHP-1 | Src homology region 2 domain-containing tyrosine phosphatase |
| SLE | Systemic lupus erythematosus |
| SNP | Single Nucleotide Polymorphism |
| TAP1 | Transporter associated with Antigen Processing 1 |
| TCR | T cell receptor |
| Th | Helper T cell |
| TLR | Toll-like receptors |
| TNF-α | Tumor necrosis factor alpha |
| Treg | Regulatory T cell |
| wt | wildtype |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef]
- Melief, C.J.; van Hall, T.; Arens, R.; Ossendorp, F.; van der Burg, S.H. Therapeutic cancer vaccines. J. Clin. Investig. 2015, 125, 3401–3412. [Google Scholar] [CrossRef] [PubMed]
- Khong, H.; Overwijk, W.W. Adjuvants for peptide-based cancer vaccines. J. Immunother. Cancer 2016, 4, 56. [Google Scholar] [CrossRef]
- Chen, K.; Wang, J.M.; Yuan, R.; Yi, X.; Li, L.; Gong, W.; Yang, T.; Li, L.; Su, S. Tissue-resident dendritic cells and diseases involving dendritic cell malfunction. Int. Immunopharmacol. 2016, 34, 1–15. [Google Scholar] [CrossRef]
- Balan, S.; Saxena, M.; Bhardwaj, N. Dendritic cell subsets and locations. Int. Rev. Cell Mol. Biol. 2019, 348, 1–68. [Google Scholar]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Reznikoff, G.; Dranoff, G.; Rock, K.L. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J. Immunol. 1997, 158, 2723–2730. [Google Scholar]
- Guery, L.; Hugues, S. New role for antigen-presenting activated pDCs in promoting Th17 cells and impacting antitumor immunity. Oncoimmunology 2015, 4, e988476. [Google Scholar] [CrossRef][Green Version]
- Mouries, J.; Moron, G.; Schlecht, G.; Escriou, N.; Dadaglio, G.; Leclerc, C. Plasmacytoid dendritic cells efficiently cross-prime naive T cells in vivo after TLR activation. Blood 2008, 112, 3713–3722. [Google Scholar] [CrossRef]
- Oberkampf, M.; Guillerey, C.; Mouriès, J.; Rosenbaum, P.; Fayolle, C.; Bobard, A.; Savina, A.; Ogier-Denis, E.; Enninga, J.; Amigorena, S.; et al. Mitochondrial reactive oxygen species regulate the induction of CD8(+) T cells by plasmacytoid dendritic cells. Nat. Commun. 2018, 9, 2241. [Google Scholar] [CrossRef] [PubMed]
- See, P.; Dutertre, C.-A.; Chen, J.; Günther, P.; McGovern, N.; Irac, S.E.; Gunawan, M.; Beyer, M.; Händler, K.; Duan, K.; et al. Mapping the human DC lineage through the integration of high-dimensional techniques. Science 2017, 356. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, A.; Lutz, K.; Winheim, E.; Krug, A.B. What Makes a pDC: Recent Advances in Understanding Plasmacytoid DC Development and Heterogeneity. Front. Immunol. 2019, 10, 1222. [Google Scholar] [CrossRef]
- Ito, T.; Amakawa, R.; Inaba, M.; Hori, T.; Ota, M.; Nakamura, K.; Takebayashi, M.; Miyaji, M.; Yoshimura, T.; Inaba, K.; et al. Plasmacytoid dendritic cells regulate Th cell responses through OX40 ligand and type I IFNs. J. Immunol. 2004, 172, 4253–4259. [Google Scholar] [CrossRef] [PubMed]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef]
- Landsverk, O.J.; Ottesen, A.H.; Berg-Larsen, A.; Appel, S.; Bakke, O. Differential regulation of MHC II and invariant chain expression during maturation of monocyte-derived dendritic cells. J. Leukoc. Biol. 2012, 91, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Luckashenak, N.; Schroeder, S.; Endt, K.; Schmidt, D.; Mahnke, K.; Bachmann, M.F.; Marconi, P.; Deeg, C.A.; Brocker, T. Constitutive crosspresentation of tissue antigens by dendritic cells controls CD8+ T cell tolerance in vivo. Immunity 2008, 28, 521–532. [Google Scholar] [CrossRef]
- Sigal, L.J.; Crotty, S.; Andino, R.; Rock, K.L. Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature 1999, 398, 77–80. [Google Scholar] [CrossRef]
- Reis e Sousa, C. Activation of dendritic cells: Translating innate into adaptive immunity. Curr. Opin. Immunol. 2004, 16, 21–25. [Google Scholar] [CrossRef]
- Van Vliet, S.J.; Garcia-Vallejo, J.J.; van Kooyk, Y. Dendritic cells and C-type lectin receptors: Coupling innate to adaptive immune responses. Immunol. Cell Biol. 2008, 86, 580–587. [Google Scholar] [CrossRef]
- Cella, M.; Sallusto, F.; Lanzavecchia, A. Origin, maturation and antigen presenting function of dendritic cells. Curr. Opin. Immunol. 1997, 9, 10–16. [Google Scholar] [CrossRef]
- Edwards, A.D.; Diebold, S.S.; Slack, E.M.C.; Tomizawa, H.; Hemmi, H.; Kaisho, T.; Akira, S.; E Sousa, C.R. Toll-like receptor expression in murine DC subsets: Lack of TLR7 expression by CD8 alpha+ DC correlates with unresponsiveness to imidazoquinolines. Eur. J. Immunol. 2003, 33, 827–833. [Google Scholar] [CrossRef]
