
Preclinical Marmoset Model for Targeting Chronic Inflammation as a Strategy to Prevent Alzheimer’s Disease


Abstract
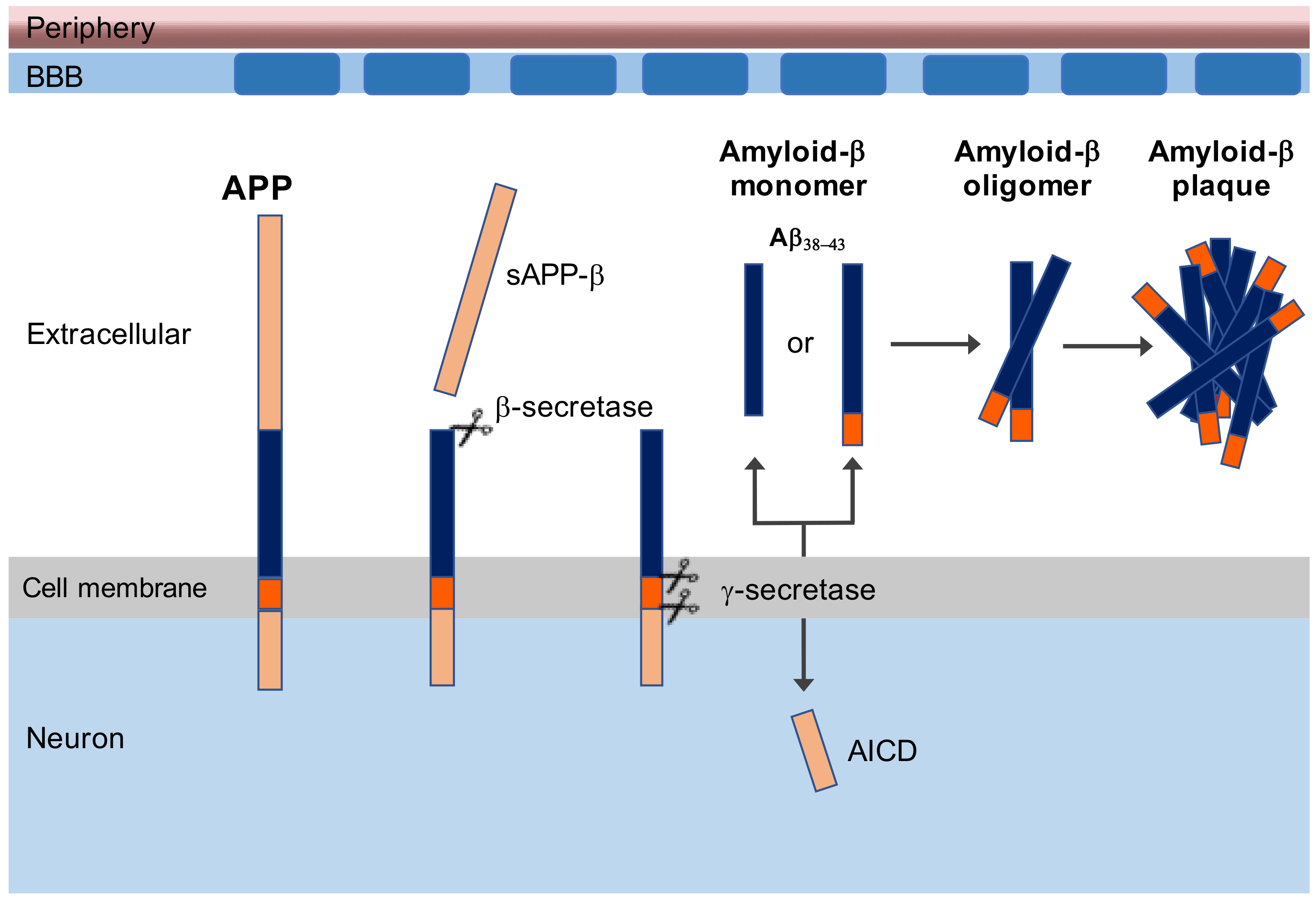
1. Introduction
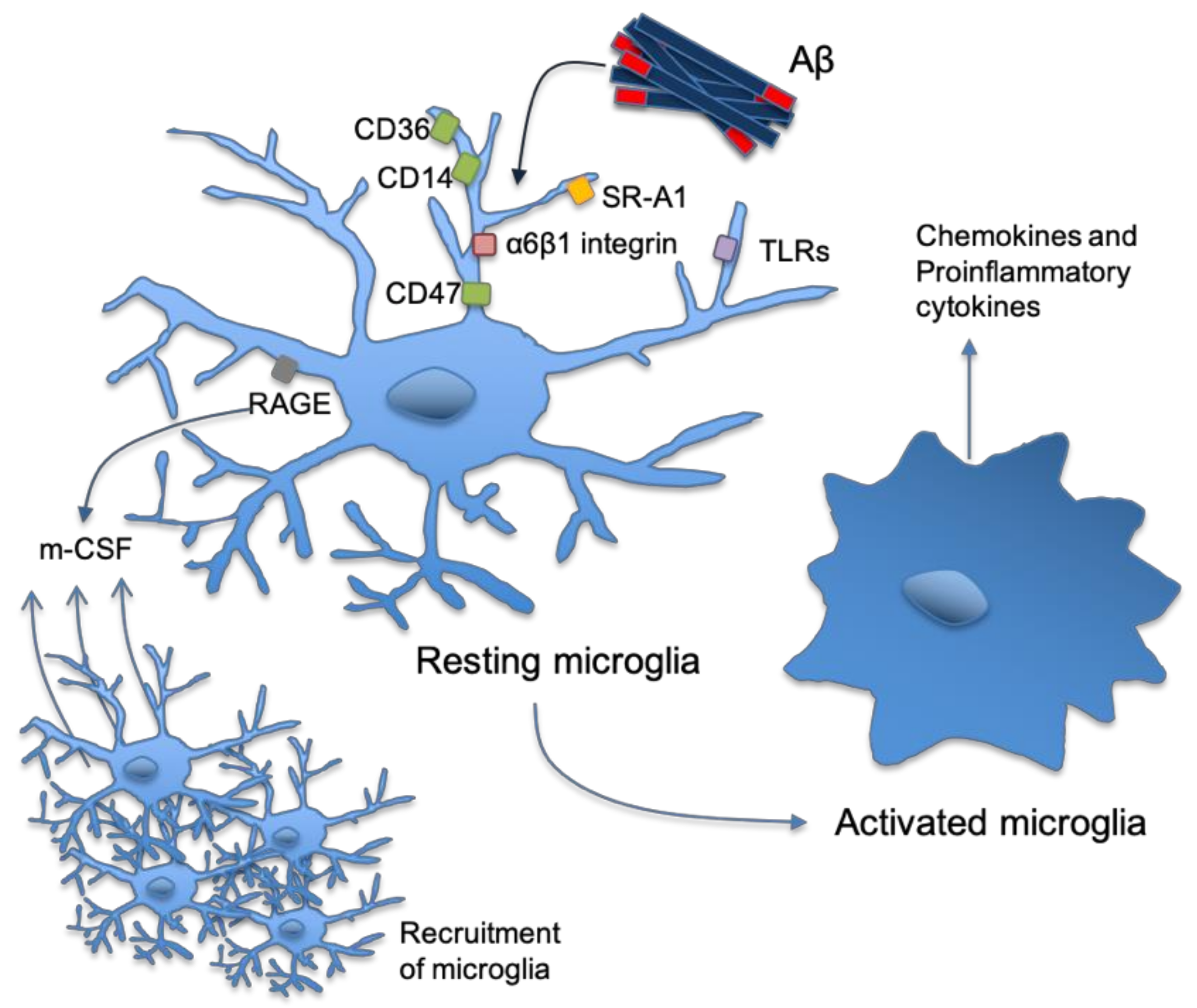
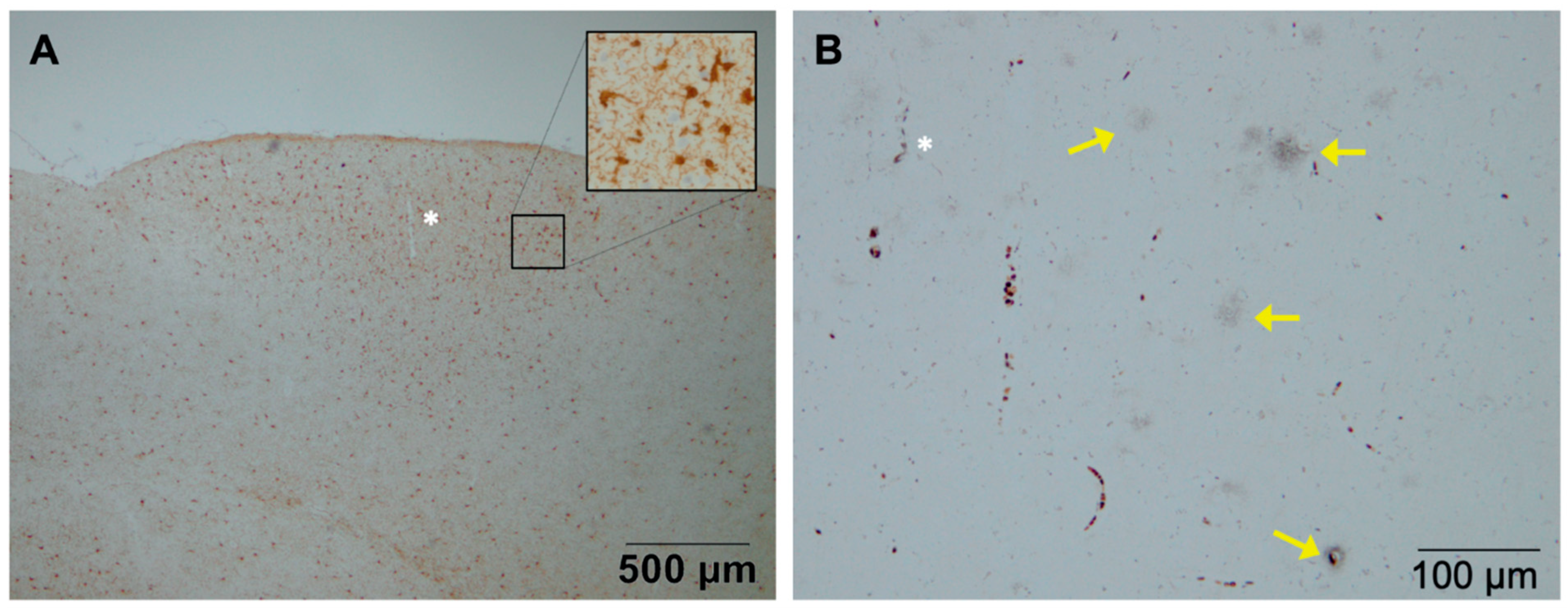
2. Astrocytes and Microglia
3. Inflammaging
4. Difficulties in Finding Targets for Treatment
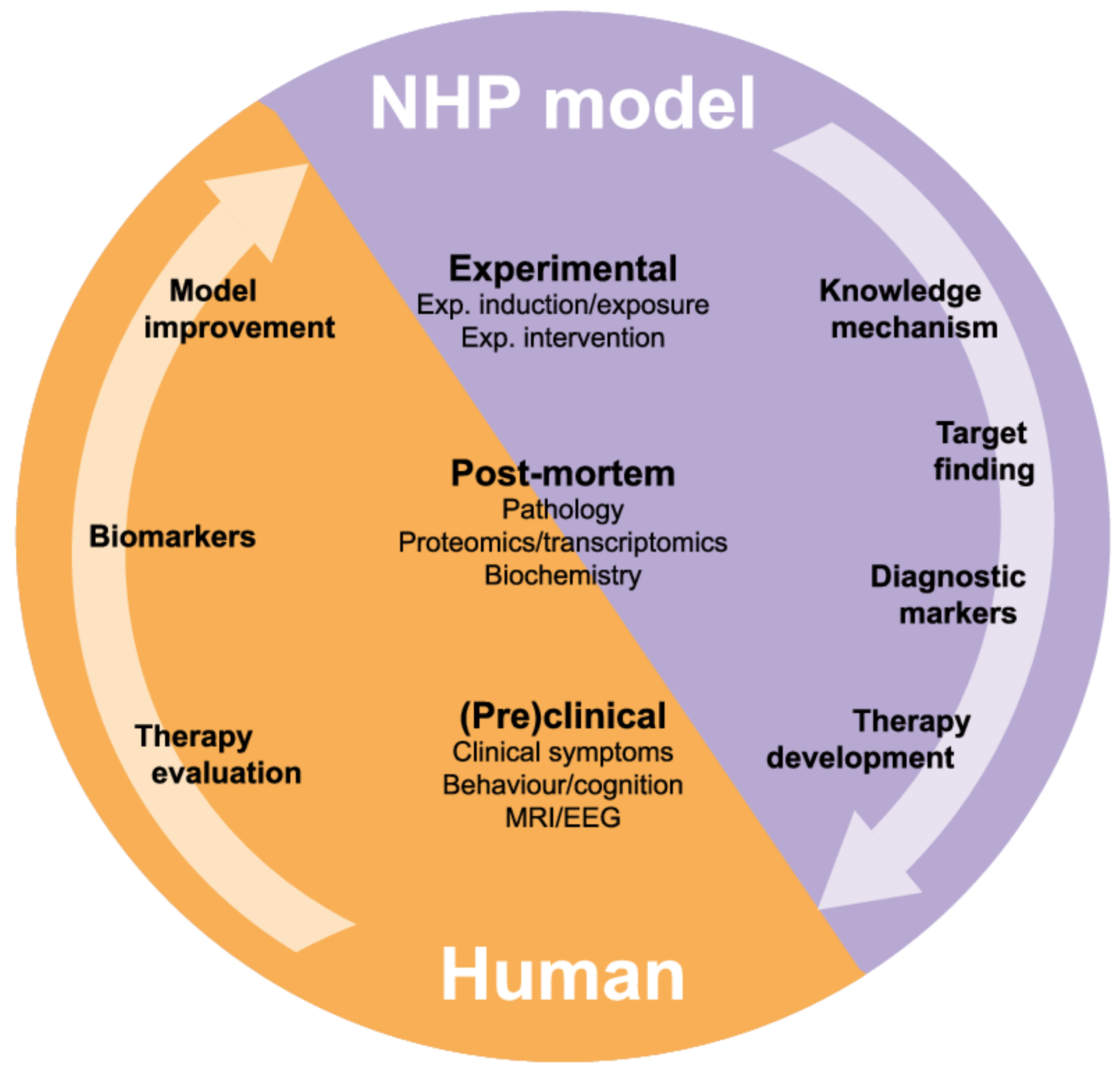
5. Research to Find Targets for Treatment: Relevance of Marmoset Models
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s disease facts and figures. Alzheimers Dement. 2016, 12, 459–509. [CrossRef] [PubMed]
- Phillips, K.A.; Watson, C.M.; Bearman, A.; Knippenberg, A.R.; Adams, J.; Ross, C.; Tardif, S.D. Age-related changes in myelin of axons of the corpus callosum and cognitive decline in common marmosets. Am. J. Primatol. 2019, 81, e22949. [Google Scholar] [CrossRef] [PubMed]
- Lacreuse, A.; Raz, N.; Schmidtke, D.; Hopkins, W.D.; Herndon, J.G. Age-related decline in executive function as a hallmark of cognitive ageing in primates: An overview of cognitive and neurobiological studies. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2020, 375, 20190618. [Google Scholar] [CrossRef] [PubMed]
- Sadoun, A.; Rosito, M.; Fonta, C.; Girard, P. Key periods of cognitive decline in a nonhuman primate model of cognitive aging, the common marmoset (Callithrix jacchus). Neurobiol. Aging 2019, 74, 1–14. [Google Scholar] [CrossRef]
- Noorbakhsh, F.; Overall, C.M.; Power, C. Deciphering complex mechanisms in neurodegenerative diseases: The advent of systems biology. Trends Neurosci. 2009, 32, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Perry, G.; Moreira, P.I.; Garrett, M.R.; Liu, Q.; Zhu, X.; Takeda, A.; Nunomura, A.; Smith, M.A. Tau phosphorylation in Alzheimer’s disease: Pathogen or protector? Trends Mol. Med. 2005, 11, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Ann. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef]
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Elliott, J.I.; Van Nostrand, W.E.; Smith, S.O. Structural conversion of neurotoxic amyloid-β (1–42) oligomers to fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561–567. [Google Scholar] [CrossRef]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Miyata, H.; Kametani, F.; Nonaka, T.; Akiyama, H.; Hisanaga, S.; Hasegawa, M. Extracellular association of APP and tau fibrils induces intracellular aggregate formation of tau. Acta Neuropathol. 2015, 129, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, D.M.; Hurban, J.; Helman, A.M.; Sudduth, T.L.; McCarty, K.L.; Beckett, T.L.; Ferrell, J.C.; Murphy, M.P.; Abner, E.L.; Schmitt, F.A.; et al. Down syndrome individuals with Alzheimer’s disease have a distinct neuroinflammatory phenotype compared to sporadic Alzheimer’s disease. Neurobiol. Aging 2015, 36, 2468–2474. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef]
- Giunta, B.; Fernandez, F.; Nikolic, W.V.; Obregon, D.; Rrapo, E.; Town, T.; Tan, J. Inflammaging as a prodrome to Alzheimer’s disease. J. Neuroinflamm. 2008, 5, 51. [Google Scholar] [CrossRef]
- Hanzel, C.E.; Pichet-Binette, A.; Pimentel, L.S.; Iulita, M.F.; Allard, S.; Ducatenzeiler, A.; Do Carmo, S.; Cuello, A.C. Neuronal driven pre-plaque inflammation in a transgenic rat model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2249–2262. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.M.; Hong, J.S. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Parthsarathy, V.; McClean, P.L.; Holscher, C.; Taylor, M.; Tinker, C.; Jones, G.; Kolosov, O.; Salvati, E.; Gregori, M.; Masserini, M.; et al. A novel retro-inverso peptide inhibitor reduces amyloid deposition, oxidation and inflammation and stimulates neurogenesis in the APPswe/PS1DeltaE9 mouse model of Alzheimer’s disease. PLoS ONE 2013, 8, e54769. [Google Scholar] [CrossRef]
- Fiala, M.; Veerhuis, R. Biomarkers of inflammation and amyloid-β phagocytosis in patients at risk of Alzheimer disease. Exp. Gerontol. 2010, 45, 57–63. [Google Scholar] [CrossRef]
