
Immunogenicity of HIV-1-Based Virus-Like Particles with Increased Incorporation and Stability of Membrane-Bound Env

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells, Enzymes, and Oligonucleotides
2.2. Monoclonal and Polyclonal Antibodies
2.3. Plasmids
2.4. Recombinant Env
2.5. Production of VLPs and Membrane-Bound Env Expression
2.6. Western Blotting
2.7. Immunization Studies
2.8. Enzyme-Linked Immunosorbent Assays (ELISAs)
2.9. Virus Fusion Assay
2.10. Neutralization Assays
3. Results
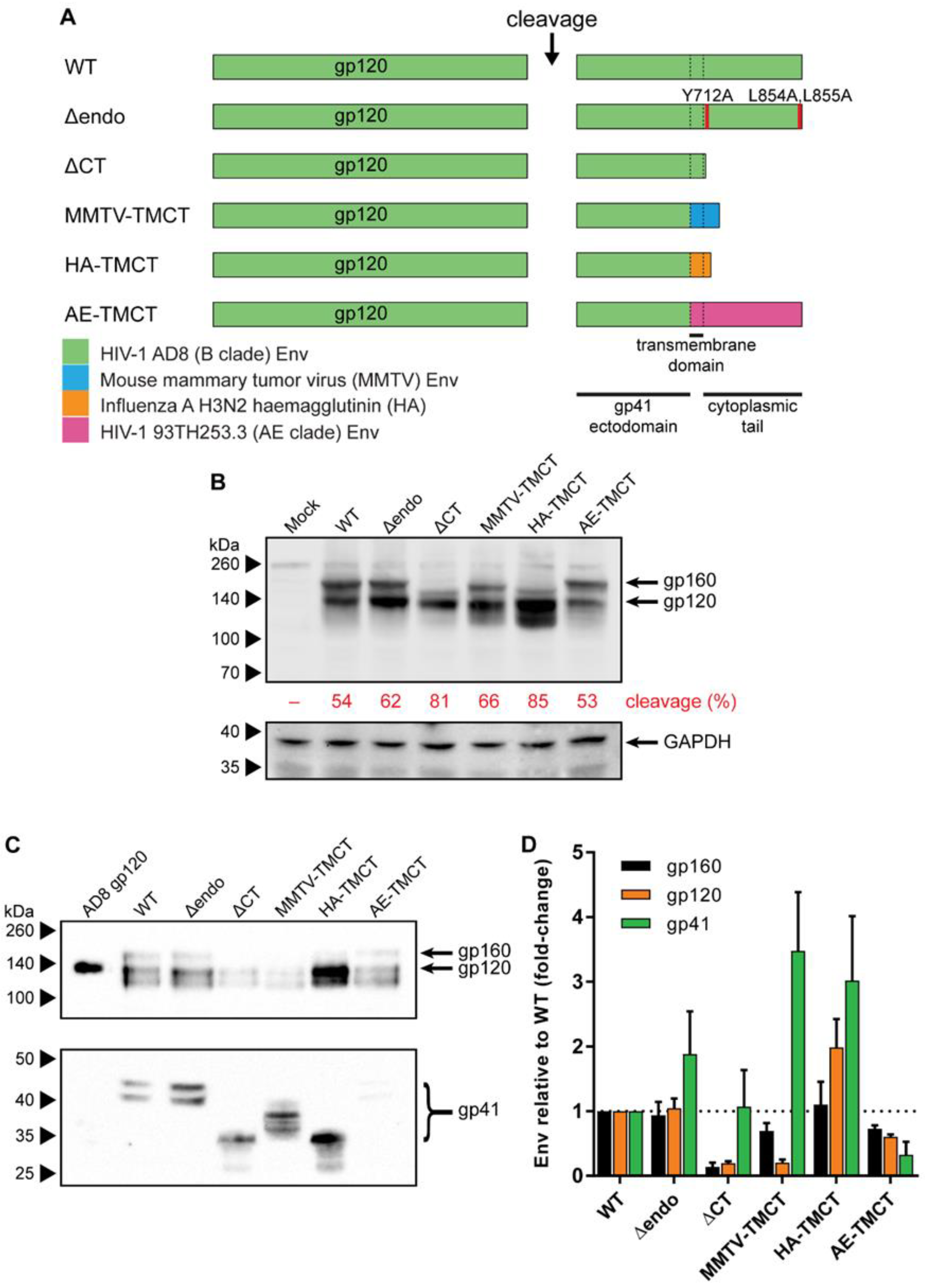
3.1. Envs with Mutated or Chimeric TM and CT Domains Have Increased Incorporation into mVLPs