- Fischetti, L.; Zhong, Z.; Pinder, C.L.; Tregoning, J.S.; Shattock, R.J. The synergistic effects of combining TLR ligand based adjuvants on the cytokine response are dependent upon p38/JNK signalling. Cytokine 2017, 99, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.; Towarowski, A.; Britsch, S.; Rothenfusser, S.; Hornung, V.; Bals, R.; Giese, T.; Engelmann, H.; Endres, S.; Krieg, A.M.; et al. Toll-like receptor expression reveals CpG DNA as a unique microbial stimulus for plasmacytoid dendritic cells which synergizes with CD40 ligand to induce high amounts of IL-12. Eur. J. Immunol. 2001, 31, 3026–3037. [Google Scholar] [CrossRef]
- Li, J.; Jiang, H.; Wen, W.; Zheng, J.; Xu, G. The dendritic cell mannose receptor mediates allergen internalization and maturation involving notch 1 signalling. Clin. Exp. Immunol. 2010, 162, 251–261. [Google Scholar] [CrossRef]
- Schreibelt, G.; Tel, J.; Sliepen, K.H.; Benitez-Ribas, D.; Figdor, C.G.; Adema, G.J.; de Vries, I.J.M. Toll-like receptor expression and function in human dendritic cell subsets: Implications for dendritic cell-based anti-cancer immunotherapy. Cancer Immunol. Immunother. 2010, 59, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Hawiger, D.; Nussenzweig, M.C. Tolerogenic dendritic cells. Annu. Rev. Immunol. 2003, 21, 685–711. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lanzavecchia, A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 1994, 179, 1109–1118. [Google Scholar] [CrossRef]
- Whiteside, T.L.; Odoux, C. Dendritic cell biology and cancer therapy. Cancer Immunol. Immunother. 2004, 53, 240–248. [Google Scholar] [CrossRef]
- Lopez, S.; Gomez, E.; Torres, M.J.; Pozo, D.; Fernandez, T.D.; Ariza, A.; Sanz, M.L.; Blanca, M.; Mayorga, C. Betalactam antibiotics affect human dendritic cells maturation through MAPK/NF-kB systems. Role in allergic reactions to drugs. Toxicol. Appl. Pharmacol. 2015, 288, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Akdis, M.; Burgler, S.; Crameri, R.; Eiwegger, T.; Fujita, H.; Gomez, E. Interleukins, from 1 to 37, and interferon-gamma: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2011, 127, 701–721. [Google Scholar] [CrossRef]
- Sporri, R.; Reis e Sousa, C. Inflammatory mediators are insufficient for full dendritic cell activation and promote expansion of CD4+ T cell populations lacking helper function. Nat. Immunol. 2005, 6, 163–170. [Google Scholar] [CrossRef]
- Riol-Blanco, L.; Sánchez-Sánchez, N.; Torres, A.; Tejedor, A.; Narumiya, S.; Corbí, A.L.; Sánchez-Mateos, P.; Rodríguez-Fernández, J.L. The chemokine receptor CCR7 activates in dendritic cells two signaling modules that independently regulate chemotaxis and migratory speed. J. Immunol. 2005, 174, 4070–4080. [Google Scholar] [CrossRef]
- Rodriguez-Fernandez, J.L.; Criado-Garcia, O. The Chemokine Receptor CCR7 Uses Distinct Signaling Modules with Biased Functionality to Regulate Dendritic Cells. Front. Immunol. 2020, 11, 528. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Dalod, M.; Chelbi, R.; Malissen, B.; Lawrence, T. Dendritic cell maturation: Functional specialization through signaling specificity and transcriptional programming. EMBO J. 2014, 33, 1104–1116. [Google Scholar] [CrossRef]
- Blanco, P.; Palucka, A.K.; Pascual, V.; Banchereau, J. Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytokine Growth Factor Rev. 2008, 19, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Obeid, J.; Hu, Y.; Slingluff, C.L., Jr. Vaccines, Adjuvants, and Dendritic Cell Activators--Current Status and Future Challenges. Semin. Oncol. 2015, 42, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, A.; Das, P.; Chakravortty, D. Engagement of TLR signaling as adjuvant: Towards smarter vaccine and beyond. Vaccine 2008, 26, 6777–6783. [Google Scholar] [CrossRef]
- Pollard, A.J.; Bijker, E.M. A guide to vaccinology: From basic principles to new developments. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- Van Duin, D.; Medzhitov, R.; Shaw, A.C. Triggering TLR signaling in vaccination. Trends Immunol. 2006, 27, 49–55. [Google Scholar] [CrossRef]
- Prince, L.R.; Whyte, M.K.; Sabroe, I.; Parker, L.C. The role of TLRs in neutrophil activation. Curr. Opin. Pharmacol. 2011, 11, 397–403. [Google Scholar] [CrossRef]