- Jaeger, L.B.; Dohgu, S.; Sultana, R.; Lynch, J.L.; Owen, J.B.; Erickson, M.A.; Shah, G.N.; Price, T.O.; Fleegal-Demotta, M.A.; Butterfield, D.A.; et al. Lipopolysaccharide alters the blood-brain barrier transport of amyloid β protein: A mechanism for inflammation in the progression of Alzheimer’s disease. Brain Behav. Immun. 2009, 23, 507–517. [Google Scholar] [CrossRef]
- Salminen, A.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Inflammation in Alzheimer’s disease: Amyloid-β oligomers trigger innate immunity defence via pattern recognition receptors. Prog. Neurobiol. 2009, 87, 181–194. [Google Scholar] [CrossRef] [PubMed]
- White, J.A.; Manelli, A.M.; Holmberg, K.H.; Van Eldik, L.J.; Ladu, M.J. Differential effects of oligomeric and fibrillar amyloid-β 1–42 on astrocyte-mediated inflammation. Neurobiol. Dis. 2005, 18, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Cuello, A.C.; Ferretti, M.T.; Iulita, M.F. Preplaque (preclinical) Abeta-induced inflammation and nerve growth factor deregulation in transgenic models of Alzheimer’s disease-like amyloid pathology. Neurodegener. Dis. 2012, 10, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Cuello, A.C.; Ferretti, M.T.; Leon, W.C.; Iulita, M.F.; Melis, T.; Ducatenzeiler, A.; Bruno, M.A.; Canneva, F. Early-stage inflammation and experimental therapy in transgenic models of the Alzheimer-like amyloid pathology. Neurodegener. Dis. 2010, 7, 96–98. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Town, T.; Abdullah, L.; Wu, Y.; Placzek, A.; Small, B.; Kroeger, J.; Crawford, F.; Richards, D.; Mullan, M. CD45 isoform alteration in CD4+ T cells as a potential diagnostic marker of Alzheimer’s disease. J. Neuroimmunol. 2002, 132, 164–172. [Google Scholar] [CrossRef]
- Lombardi, V.R.; Garcia, M.; Rey, L.; Cacabelos, R. Characterization of cytokine production, screening of lymphocyte subset patterns and in vitro apoptosis in healthy and Alzheimer’s disease (AD) individuals. J. Neuroimmunol. 1999, 97, 163–171. [Google Scholar] [CrossRef]
- Rezai-Zadeh, K.; Gate, D.; Town, T. CNS infiltration of peripheral immune cells: D-Day for neurodegenerative disease? J. Neuroimm. Pharmacol. 2009, 4, 462–475. [Google Scholar] [CrossRef]
- Eikelenboom, P.; Bate, C.; Van Gool, W.A.; Hoozemans, J.J.; Rozemuller, J.M.; Veerhuis, R.; Williams, A. Neuroinflammation in Alzheimer’s disease and prion disease. Glia 2002, 40, 232–239. [Google Scholar] [CrossRef]
- Lee, J.W.; Lee, Y.K.; Yuk, D.Y.; Choi, D.Y.; Ban, S.B.; Oh, K.W.; Hong, J.T. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflamm. 2008, 5, 37. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Muzikansky, A.; Gomez-Isla, T.; Growdon, J.H.; Betensky, R.A.; Frosch, M.P.; Hyman, B.T. Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2013, 72, 462–471. [Google Scholar] [CrossRef]
- Li, Y.; Tan, M.S.; Jiang, T.; Tan, L. Microglia in Alzheimer’s disease. Biomed. Res. Int. 2014, 2014, 437483. [Google Scholar] [CrossRef] [PubMed]
- Marlatt, M.W.; Bauer, J.; Aronica, E.; van Haastert, E.S.; Hoozemans, J.J.; Joels, M.; Lucassen, P.J. Proliferation in the Alzheimer hippocampus is due to microglia, not astroglia, and occurs at sites of amyloid deposition. Neural Plast. 2014, 2014, 693851. [Google Scholar] [CrossRef] [PubMed]
- Philippens, I.H.; Hart, B.A.; Torres, G. The MPTP marmoset model of parkinsonism: A multi-purpose non-human primate model for neurodegenerative diseases. Drug Discov. Today 2010, 15, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; O’Connor, T.; Vassar, R. The contribution of activated astrocytes to Aβ production: Implications for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2011, 8, 150. [Google Scholar] [CrossRef]
- Kraft, A.W.; Hu, X.; Yoon, H.; Yan, P.; Xiao, Q.; Wang, Y.; Gil, S.C.; Brown, J.; Wilhelmsson, U.; Restivo, J.L.; et al. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 2013, 27, 187–198. [Google Scholar] [CrossRef]