3.2. Introducing Stabilizing Env Mutations to Anchor gp120 to VLP Membrane Improves Env Retention
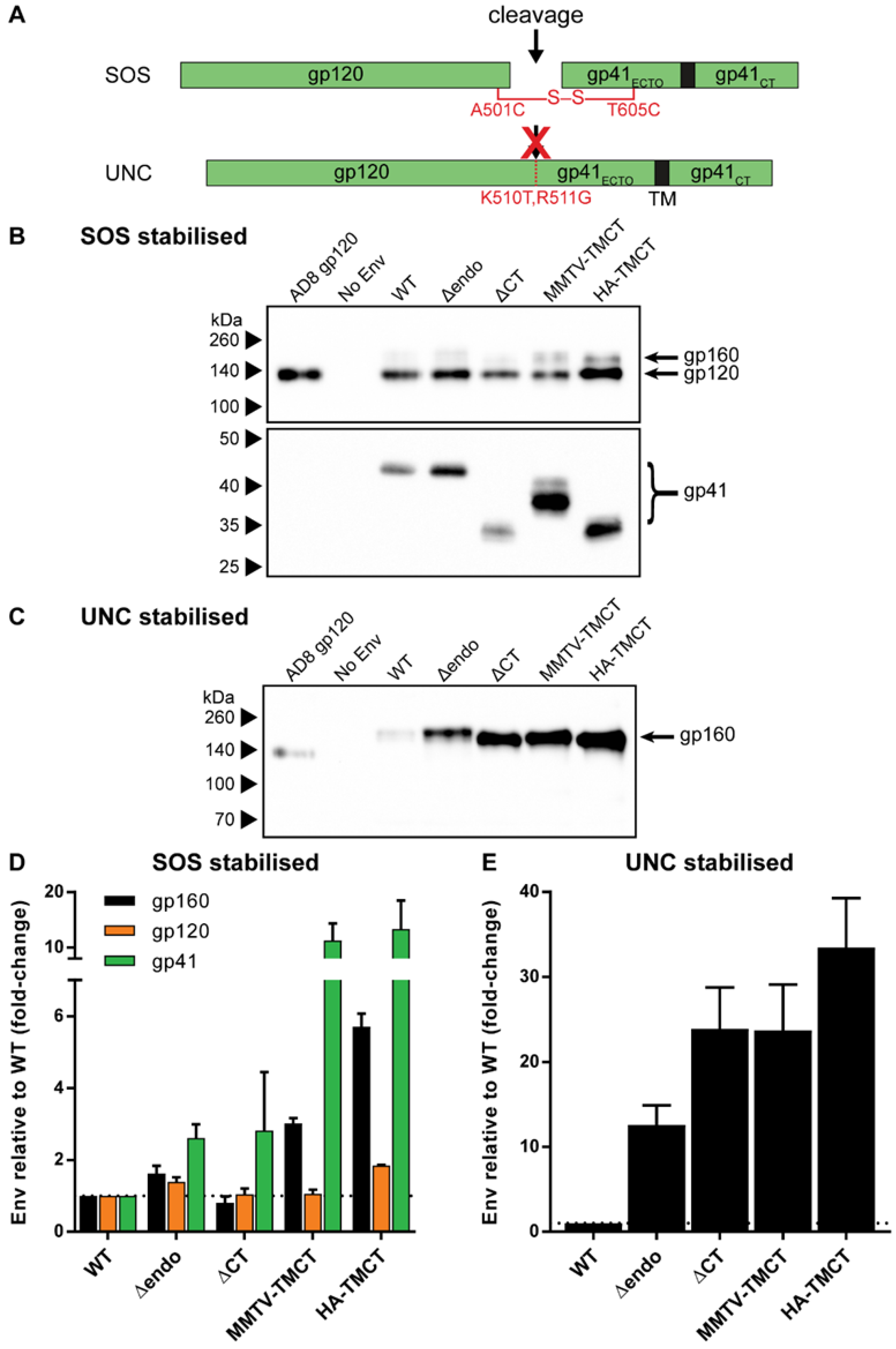
3.3. Cleaved and Uncleaved VLP-Associated Env Antigenicity Is Enhanced with Introduction of the SOSIP Mutation
3.4. Env Antigenicity but Not Function Is Preserved When Combining SOS and SOSIP Stabilization with HA TM and CT Domain