- Hornung, V.; Rothenfusser, S.; Britsch, S.; Krug, A.; Jahrsdorfer, B.; Giese, T. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol. 2002, 168, 4531–4537. [Google Scholar] [CrossRef] [PubMed]
- Krutzik, S.R.; Tan, B.; Li, H.; Ochoa, M.T.; Liu, P.T.; Sharfstein, S.E.; Graeber, T.G.; Sieling, P.A.; Liu, Y.-J.; Rea, T.H.; et al. TLR activation triggers the rapid differentiation of monocytes into macrophages and dendritic cells. Nat. Med. 2005, 11, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Pegu, A.; Qin, S.; Fallert Junecko, B.A.; Nisato, R.E.; Pepper, M.S.; Reinhart, T.A. Human lymphatic endothelial cells express multiple functional TLRs. J. Immunol. 2008, 180, 3399–3405. [Google Scholar] [CrossRef]
- Li, J.K.; Balic, J.J.; Yu, L.; Jenkins, B. TLR Agonists as Adjuvants for Cancer Vaccines. Adv. Exp. Med. Biol. 2017, 1024, 195–212. [Google Scholar]
- Lu, B.L.; Williams, G.M.; Verdon, D.J.; Dunbar, P.R.; Brimble, M.A. Synthesis and Evaluation of Novel TLR2 Agonists as Potential Adjuvants for Cancer Vaccines. J. Med. Chem. 2020, 63, 2282–2291. [Google Scholar] [CrossRef]
- Carter, D.; Duthie, M.S.; Reed, S.G. Adjuvants. Curr. Top. Microbiol. Immunol. 2020, 428, 103–127. [Google Scholar] [PubMed]
- Del Giudice, G.; Rappuoli, R.; Didierlaurent, A.M. Correlates of adjuvanticity: A review on adjuvants in licensed vaccines. Semin. Immunol. 2018, 39, 14–21. [Google Scholar] [CrossRef]
- Ho, N.I.; Veld, L.G.M.H.I.; Raaijmakers, T.K.; Adema, G.J. Adjuvants Enhancing Cross-Presentation by Dendritic Cells: The Key to More Effective Vaccines? Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Cuzzubbo, S.; Mangsbo, S.; Nagarajan, D.; Habra, K.; Pockley, A.G.; McArdle, S.E.B. Cancer Vaccines: Adjuvant Potency, Importance of Age, Lifestyle, and Treatments. Front. Immunol. 2020, 11, 615240. [Google Scholar] [CrossRef] [PubMed]
- Regnault, A.; Lankar, D.; Lacabanne, V.; Rodriguez, A.; Thery, C.; Rescigno, M.; Saito, T.; Verbeek, S.; Bonnerot, C.; Ricciardi-Castagnoli, P.; et al. Fcgamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J. Exp. Med. 1999, 189, 371–380. [Google Scholar] [CrossRef]
- Junker, F.; Gordon, J.; Qureshi, O. Fc Gamma Receptors and Their Role in Antigen Uptake, Presentation, and T Cell Activation. Front. Immunol. 2020, 11, 1393. [Google Scholar] [CrossRef] [PubMed]
- Junker, F.; Krishnarajah, S.; Qureshi, O.; Humphreys, D.; Fallah-Arani, F. A simple method for measuring immune complex-mediated, Fc gamma receptor dependent antigen-specific activation of primary human T cells. J. Immunol. Methods 2018, 454, 32–39. [Google Scholar] [CrossRef]
- Ravetch, J.V.; Bolland, S. IgG Fc receptors. Annu. Rev. Immunol. 2001, 19, 275–290. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S.; Daëron, M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009, 113, 3716–3725. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, X.; Masuda, E.; Redecha, P.B.; Blank, M.C.; Pricop, L. Regulated expression of FcgammaR in human dendritic cells controls cross-presentation of antigen-antibody complexes. J. Immunol. 2006, 177, 8440–8447. [Google Scholar] [CrossRef] [PubMed]
- Boruchov, A.M.; Heller, G.; Veri, M.C.; Bonvini, E.; Ravetch, J.V.; Young, J.W. Activating and inhibitory IgG Fc receptors on human DCs mediate opposing functions. J. Clin. Investig. 2005, 115, 2914–2923. [Google Scholar] [CrossRef] [PubMed]
- Flores, M.; Desai, D.D.; Downie, M.; Liang, B.; Reilly, M.P.; McKenzie, S.E.; Clynes, R. Dominant expression of the inhibitory FcgammaRIIB prevents antigen presentation by murine plasmacytoid dendritic cells. J. Immunol. 2009, 183, 7129–7139. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Beenhakker, N.; Koopman, G.; Hart, B.; Mudde, G.C.; de Vries, I.J. Targeted delivery of CpG ODN to CD32 on human and monkey plasmacytoid dendritic cells augments IFNalpha secretion. Immunobiology 2012, 217, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda-Toepfer, J.A.; Pichler, J.; Fink, K.; Sevo, M.; Wildburger, S.; Mudde-Boer, L.C.; Taus, C.; Mudde, G.C.; Sepulveda, T.J.A. TLR9-mediated activation of dendritic cells by CD32 targeting for the generation of highly immunostimulatory vaccines. Hum. Vaccin. Immunother. 2019, 15, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Means, T.K.; Latz, E.; Hayashi, F.; Murali, M.R.; Golenbock, D.T.; Luster, A.D. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J. Clin. Investig. 2005, 115, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Bruhns, P.; Saeys, Y.; Hammad, H.; Lambrecht, B.N. The function of Fcgamma receptors in dendritic cells and macrophages. Nat. Rev. Immunol. 2014, 14, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Bruhns, P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 2012, 119, 5640–5649. [Google Scholar] [CrossRef]