- Narayan, P.; Holmstrom, K.M.; Kim, D.H.; Whitcomb, D.J.; Wilson, M.R.; St George-Hyslop, P.; Wood, N.W.; Dobson, C.M.; Cho, K.; Abramov, A.Y.; et al. Rare individual amyloid-β oligomers act on astrocytes to initiate neuronal damage. Biochemistry 2014, 53, 2442–2453. [Google Scholar] [CrossRef]
- Thal, D.R. The role of astrocytes in amyloid beta-protein toxicity and clearance. Exp. Neurol. 2012, 236, 1–5. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Latta, C.H.; Brothers, H.M.; Wilcock, D.M. Neuroinflammation in Alzheimer’s disease: A source of heterogeneity and target for personalized therapy. Neuroscience 2015, 302, 103–111. [Google Scholar] [CrossRef]
- Karperien, A.; Ahammer, H.; Jelinek, H.F. Quantitating the subtleties of microglial morphology with fractal analysis. Front. Cell Neurosci. 2013, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Serpente, M.; Bonsi, R.; Scarpini, E.; Galimberti, D. Innate immune system and inflammation in Alzheimer’s disease: From pathogenesis to treatment. Neuroimmunomodulation 2014, 21, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Du Yan, S.; Zhu, H.; Fu, J.; Yan, S.F.; Roher, A.; Tourtellotte, W.W.; Rajavashisth, T.; Chen, X.; Godman, G.C.; Stern, D.; et al. Amyloid-β peptide-receptor for advanced glycation endproduct interaction elicits neuronal expression of macrophage-colony stimulating factor: A proinflammatory pathway in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1997, 94, 5296–5301. [Google Scholar] [CrossRef] [PubMed]
- Doens, D.; Fernandez, P.L. Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2014, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Sondag, C.M.; Dhawan, G.; Combs, C.K. Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J. Neuroinflamm. 2009, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Maezawa, I.; Zimin, P.I.; Wulff, H.; Jin, L.W. Amyloid-β protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J. Biol. Chem. 2011, 286, 3693–3706. [Google Scholar] [CrossRef]
- Frautschy, S.A.; Yang, F.; Irrizarry, M.; Hyman, B.; Saido, T.C.; Hsiao, K.; Cole, G.M. Microglial response to amyloid plaques in APPsw transgenic mice. Am. J. Pathol. 1998, 152, 307–317. [Google Scholar] [PubMed]
- Walker, D.G.; Lue, L.F. Investigations with cultured human microglia on pathogenic mechanisms of Alzheimer’s disease and other neurodegenerative diseases. J. Neurosci. Res. 2005, 81, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Yu, Q.; Arancio, O.; Chen, D.; Gore, S.S.; Yan, S.S.; Yan, S.F. RAGE mediates Aβ accumulation in a mouse model of Alzheimer’s disease via modulation of β and γ-secretase activity. Hum. Mol. Genet. 2018, 27, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Hu, X.; Qian, L.; Chen, S.H.; Zhou, H.; Wilson, B.; Miller, D.S.; Hong, J.S. Microglial MAC1 receptor and PI3K are essential in mediating β-amyloid peptide-induced microglial activation and subsequent neurotoxicity. J. Neuroinflamm. 2011, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Sudduth, T.L.; Schmitt, F.A.; Nelson, P.T.; Wilcock, D.M. Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1051–1059. [Google Scholar] [CrossRef]
- Minhas, P.S.; Latif-Hernandez, A.; McReynolds, M.R.; Durairaj, A.S.; Wang, Q.; Rubin, A.; Joshi, A.U.; He, J.Q.; Gauba, E.; Liu, L.; et al. Restoring metabolism of myeloid cells reverses cognitive decline in ageing. Nature 2021, 590, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Montine, T.J.; Sidell, K.R.; Crews, B.C.; Markesbery, W.R.; Marnett, L.J.; Roberts, L.J., 2nd; Morrow, J.D. Elevated CSF prostaglandin E2 levels in patients with probable AD. Neurology 1999, 53, 1495–1498. [Google Scholar] [CrossRef] [PubMed]
- Njie, E.G.; Boelen, E.; Stassen, F.R.; Steinbusch, H.W.; Borchelt, D.R.; Streit, W.J. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol. Aging 2012, 33, 195.e1–195.e12. [Google Scholar] [CrossRef] [PubMed]
- Solito, E.; Sastre, M. Microglia function in Alzheimer’s disease. Front. Pharmacol. 2012, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C. Microglia and neurodegeneration: The role of systemic inflammation. Glia 2013, 61, 71–90. [Google Scholar] [CrossRef]