- Kerntke, C.; Nimmerjahn, F.; Biburger, M. There Is (Scientific) Strength in Numbers: A Comprehensive Quantitation of Fc Gamma Receptor Numbers on Human and Murine Peripheral Blood Leukocytes. Front. Immunol. 2020, 11, 118. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Rowley, T.F.; Junker, F.; Peters, S.J.; Crilly, S.; Compson, J.; Eddleston, A.; Björkelund, H.; Greenslade, K.; Parkinson, M.; et al. Multivalent Fcgamma-receptor engagement by a hexameric Fc-fusion protein triggers Fcgamma-receptor internalisation and modulation of Fcgamma-receptor functions. Sci. Rep. 2017, 7, 17049. [Google Scholar] [CrossRef]
- Lehmann, C.H.K.; Baranska, A.; Heidkamp, G.F.; Heger, L.; Neubert, K.; Luhr, J.J.; et al. DC subset-specific induction of T cell responses upon antigen uptake via Fcgamma receptors in vivo. J. Exp. Med. 2017, 214, 1509–1528. [Google Scholar] [CrossRef] [PubMed]
- Harper, M.; Lema, F.; Boulot, G.; Poljak, R.J. Antigen specificity and cross-reactivity of monoclonal anti-lysozyme antibodies. Mol. Immunol. 1987, 24, 97–108. [Google Scholar] [CrossRef]
- Van Vugt, M.J.; Heijnen, I.A.; Capel, P.J.; Park, S.Y.; Ra, C.; Saito, T. FcR gamma-chain is essential for both surface expression and function of human Fc gamma RI (CD64) in vivo. Blood 1996, 87, 3593–3599. [Google Scholar] [CrossRef] [PubMed]
- Van Montfoort, N.; Mangsbo, S.M.; Camps, M.G.M.; Van Maren, W.W.C.; Verhaart, I.E.C.; Waisman, A.; Drijfhout, J.W.; Melief, C.J.M.; Verbeek, J.S.; Ossendorp, F. Circulating specific antibodies enhance systemic cross-priming by delivery of complexed antigen to dendritic cells in vivo. Eur. J. Immunol. 2012, 42, 598–606. [Google Scholar] [CrossRef]
- Palucka, K.; Banchereau, J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013, 39, 38–48. [Google Scholar] [CrossRef]
- Spitzer, M.H.; Carmi, Y.; Reticker-Flynn, N.E.; Kwek, S.S.; Madhireddy, D.; Martins, M.M. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell 2017, 168, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef]
- Tagliamonte, M.; Petrizzo, A.; Tornesello, M.L.; Buonaguro, F.M.; Buonaguro, L. Antigen-specific vaccines for cancer treatment. Hum. Vaccin. Immunother. 2014, 10, 3332–3346. [Google Scholar] [CrossRef]
- Yang, L.; Carbone, D.P. Tumor-host immune interactions and dendritic cell dysfunction. Adv. Cancer Res. 2004, 92, 13–27. [Google Scholar]
- Bol, K.F.; Schreibelt, G.; Gerritsen, W.R.; de Vries, I.J.; Figdor, C.G. Dendritic Cell-Based Immunotherapy: State of the Art and Beyond. Clin. Cancer Res. 2016, 22, 1897–1906. [Google Scholar] [CrossRef]
- Chung, E.H. Vaccine allergies. Clin. Exp. Vaccine Res. 2014, 3, 50–57. [Google Scholar] [CrossRef]
- Blankenstein, T.; Coulie, P.G.; Gilboa, E.; Jaffee, E.M. The determinants of tumour immunogenicity. Nat. Rev. Cancer 2012, 12, 307–313. [Google Scholar] [CrossRef]
- Gilboa, E. The makings of a tumor rejection antigen. Immunity 1999, 11, 263–270. [Google Scholar] [CrossRef]
- Bishop, M.R.; Fowler, D.H.; Marchigiani, D.; Castro, K.; Kasten-Sportes, C.; Steinberg, S.M.; Gea-Banacloche, J.C.; Dean, R.; Chow, C.K.; Carter, C.; et al. Allogeneic lymphocytes induce tumor regression of advanced metastatic breast cancer. J. Clin. Oncol. 2004, 22, 3886–3892. [Google Scholar] [CrossRef]
- Carmi, Y.; Spitzer, M.H.; Linde, I.L.; Burt, B.M.; Prestwood, T.R.; Perlman, N.; Davidson, M.G.; Kenkel, J.A.; Segal, E.; Pusapati, G.V.; et al. Allogeneic IgG combined with dendritic cell stimuli induce antitumour T-cell immunity. Nature 2015, 521, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Kayaga, J.; E Souberbielle, B.; Sheikh, N.; Morrow, W.J.W.; Scott-Taylor, T.; Vile, R.; Dalgleish, A.G. Anti-tumour activity against B16-F10 melanoma with a GM-CSF secreting allogeneic tumour cell vaccine. Gene Ther. 1999, 6, 1475–1481. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mellman, I.; Plutner, H.; Ukkonen, P. Internalization and rapid recycling of macrophage Fc receptors tagged with monovalent antireceptor antibody: Possible role of a prelysosomal compartment. J. Cell Biol. 1984, 98, 1163–1169. [Google Scholar] [CrossRef] [PubMed]
- Pham, G.H.; Iglesias, B.V.; Gosselin, E.J. Fc receptor-targeting of immunogen as a strategy for enhanced antigen loading, vaccination, and protection using intranasally administered antigen-pulsed dendritic cells. Vaccine 2014, 32, 5212–5220. [Google Scholar] [CrossRef]
- Schuurhuis, D.H.; Ioan-Facsinay, A.; Nagelkerken, B.; Van Schip, J.J.; Sedlik, C.; Melief, C.J.M.; Verbeek, J.S.; Ossendorp, F. Antigen-antibody immune complexes empower dendritic cells to efficiently prime specific CD8+ CTL responses in vivo. J. Immunol. 2002, 168, 2240–2246. [Google Scholar] [CrossRef]
- Flamand, V.; Sornasse, T.; Thielemans, K.; Demanet, C.; Bakkus, M.; Bazin, H. Murine dendritic cells pulsed in vitro with tumor antigen induce tumor resistance in vivo. Eur. J. Immunol. 1994, 24, 605–610. [Google Scholar] [CrossRef]
- Inaba, K.; Metlay, J.P.; Crowley, M.T.; Steinman, R.M. Dendritic cells pulsed with protein antigens in vitro can prime antigen-specific, MHC-restricted T cells in situ. J. Exp. Med. 1990, 172, 631–640. [Google Scholar] [CrossRef]
- Bournazos, S.; Ravetch, J.V. Fcgamma Receptor Function and the Design of Vaccination Strategies. Immunity 2017, 47, 224–233. [Google Scholar] [CrossRef]
- Wang, X.Y.; Wang, B.; Wen, Y.M. From therapeutic antibodies to immune complex vaccines. NPJ Vaccines 2019, 4, 2. [Google Scholar] [CrossRef]
- Rafiq, K.; Bergtold, A.; Clynes, R. Immune complex-mediated antigen presentation induces tumor immunity. J. Clin. Investig. 2002, 110, 71–79. [Google Scholar] [CrossRef]
- Schuurhuis, D.H.; Van Montfoort, N.; Ioan-Facsinay, A.; Jiawan, R.; Camps, M.; Nouta, J.; Melief, C.J.M.; Verbeek, J.S.; Ossendorp, F. Immune complex-loaded dendritic cells are superior to soluble immune complexes as antitumor vaccine. J. Immunol. 2006, 176, 4573–4580. [Google Scholar] [CrossRef]
- Kim, D.-S.; Kang, Y.J.; Lee, K.J.; Qiao, L.; Ko, K.; Kim, D.H.; Myeung, S.C.; Ko, K. A Plant-Derived Antigen-Antibody Complex Induces Anti-Cancer Immune Responses by Forming a Large Quaternary Structure. Int. J. Mol. Sci. 2020, 21, 5603. [Google Scholar] [CrossRef]
- Gholizadeh, Z.; Tavakkol-Afshari, J.; Nikpoor, A.R.; Jalali, S.A.; Jaafari, M.R. Enhanced immune response induced by P5 HER2/neu-derived peptide-pulsed dendritic cells as a preventive cancer vaccine. J. Cell Mol. Med. 2018, 22, 558–567. [Google Scholar] [CrossRef]
- Dintzis, H.M.; Dintzis, R.Z.; Vogelstein, B. Molecular determinants of immunogenicity: The immunon model of immune response. Proc. Natl. Acad. Sci USA 1976, 73, 3671–3675. [Google Scholar] [CrossRef]
- Moyer, T.J.; Zmolek, A.C.; Irvine, D.J. Beyond antigens and adjuvants: Formulating future vaccines. J. Clin. Investig. 2016, 126, 799–808. [Google Scholar] [CrossRef]
- Finn, O.J. Cancer vaccines: Between the idea and the reality. Nat. Rev. Immunol. 2003, 3, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Landsteiner, K.; Jacobs, J. Studies on the Sensitization of Animals with Simple Chemical Compounds. J. Exp. Med. 1935, 61, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; You, F.; Vlahov, I.; Westrick, E.; Fan, M.; Low, P.S.; Leamon, C.P. Folate-targeted dinitrophenyl hapten immunotherapy: Effect of linker chemistry on antitumor activity and allergic potential. Mol. Pharm. 2007, 4, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Schrand, B.; Clark, E.; Levay, A.; Capote, A.R.; Martinez, O.; Brenneman, R.; Castro, I.; Gilboa, E. Hapten-mediated recruitment of polyclonal antibodies to tumors engenders antitumor immunity. Nat. Commun. 2018, 9, 3348. [Google Scholar] [CrossRef] [PubMed]
- Lux, A.; Yu, X.; Scanlan, C.N.; Nimmerjahn, F. Impact of immune complex size and glycosylation on IgG binding to human FcgammaRs. J. Immunol. 2013, 190, 4315–4323. [Google Scholar] [CrossRef]
- Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Mantovani, A.; Lambris, J.D. Complement in cancer: Untangling an intricate relationship. Nat. Rev. Immunol. 2018, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Usman, N.; Annamaraju, P. Type III Hypersensitivity Reaction; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Kolev, M.; Markiewski, M.M. Targeting complement-mediated immunoregulation for cancer immunotherapy. Semin. Immunol. 2018, 37, 85–97. [Google Scholar] [CrossRef]