- Perry, V.H.; Holmes, C. Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 2014, 10, 217–224. [Google Scholar] [CrossRef]
- Blasko, I.; Stampfer-Kountchev, M.; Robatscher, P.; Veerhuis, R.; Eikelenboom, P.; Grubeck-Loebenstein, B. How chronic inflammation can affect the brain and support the development of Alzheimer’s disease in old age: The role of microglia and astrocytes. Aging Cell. 2004, 3, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Rodriguez, L.; Lopez-Hoyos, M.; Munoz-Cacho, P.; Martinez-Taboada, V.M. Aging is associated with circulating cytokine dysregulation. Cell Immunol. 2012, 273, 124–132. [Google Scholar] [CrossRef]
- Conde, J.R.; Streit, W.J. Microglia in the aging brain. J. Neuropathol. Exp. Neurol. 2006, 65, 199–203. [Google Scholar] [CrossRef]
- Baron, R.; Babcock, A.A.; Nemirovsky, A.; Finsen, B.; Monsonego, A. Accelerated microglial pathology is associated with Aβ plaques in mouse models of Alzheimer’s disease. Aging Cell. 2014, 13, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J. Microglial senescence: Does the brain’s immune system have an expiration date? Trends Neurosci. 2006, 29, 506–510. [Google Scholar] [CrossRef]
- Soulet, D.; Rivest, S. Microglia. Curr. Biol. 2008, 18, R506–R508. [Google Scholar] [CrossRef] [PubMed]
- Kamboh, M.I.; Sanghera, D.K.; Ferrell, R.E.; DeKosky, S.T. APOE*4-associated Alzheimer’s disease risk is modified by α 1-antichymotrypsin polymorphism. Nat. Genet. 1995, 10, 486–488. [Google Scholar] [CrossRef]
- Papassotiropoulos, A.; Bagli, M.; Jessen, F.; Bayer, T.A.; Maier, W.; Rao, M.L.; Heun, R. A genetic variation of the inflammatory cytokine interleukin-6 delays the initial onset and reduces the risk for sporadic Alzheimer’s disease. Ann. Neurol. 1999, 45, 666–668. [Google Scholar] [CrossRef]
- McCusker, S.M.; Curran, M.D.; Dynan, K.B.; McCullagh, C.D.; Urquhart, D.D.; Middleton, D.; Patterson, C.C.; McIlroy, S.P.; Passmore, A.P. Association between polymorphism in regulatory region of gene encoding tumour necrosis factor alpha and risk of Alzheimer’s disease and vascular dementia: A case-control study. Lancet 2001, 357, 436–439. [Google Scholar] [CrossRef]
- Donnelly, R.J.; Friedhoff, A.J.; Beer, B.; Blume, A.J.; Vitek, M.P. Interleukin-1 stimulates the beta-amyloid precursor protein promoter. Cell. Mol. Neurobiol. 1990, 10, 485–495. [Google Scholar] [CrossRef]
- Goldgaber, D.; Harris, H.W.; Hla, T.; Maciag, T.; Donnelly, R.J.; Jacobsen, J.S.; Vitek, M.P.; Gajdusek, D.C. Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc. Natl. Acad. Sci. USA 1989, 86, 7606–7610. [Google Scholar] [CrossRef]
- Mrak, R.E.; Griffin, W.S. Interleukin-1, neuroinflammation, and Alzheimer’s disease. Neurobiol. Aging 2001, 22, 903–908. [Google Scholar] [CrossRef]
- Swomley, A.M.; Forster, S.; Keeney, J.T.; Triplett, J.; Zhang, Z.; Sultana, R.; Butterfield, D.A. Abeta, oxidative stress in Alzheimer disease: Evidence based on proteomics studies. Biochim. Biophys. Acta 2014, 1842, 1248–1257. [Google Scholar] [CrossRef]
- Dilger, R.N.; Johnson, R.W. Aging, microglial cell priming, and the discordant central inflammatory response to signals from the peripheral immune system. J. Leukoc. Biol. 2008, 84, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Bamberger, M.E.; Harris, M.E.; McDonald, D.R.; Husemann, J.; Landreth, G.E. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J. Neurosci. 2003, 23, 2665–2674. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Zhong, Z.; Lindholm, K.; Berning, L.; Lee, W.; Lemere, C.; Staufenbiel, M.; Li, R.; Shen, Y. Deletion of tumor necrosis factor death receptor inhibits amyloid β generation and prevents learning and memory deficits in Alzheimer’s mice. J. Cell. Biol. 2007, 178, 829–841. [Google Scholar] [CrossRef]