- Hegde, G.V.; Meyers-Clark, E.; Joshi, S.S.; Sanderson, S.D. A conformationally-biased, response-selective agonist of C5a acts as a molecular adjuvant by modulating antigen processing and presentation activities of human dendritic cells. Int. Immunopharmacol. 2008, 8, 819–827. [Google Scholar] [CrossRef]
- Floreani, A.A.; Gunselman, S.J.; Heires, A.J.; Hauke, R.J.; Tarantolo, S.; Jackson, J.D. Novel C5a agonist-based dendritic cell vaccine in a murine model of melanoma. Cell Cycle 2007, 6, 2835–2839. [Google Scholar] [CrossRef]
- Ortiz, D.F.; Lansing, J.C.; Rutitzky, L.; Kurtagic, E.; Prod’homme, T.; Choudhury, A. Elucidating the interplay between IgG-Fc valency and FcgammaR activation for the design of immune complex inhibitors. Sci. Transl. Med. 2016, 8, 365ra158. [Google Scholar] [CrossRef]
- Jain, A.; Olsen, H.S.; Vyzasatya, R.; Burch, E.; Sakoda, Y.; Merigeon, E.Y.; Cai, L.; Lu, C.; Tan, M.; Tamada, K.; et al. Fully recombinant IgG2a Fc multimers (stradomers) effectively treat collagen-induced arthritis and prevent idiopathic thrombocytopenic purpura in mice. Arthritis Res. Ther. 2012, 14, R192. [Google Scholar] [CrossRef]
- Spirig, R.; Campbell, I.K.; Koernig, S.; Chen, C.G.; Lewis, B.J.B.; Butcher, R.; Muir, I.; Taylor, S.; Chia, J.; Leong, D.; et al. rIgG1 Fc Hexamer Inhibits Antibody-Mediated Autoimmune Disease via Effects on Complement and FcgammaRs. J. Immunol. 2018, 200, 2542–2553. [Google Scholar] [CrossRef]
- Mekhaiel, D.N.A.; Czajkowsky, D.M.; Andersen, J.T.; Shi, J.; El-Faham, M.; Doenhoff, M.; McIntosh, R.S.; Sandlie, I.; He, J.; Hu, J.; et al. Polymeric human Fc-fusion proteins with modified effector functions. Sci. Rep. 2011, 1, 124. [Google Scholar] [CrossRef]
- Rowley, T.F.; Peters, S.J.; Aylott, M.; Griffin, R.; Davies, N.L.; Healy, L.J.; et al. Engineered hexavalent Fc proteins with enhanced Fc-gamma receptor avidity provide insights into immune-complex interactions. Commun. Biol. 2018, 1, 146. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-Y.; Van Dolleweerd, C.; Copland, A.; Paul, M.J.; Hofmann, S.; Webster, G.R.; Julik, E.; Ceballos-Olvera, I.; Valle, J.R.-D.; Yang, M.-S.; et al. Molecular engineering and plant expression of an immunoglobulin heavy chain scaffold for delivery of a dengue vaccine candidate. Plant Biotechnol. J. 2017, 15, 1590–1601. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Copland, A.; Nayak, K.; Chandele, A.; Ahmed, M.S.; Zhang, Q.; Diogo, G.R.; Paul, M.J.; Hofmann, S.; Yang, M.; et al. Plant-expressed Fc-fusion protein tetravalent dengue vaccine with inherent adjuvant properties. Plant Biotechnol. J. 2018, 16, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Hussain, K.; Hargreaves, C.E.; Rowley, T.F.; Sopp, J.M.; Latham, K.V.; Bhatta, P.; Sherington, J.; Cutler, M.; Humphreys, P.; Glennie, M.J.; et al. Impact of Human FcgammaR Gene Polymorphisms on IgG-Triggered Cytokine Release: Critical Importance of Cell Assay Format. Front. Immunol. 2019, 10, 390. [Google Scholar] [CrossRef]
- Bulliard, Y.; Jolicoeur, R.; Windman, M.; Rue, S.M.; Ettenberg, S.; Knee, D.A.; Wilson, A.S.; Dranoff, G.; Brogdon, J.F. Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J. Exp. Med. 2013, 210, 1685–1693. [Google Scholar] [CrossRef]
- Stapleton, N.M.; Einarsdottir, H.K.; Stemerding, A.M.; Vidarsson, G. The multiple facets of FcRn in immunity. Immunol. Rev. 2015, 268, 253–268. [Google Scholar] [CrossRef]
- Santana-Magal, N.; Rasoulouniriana, D.; Saperia, C.; Gutwillig, A.; Rider, P.; Engleman, E.G.; Carmi, Y. Isolation Protocol of Mouse Monocyte-derived Dendritic Cells and Their Subsequent In Vitro Activation with Tumor Immune Complexes. J. Vis. Exp. 2018. [Google Scholar] [CrossRef]
- Wimmers, F.; Schreibelt, G.; Skold, A.E.; Figdor, C.G.; De Vries, I.J. Paradigm Shift in Dendritic Cell-Based Immunotherapy: From in vitro Generated Monocyte-Derived DCs to Naturally Circulating DC Subsets. Front. Immunol. 2014, 5, 165. [Google Scholar] [CrossRef]
- Guilliams, M.; Dutertre, C.-A.; Scott, C.L.; McGovern, N.; Sichien, D.; Chakarov, S.; Van Gassen, S.; Chen, J.; Poidinger, M.; De Prijck, S.; et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 2016, 45, 669–684. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.B.; Rahman, M.J.; Tarbell, K.V. Flow cytometric gating for spleen monocyte and DC subsets: Differences in autoimmune NOD mice and with acute inflammation. J. Immunol. Methods 2016, 432, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, C.-A.; Becht, E.; Irac, S.E.; Khalilnezhad, A.; Narang, V.; Khalilnezhad, S.; Ng, P.Y.; Hoogen, L.L.V.D.; Leong, J.Y.; Lee, B.; et al. Single-Cell Analysis of Human Mononuclear Phagocytes Reveals Subset-Defining Markers and Identifies Circulating Inflammatory Dendritic Cells. Immunity 2019, 51, 573–589. [Google Scholar] [CrossRef] [PubMed]
- McGovern, N.; Chan, J.K.; Ginhoux, F. Dendritic cells in humans—From fetus to adult. Int. Immunol. 2015, 27, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Mogilenko, D.A.; Shpynov, O.; Andhey, P.S.; Arthur, L.; Swain, A.; Esaulova, E.; Brioschi, S.; Shchukina, I.; Kerndl, M.; Bambouskova, M.; et al. Comprehensive Profiling of an Aging Immune System Reveals Clonal GZMK(+) CD8(+) T Cells as Conserved Hallmark of Inflammaging. Immunity 2020. [Google Scholar] [CrossRef]
- Nimmerjahn, F. Translating Inhibitory Fc Receptor Biology into Novel Therapeutic Approaches. J. Clin. Immunol. 2016, 36 (Suppl. 1), 83–87. [Google Scholar] [CrossRef]
- Schnurr, M.; Chen, Q.; Shin, A.; Chen, W.; Toy, T.; Jenderek, C.; Green, S.; Miloradovic, L.; Drane, D.; Davis, I.D.; et al. Tumor antigen processing and presentation depend critically on dendritic cell type and the mode of antigen delivery. Blood 2005, 105, 2465–2472. [Google Scholar] [CrossRef]
- Ridge, J.P.; Di Rosa, F.; Matzinger, P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T− killer cell. Nature 1998, 393, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Robinett, R.A.; Guan, N.; Lux, A.; Biburger, M.; Nimmerjahn, F.; Meyer, A.S. Dissecting FcgammaR Regulation through a Multivalent Binding Model. Cell Syst. 2018, 7, 41–48. [Google Scholar] [CrossRef]
- Prue, R.L.; Vari, F.; Radford, K.J.; Tong, H.; Hardy, M.Y.; D’Rozario, R.; Waterhouse, N.J.; Rossetti, T.; Coleman, R.; Tracey, C.; et al. A phase I clinical trial of CD1c (BDCA-1)+ dendritic cells pulsed with HLA-A*0201 peptides for immunotherapy of metastatic hormone refractory prostate cancer. J. Immunother. 2015, 38, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Westdorp, H.; Creemers, J.H.A.; Van Oort, I.M.; Schreibelt, G.; Gorris, M.A.J.; Mehra, N.; Simons, M.; De Goede, A.L.; Van Rossum, M.M.; Croockewit, A.J.; et al. Blood-derived dendritic cell vaccinations induce immune responses that correlate with clinical outcome in patients with chemo-naive castration-resistant prostate cancer. J. Immunother. Cancer 2019, 7, 302. [Google Scholar] [CrossRef] [PubMed]
- Nagelkerke, S.Q.; Schmidt, D.E.; de Haas, M.; Kuijpers, T.W. Genetic Variation in Low-To-Medium-Affinity Fcγ Receptors: Functional Consequences, Disease Associations, and Opportunities for Personalized Medicine. Front. Immunol. 2019, 10, 2237. [Google Scholar] [CrossRef] [PubMed]
- Poulart, V.; Jou, Y.M.; Delmonte, T.; Robbins, M. FC-gamma receptor polymorphisms and progression-free survival: Analysis of three clinical trials of elotuzumab in multiple myeloma. Haematologica 2016, 101, 529–530. [Google Scholar]
- Cartin-Ceba, R.; Indrakanti, D.; Fervenza, F.; Hoffman, G.; Kallenberg, C.; Langford, C. Association of Fc gamma receptor polymorphisms and outcomes in patients with ANCA-associated vasculitis treated with rituximab. Nephron 2015, 129, 96. [Google Scholar]
- Tout, M.; Gagez, A.L.; Leprêtre, S.; Gouilleux-Gruart, V.; Azzopardi, N.; Delmer, A.; et al. Influence of FCGR3A-158V/F Genotype and Baseline CD20 Antigen Count on Target-Mediated Elimination of Rituximab in Patients with Chronic Lymphocytic Leukemia: A Study of FILO Group. Clin. Pharmacokinet. 2017, 56, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Kenkre, V.P.; Hong, F.; Cerhan, J.R.; Lewis, M.; Sullivan, L.; Williams, M.E.; et al. Fc Gamma Receptor 3A and 2A Polymorphisms Do Not Predict Response to Rituximab in Follicular Lymphoma. Clin. Cancer Res. 2016, 22, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Strefford, J.C.; Nowicka, M.; Hargreaves, C.; Iriyama, C.; Latham, K.V.; Ganderton, R.; Parker, H.; Potter, K.N.; Knapp, A.; Mir, F.; et al. Prognostic Impact of Germ-Line FCGR2A (H131R), FCGR3A (F158V), and FCGR2B (I232T) Single Nucleotide Polymorphisms in Lymphoma Patients Treated with Obinutuzumab or Rituximab in Combination with Chemotherapy: Results from the Phase III GALLIUM and GOYA Clinical Trials. Blood 2018, 132. [Google Scholar] [CrossRef]