- Johnston, H.; Boutin, H.; Allan, S.M. Assessing the contribution of inflammation in models of Alzheimer’s disease. Biochem. Soc. Trans. 2011, 39, 886–890. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease--a double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar] [CrossRef]
- Glezer, I.; Simard, A.R.; Rivest, S. Neuroprotective role of the innate immune system by microglia. Neuroscience 2007, 147, 867–883. [Google Scholar] [CrossRef]
- Soulet, D.; Rivest, S. Bone-marrow-derived microglia: Myth or reality? Curr. Opin. Pharmacol. 2008, 8, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.P.; Rivest, S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 2006, 49, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Guan, P.P.; Wang, T.; Yu, X.; Guo, J.J.; Wang, Z.Y. Aggravation of Alzheimer’s disease due to the COX-2-mediated reciprocal regulation of IL-1 β and Aβ between glial and neuron cells. Aging Cell. 2014, 13, 605–615. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. Anti-inflammatory drugs in the fight against Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1996, 777, 213–220. [Google Scholar] [CrossRef] [PubMed]
- in’t Veld, B.A.; Launer, L.J.; Hoes, A.W.; Ott, A.; Hofman, A.; Breteler, M.M.; Stricker, B.H. NSAIDs and incident Alzheimer’s disease. The Rotterdam Study. Neurobiol. Aging 1998, 19, 607–611. [Google Scholar] [CrossRef]
- Launer, L. Nonsteroidal anti-inflammatory drug use and the risk for Alzheimer’s disease: Dissecting the epidemiological evidence. Drugs 2003, 63, 731–739. [Google Scholar] [CrossRef]
- Cote, S.; Carmichael, P.H.; Verreault, R.; Lindsay, J.; Lefebvre, J.; Laurin, D. Nonsteroidal anti-inflammatory drug use and the risk of cognitive impairment and Alzheimer’s disease. Alzheimers Dement. 2012, 8, 219–226. [Google Scholar] [CrossRef]
- Weekman, E.M.; Sudduth, T.L.; Abner, E.L.; Popa, G.J.; Mendenhall, M.D.; Brothers, H.M.; Braun, K.; Greenstein, A.; Wilcock, D.M. Transition from an M1 to a mixed neuroinflammatory phenotype increases amyloid deposition in APP/PS1 transgenic mice. J. Neuroinflamm. 2014, 11, 127. [Google Scholar] [CrossRef]
- Philipson, O.; Lord, A.; Gumucio, A.; O’Callaghan, P.; Lannfelt, L.; Nilsson, L.N. Animal models of amyloid-beta-related pathologies in Alzheimer’s disease. FEBS J. 2010, 277, 1389–1409. [Google Scholar] [CrossRef]
- Mandavilli, A. The amyloid code. Nat. Med. 2006, 12, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Egan, K.J.; Vesterinen, H.M.; Beglopoulos, V.; Sena, E.S.; Macleod, M.R. From a mouse: Systematic analysis reveals limitations of experiments testing interventions in Alzheimer’s disease mouse models. Evid. Based Preclin. Med. 2016, 3, e00015. [Google Scholar] [CrossRef]
- Gotz, J.; Gotz, N.N. Animal models for Alzheimer’s disease and frontotemporal dementia: A perspective. ASN Neuro 2009, 1, e00019. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Carrion, R., Jr.; Patterson, J.L. An animal model that reflects human disease: The common marmoset (Callithrix jacchus). Curr. Opin. Virol. 2012, 2, 357–362. [Google Scholar] [CrossRef]
- Ross, C.N.; Davis, K.; Dobek, G.; Tardif, S.D. Aging Phenotypes of Common Marmosets (Callithrix jacchus). J. Aging Res. 2012, 2012, 567143. [Google Scholar] [CrossRef]
- Latimer, C.S.; Shively, C.A.; Keene, C.D.; Jorgensen, M.J.; Andrews, R.N.; Register, T.C.; Montine, T.J.; Wilson, A.M.; Neth, B.J.; Mintz, A.; et al. A nonhuman primate model of early Alzheimer’s disease pathologic change: Implications for disease pathogenesis. Alzheimers Dement. 2019, 15, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C.; Cork, L.C. The neurobiology of aging in nonhuman primates. In Alzheimer Disease; Terry, R.D., Katzman, R., Bick, K.L., Sisodia, S.S., Eds.; Lippincott Williams & Wilkin: Philadelphia, PA, USA, 1999; pp. 233–243. [Google Scholar]
- Mansfield, K. Marmoset models commonly used in biomedical research. Comp. Med. 2003, 53, 383–392. [Google Scholar] [PubMed]