- Shepshelovich, D.; Townsend, A.R.; Espin-Garcia, O.; Latifovic, L.; O’Callaghan, C.J.; Jonker, D.J.; Tu, N.; Chen, E.; Morgen, E.; Price, T.J.; et al. Fc-gamma receptor polymorphisms, cetuximab therapy, and overall survival in the CCTG CO.20 trial of metastatic colorectal cancer. Cancer Med. 2018, 7, 5478–5487. [Google Scholar] [CrossRef]
- Capuano, C.; Pighi, C.; Molfetta, R.; Paolini, R.; Battella, S.; Palmieri, G.; Giannini, G.; Belardinilli, F.; Santoni, A.; Galandrini, R. Obinutuzumab-mediated high-affinity ligation of FcgammaRIIIA/CD16 primes NK cells for IFNgamma production. Oncoimmunology 2017, 6, e1290037. [Google Scholar] [CrossRef]
- Hilchey, S.P.; Hyrien, O.; Mosmann, T.R.; Livingstone, A.M.; Friedberg, J.W.; Young, F.; Fisher, R.I.; Kelleher, J.R.J.; Bankert, R.B.; Bernstein, S.H. Rituximab immunotherapy results in the induction of a lymphoma idiotype-specific T-cell response in patients with follicular lymphoma: Support for a “vaccinal effect” of rituximab. Blood 2009, 113, 3809–3812. [Google Scholar] [CrossRef] [PubMed]
- Dixon, K.J.; Wu, J.; Walcheck, B. Engineering Anti-Tumor Monoclonal Antibodies and Fc Receptors to Enhance ADCC by Human NK Cells. Cancers 2021, 13, 312. [Google Scholar] [CrossRef] [PubMed]
- Vankemmelbeke, M.; McIntosh, R.S.; Chua, J.X.; Kirk, T.; Daniels, I.; Patsalidou, M.; Moss, R.; Parsons, T.; Scott, D.; Harris, G.; et al. Engineering the Human Fc Region Enables Direct Cell Killing by Cancer Glycan-Targeting Antibodies without the Need for Immune Effector Cells or Complement. Cancer Res. 2020, 80, 3399–3412. [Google Scholar] [CrossRef] [PubMed]
- DiLillo, D.J.; Ravetch, J.V. Differential Fc-Receptor Engagement Drives an Anti-tumor Vaccinal Effect. Cell 2015, 161, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Li, S.S.; Gilbert, P.B.; Carpp, L.N.; Pyo, C.-W.; Janes, H.; Fong, Y.; Shen, X.; Neidich, S.D.; Goodman, D.; DeCamp, A.; et al. Fc Gamma Receptor Polymorphisms Modulated the Vaccine Effect on HIV-1 Risk in the HVTN 505 HIV Vaccine Trial. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Qiao, J.; Al-Tamimi, M.; Baker, R.I.; Andrews, R.K.; Gardiner, E.E. The platelet Fc receptor, FcgammaRIIa. Immunol. Rev. 2015, 268, 241–252. [Google Scholar] [CrossRef]
- Ben Mkaddem, S.; Benhamou, M.; Monteiro, R.C. Understanding Fc Receptor Involvement in Inflammatory Diseases: From Mechanisms to New Therapeutic Tools. Front. Immunol. 2019, 10, 811. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.M.; Ottensmeier, C.H.; Mulatero, C.; Lorigan, P.; Plummer, R.; Pandha, H.; Elsheikh, S.; Hadjimichael, E.; Villasanti, N.; Adams, S.E.; et al. Targeting gp100 and TRP-2 with a DNA vaccine: Incorporating T cell epitopes with a human IgG1 antibody induces potent T cell responses that are associated with favourable clinical outcome in a phase I/II trial. Oncoimmunology 2018, 7, e1433516. [Google Scholar] [CrossRef]
- Fletcher, E.A.K.; Van Maren, W.; Cordfunke, R.; Dinkelaar, J.; Codee, J.D.C.; Van Der Marel, G.; Melief, C.J.M.; Ossendorp, F.; Drijfhout, J.W.; Mangsbo, S.M. Formation of Immune Complexes with a Tetanus-Derived B Cell Epitope Boosts Human T Cell Responses to Covalently Linked Peptides in an Ex Vivo Blood Loop System. J. Immunol. 2018, 201, 87–97. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alcaide, E.G.; Krishnarajah, S.; Junker, F. Dendritic Cell Tumor Vaccination via Fc Gamma Receptor Targeting: Lessons Learned from Pre-Clinical and Translational Studies. Vaccines 2021, 9, 409. https://doi.org/10.3390/vaccines9040409
Alcaide EG, Krishnarajah S, Junker F. Dendritic Cell Tumor Vaccination via Fc Gamma Receptor Targeting: Lessons Learned from Pre-Clinical and Translational Studies. Vaccines. 2021; 9(4):409. https://doi.org/10.3390/vaccines9040409
Chicago/Turabian StyleAlcaide, Enrique Gómez, Sinduya Krishnarajah, and Fabian Junker. 2021. "Dendritic Cell Tumor Vaccination via Fc Gamma Receptor Targeting: Lessons Learned from Pre-Clinical and Translational Studies" Vaccines 9, no. 4: 409. https://doi.org/10.3390/vaccines9040409
APA StyleAlcaide, E. G., Krishnarajah, S., & Junker, F. (2021). Dendritic Cell Tumor Vaccination via Fc Gamma Receptor Targeting: Lessons Learned from Pre-Clinical and Translational Studies. Vaccines, 9(4), 409. https://doi.org/10.3390/vaccines9040409