- Geula, C.; Nagykery, N.; Wu, C.K. Amyloid-β deposits in the cerebral cortex of the aged common marmoset (Callithrix jacchus): Incidence and chemical composition. Acta Neuropathol. 2002, 103, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Rosen, R.F.; Walker, L.C.; Levine, H. 3rd. PIB binding in aged primate brain: Enrichment of high-affinity sites in humans with Alzheimer’s disease. Neurobiol. Aging 2011, 32, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Paspalas, C.D.; Carlyle, B.C.; Leslie, S.; Preuss, T.M.; Crimins, J.L.; Huttner, A.J.; van Dyck, C.H.; Rosene, D.L.; Nairn, A.C.; Arnsten, A.F.T. The aged rhesus macaque manifests Braak stage III/IV Alzheimer’s-like pathology. Alzheimers Dement. 2018, 14, 680–691. [Google Scholar] [CrossRef]
- Rodriguez-Callejas, J.D.; Fuchs, E.; Perez-Cruz, C. Evidence of tau hyperphosphorylation and dystrophic microglia in the common marmoset. Front. Aging Neurosci. 2016, 8, 315. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. Alzheimer’s disease: Pathogenesis and prevention. Alzheimers Dement. 2012, 8, 227–233. [Google Scholar] [CrossRef]
- Sharma, G.; Huo, A.; Kimura, T.; Shiozawa, S.; Kobayashi, R.; Sahara, N.; Ishibashi, M.; Ishigaki, S.; Saito, T.; Ando, K.; et al. Tau isoform expression and phosphorylation in marmoset brains. J. Biol. Chem. 2019, 294, 11433–11444. [Google Scholar] [CrossRef]
- Beckman, D.; Chakrabarty, P.; Ott, S.; Dao, A.; Zhou, E.; Janssen, W.G.; Donis-Cox, K.; Muller, S.; Kordower, J.H.; Morrison, J.H. A novel tau-based rhesus monkey model of Alzheimer’s pathogenesis. Alzheimers Dement. 2021. [Google Scholar] [CrossRef]
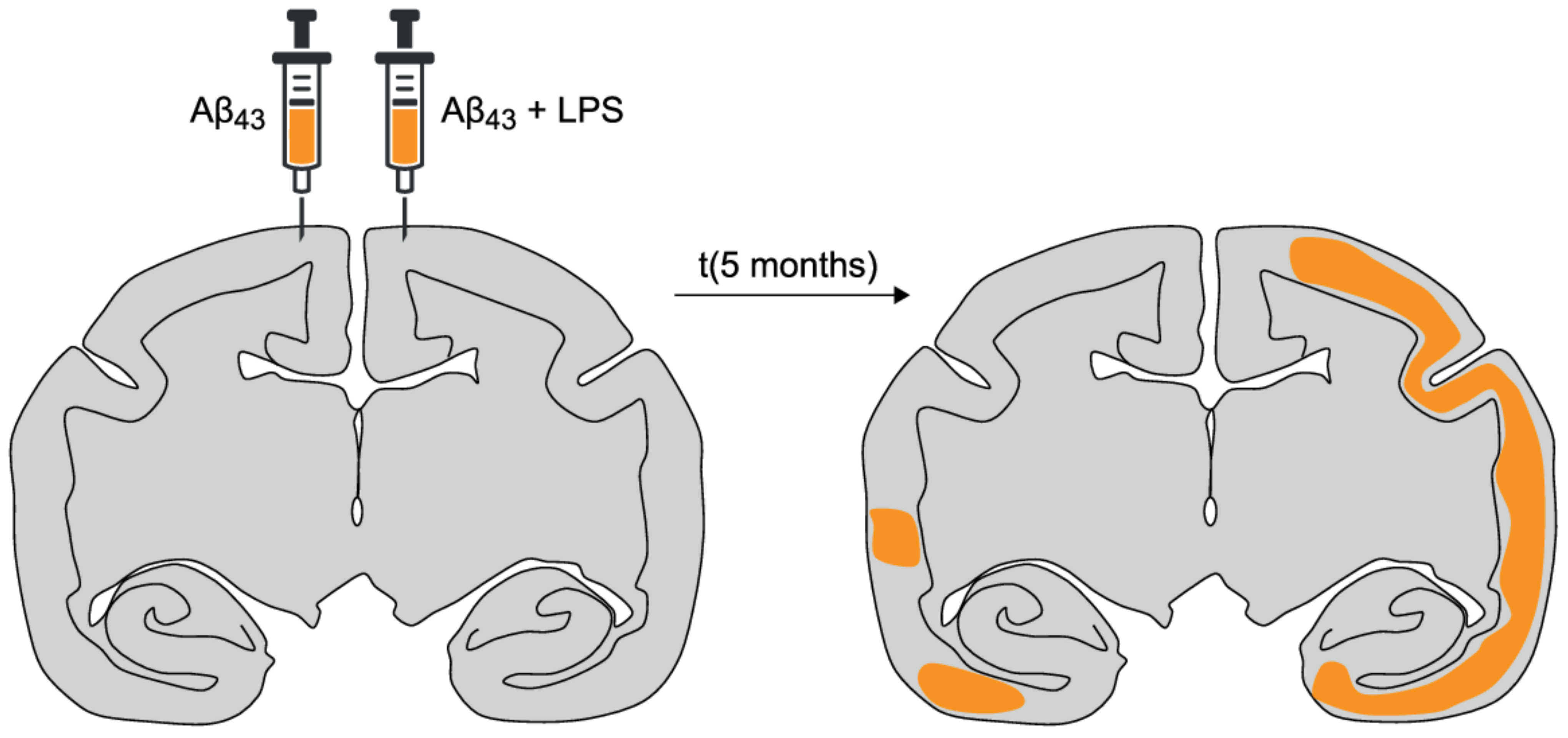
- Philippens, I.H.; Ormel, P.R.; Baarends, G.; Johansson, M.; Remarque, E.J.; Doverskog, M. Acceleration of Amyloidosis by Inflammation in the Amyloid-β marmoset monkey model of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 55, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, E.; Suemizu, H.; Shimada, A.; Hanazawa, K.; Oiwa, R.; Kamioka, M.; Tomioka, I.; Sotomaru, Y.; Hirakawa, R.; Eto, T.; et al. Generation of transgenic non-human primates with germline transmission. Nature 2009, 459, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Maclean, C.J.; Baker, H.F.; Ridley, R.M.; Mori, H. Naturally occurring and experimentally induced beta-amyloid deposits in the brains of marmosets (Callithrix jacchus). J. Neural Transm. 2000, 107, 799–814. [Google Scholar] [CrossRef] [PubMed]
- Ridley, R.M.; Baker, H.F.; Windle, C.P.; Cummings, R.M. Very long term studies of the seeding of beta-amyloidosis in primates. J. Neural Transm. 2006, 113, 1243–1251. [Google Scholar] [CrossRef]
- Griffin, W.S.; Sheng, J.G.; Royston, M.C.; Gentleman, S.M.; McKenzie, J.E.; Graham, D.I.; Roberts, G.W.; Mrak, R.E. Glial-neuronal interactions in Alzheimer’s disease: The potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. 1998, 8, 65–72. [Google Scholar] [CrossRef]
- Logan, A.C.; Khan, K.N. Clinical pathologic changes in two marmosets with wasting syndrome. Toxicol. Pathol. 1996, 24, 707–709. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, E.; Okano, Y.; Niimi, K.; Takahashi, E. Detection of calprotectin and apoptotic activity in the colon of marmosets with chronic diarrhea. J. Vet. Med. Sci. 2013, 75, 1633–1636. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Van Dam, D.; De Deyn, P.P. Non human primate models for Alzheimer’s disease-related research and drug discovery. Expert Opin. Drug Discov. 2017, 12, 187–200. [Google Scholar] [CrossRef]
- Colman, R.J. Non-human primates as a model for aging. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2733–2741. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Busquets, O.; Ettcheto, M.; Sanchez-Lopez, E.; Pallas, M.; Beas-Zarate, C.; Marin, M.; Casadesus, G.; Olloquequi, J.; Auladell, C.; et al. Experimental Models for Aging and their Potential for Novel Drug Discovery. Curr. Neuropharmacol. 2018, 16, 1466–1483. [Google Scholar] [CrossRef]
- Kaas, J.H. The evolution of brains from early mammals to humans. Wiley Interdiscip. Rev. Cogn. Sci. 2013, 4, 33–45. [Google Scholar] [CrossRef]
- Didier, E.S.; MacLean, A.G.; Mohan, M.; Didier, P.J.; Lackner, A.A.; Kuroda, M.J. Contributions of nonhuman primates to research on aging. Vet. Pathol. 2016, 53, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Weerts, E.M.; Fantegrossi, W.E.; Goodwin, A.K. The value of nonhuman primates in drug abuse research. Exp. Clin. Psychopharmacol. 2007, 15, 309–327. [Google Scholar] [CrossRef] [PubMed]
- t Hart, B.A.; Abbott, D.H.; Nakamura, K.; Fuchs, E. The marmoset monkey: A multi-purpose preclinical and translational model of human biology and disease. Drug Discov. Today 2012, 17, 1160–1165. [Google Scholar] [CrossRef] [PubMed]
- Barkholt, P.; Sanchez-Guajardo, V.; Kirik, D.; Romero-Ramos, M. Long-term polarization of microglia upon alpha-synuclein overexpression in nonhuman primates. Neuroscience 2012, 208, 85–96. [Google Scholar] [CrossRef]
- Antunes, S.G.; de Groot, N.G.; Brok, H.; Doxiadis, G.; Menezes, A.A.; Otting, N.; Bontrop, R.E. The common marmoset: A new world primate species with limited Mhc class II variability. Proc. Natl. Acad. Sci. USA 1998, 95, 11745–11750. [Google Scholar] [CrossRef]
- Perlmutter, L.S.; Scott, S.A.; Barron, E.; Chui, H.C. MHC class II-positive microglia in human brain: Association with Alzheimer lesions. J. Neurosci. Res. 1992, 33, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, R.A.; Rogers, J.; Katze, M.G.; Bumgarner, R.; Weinstock, G.M.; Mardis, E.R.; Remington, K.A.; Strausberg, R.L.; Venter, J.C.; Wilson, R.K.; et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science 2007, 316, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Marmoset Genome, S.; Analysis, C. The common marmoset genome provides insight into primate biology and evolution. Nat. Genet. 2014, 46, 850–857. [Google Scholar] [CrossRef]
- Austad, S.N. Comparative biology of aging. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Tardif, S.D.; Mansfield, K.G.; Ratnam, R.; Ross, C.N.; Ziegler, T.E. The marmoset as a model of aging and age-related diseases. ILAR J. 2011, 52, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Finch, C.E.; Austad, S.N. Primate aging in the mammalian scheme: The puzzle of extreme variation in brain aging. Age 2012, 34, 1075–1091. [Google Scholar] [CrossRef] [PubMed]
- Quint, D.J.; Buckham, S.P.; Bolton, E.J.; Solari, R.; Champion, B.R.; Zanders, E.D. Immunoregulation in the common marmoset, Calithrix jaccus: Functional properties of T and B lymphocytes and their response to human interleukins 2 and 4. Immunology 1990, 69, 616–621. [Google Scholar]
- Plaza, D.F.; Gomez, M.F.; Patarroyo, M.A. NHP-immunome: A translational research-oriented database of non-human primate immune system proteins. Cell. Immunol. 2020, 347, 103999. [Google Scholar] [CrossRef] [PubMed]
- Mietsch, M.; Paque, K.; Drummer, C.; Stahl-Hennig, C.; Roshani, B. The aging common marmoset’s immune system: From junior to senior. Am. J. Primatol. 2020, 82, e23128. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, C.; Ward, J.; Vallender, E.J. Naturally occurring, physiologically normal, primate chimeras. Chimerism 2012, 3, 43–44. [Google Scholar] [CrossRef] [PubMed]
- Okano, H.; Sasaki, E.; Yamamori, T.; Iriki, A.; Shimogori, T.; Yamaguchi, Y.; Kasai, K.; Miyawaki, A. Brain/MINDS: A Japanese national brain project for marmoset neuroscience. Neuron 2016, 92, 582–590. [Google Scholar] [CrossRef]
- Philippens, I.H.C.H.M.; Verhave, P.S. Preclinical Solutions for insight in premotor Parkinson. In Parkinson’s Disease; Rana, A.Q., Ed.; IntechOpen Ltd: London, UK, 2011; ISBN 978-953-307-464-1. [Google Scholar]
- Philippens, I.H.C.H.M. Marmosets in neurologic disease research: Parkinson’s disease. In The Common Marmoset in Captivity and Biomedical Research; Marini, R.P., Wachtman, L.M., Tardif, S.D., Mansfield, K., Fox, J.G., Eds.; Academic Press, Elsevier: London, UK, 2019. [Google Scholar]
- Kobayashi, R.; Takahashi-Fujigasaki, J.; Shiozawa, S.; Hara-Miyauchi, C.; Inoue, T.; Okano, H.J.; Sasaki, E.; Okano, H. alpha-Synuclein aggregation in the olfactory bulb of middle-aged common marmoset. Neurosci. Res. 2016, 106, 55–61. [Google Scholar] [CrossRef]
- Philippens, I.H.; Melchers, B.P.; Roeling, T.A.; Bruijnzeel, P.L. Behavioral test systems in marmoset monkeys. Behav. Res. Methods Instrum. Comput. 2000, 32, 173–179. [Google Scholar] [CrossRef]
- Verhave, P.S.; Vanwersch, R.A.; van Helden, H.P.; Smit, A.B.; Philippens, I.H. Two new test methods to quantify motor deficits in a marmoset model for Parkinson’s disease. Behav. Brain Res. 2009, 200, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Kwan, C.; Frouni, I.; Nuara, S.G.; Belliveau, S.; Kang, W.; Hamadjida, A.; Bedard, D.; Beaudry, F.; Panisset, M.; Gourdon, J.C.; et al. Combined 5-HT2A and mGlu2 modulation for the treatment of dyskinesia and psychosis in Parkinson’s disease. Neuropharmacology 2021, 186, 108465. [Google Scholar] [CrossRef]
- Verhave, P.S.; Jongsma, M.J.; Van den Berg, R.M.; Vis, J.C.; Vanwersch, R.A.; Smit, A.B.; Van Someren, E.J.; Philippens, I.H. REM sleep behavior disorder in the marmoset MPTP model of early Parkinson disease. Sleep 2011, 34, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Verhave, P.S.; Jongsma, M.J.; Van Den Berg, R.M.; Vanwersch, R.A.; Smit, A.B.; Philippens, I.H. Neuroprotective effects of riluzole in early phase Parkinson’s disease on clinically relevant parameters in the marmoset MPTP model. Neuropharmacology 2012, 62, 1700–1707. [Google Scholar] [CrossRef]
- Franke, S.K.; van Kesteren, R.E.; Hofman, S.; Wubben, J.A.; Smit, A.B.; Philippens, I.H. Individual and familial susceptibility to MPTP in a common marmoset model for Parkinson’s disease. Neurodegener. Dis. 2016, 16, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Franke, S.K.; van Kesteren, R.E.; Wubben, J.A.; Hofman, S.; Paliukhovich, I.; van der Schors, R.C.; van Nierop, P.; Smit, A.B.; Philippens, I.H. Progression and recovery of Parkinsonism in a chronic progressive MPTP-induction model in the marmoset without persistent molecular and cellular damage. Neuroscience 2016, 312, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Philippens, I.; Wubben, J.A.; Franke, S.K.; Hofman, S.; Langermans, J.A.M. Involvement of the red nucleus in the compensation of parkinsonism may explain why primates can develop stable Parkinson’s disease. Sci. Rep. 2019, 9, 880. [Google Scholar] [CrossRef] [PubMed]
- Bertini, G.; Colavito, V.; Tognoli, C.; Seke Etet, P.F.; Bentivoglio, M. The aging brain, neuroinflammatory signaling and sleep-wake regulation. Ital. J. Anat. Embryol. 2010, 115, 31–38. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aspect | Mice | Marmoset | Human |
|---|---|---|---|
| Evolutionary distance (million years) | 65 | 35 | 0 |
| Life expectancy (years) | 2 | 10–15 | 80 |
| Reproduction (number per year) | 50 | 2–4 | 1 |
| Genetic diversity | inbred | outbred | outbred |
| Environment | standardized | standardized | variable |
| Main sense | smell | vision | vision |
| Cortical thickness (mm) | 1.21 | 2.37 | 2.62 |
| Sleep pattern | nocturnal | diurnal | diurnal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Philippens, I.H.C.H.M.; Langermans, J.A.M. Preclinical Marmoset Model for Targeting Chronic Inflammation as a Strategy to Prevent Alzheimer’s Disease. Vaccines 2021, 9, 388. https://doi.org/10.3390/vaccines9040388
Philippens IHCHM, Langermans JAM. Preclinical Marmoset Model for Targeting Chronic Inflammation as a Strategy to Prevent Alzheimer’s Disease. Vaccines. 2021; 9(4):388. https://doi.org/10.3390/vaccines9040388
Chicago/Turabian StylePhilippens, Ingrid H. C. H. M., and Jan A. M. Langermans. 2021. "Preclinical Marmoset Model for Targeting Chronic Inflammation as a Strategy to Prevent Alzheimer’s Disease" Vaccines 9, no. 4: 388. https://doi.org/10.3390/vaccines9040388
APA StylePhilippens, I. H. C. H. M., & Langermans, J. A. M. (2021). Preclinical Marmoset Model for Targeting Chronic Inflammation as a Strategy to Prevent Alzheimer’s Disease. Vaccines, 9(4), 388. https://doi.org/10.3390/vaccines9040388