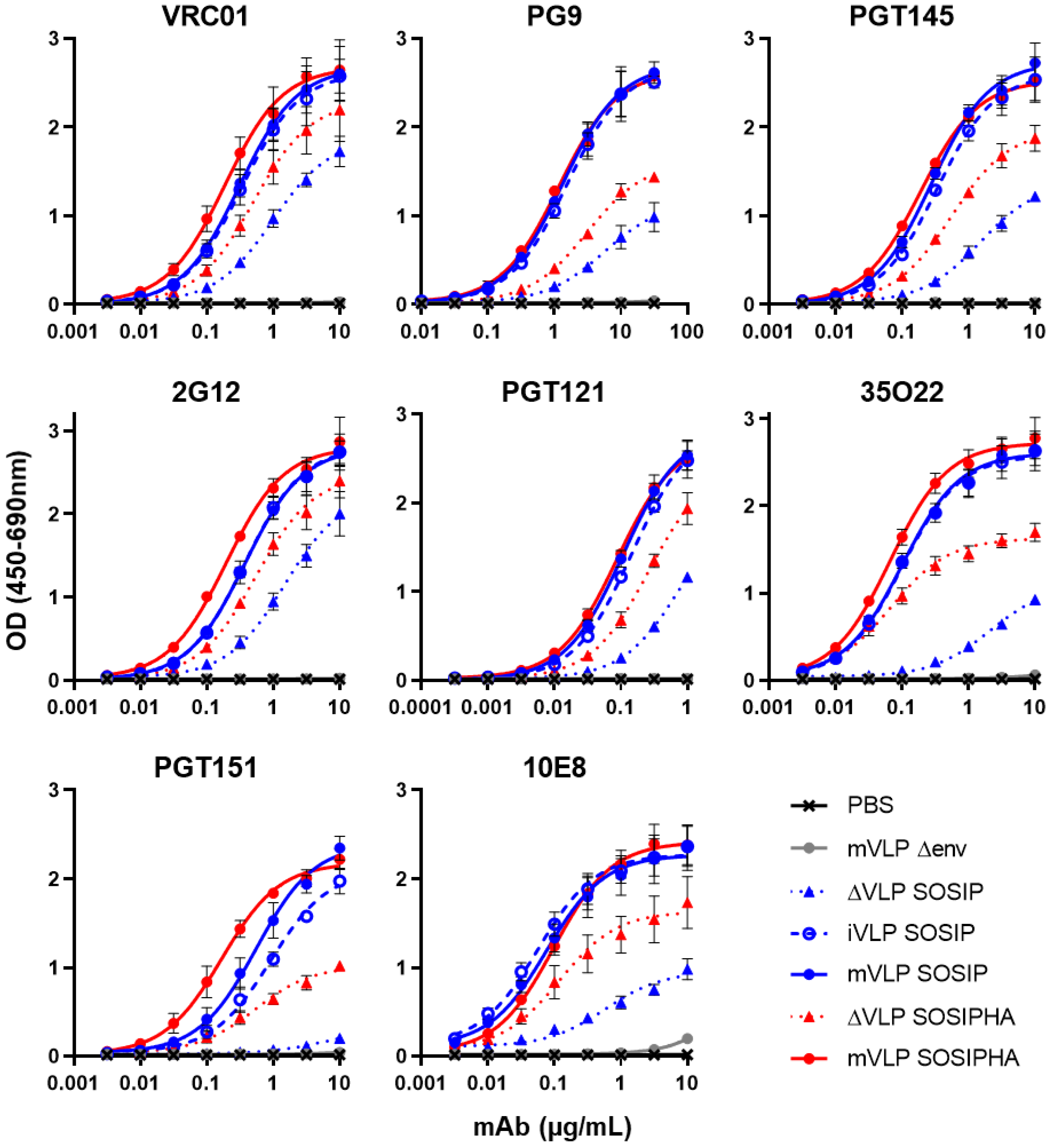
3.5. Suspension Cell Expression of VLPs Preserve bNAb Recognition and Can Reduce Contaminating Non-VLP Particles
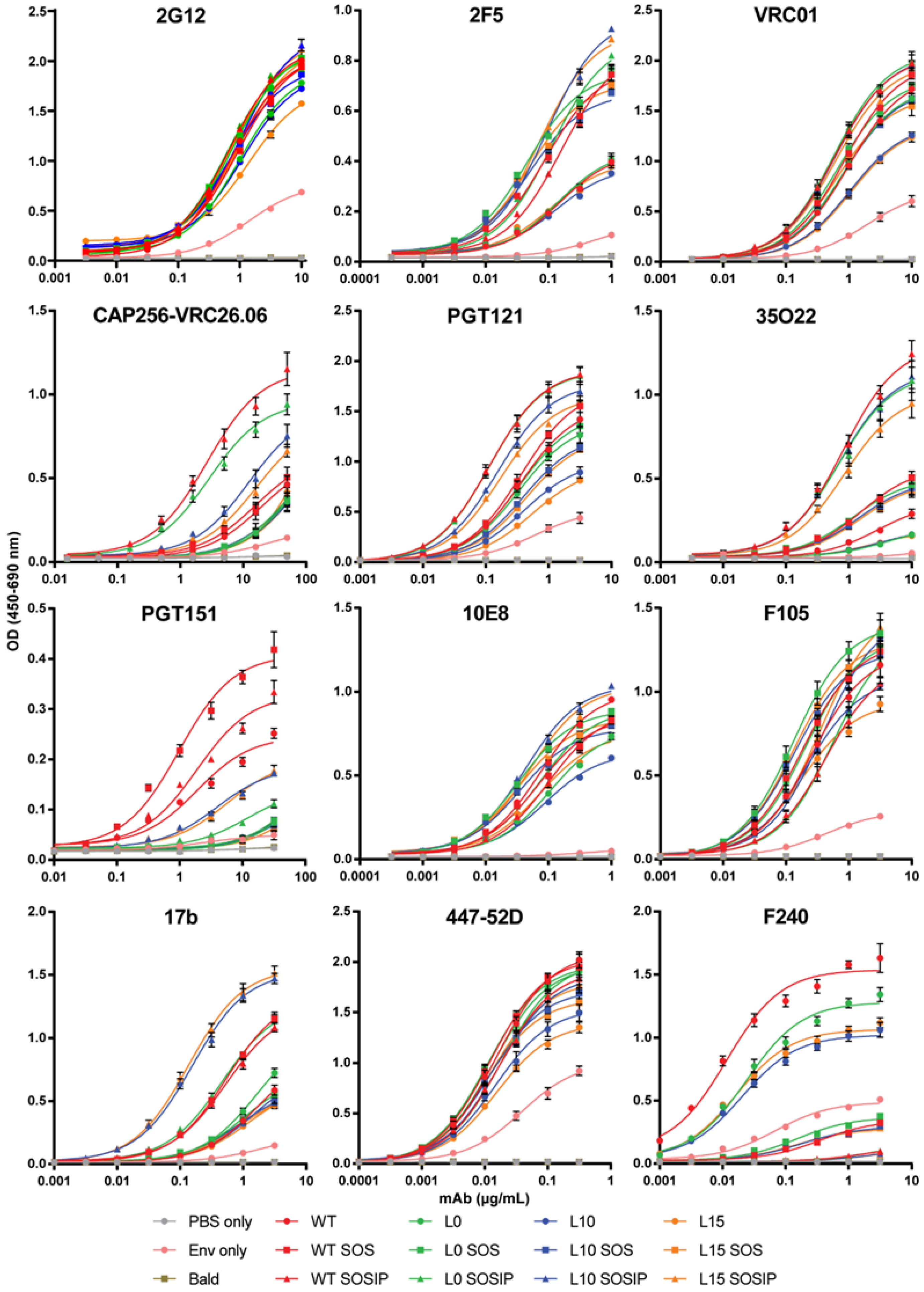
3.6. Neutralizing Antibody Epitope Exposure Is Preserved on SOSIP-Stabilized Env When Expressed on iVLPs and mVLPs with HA TM and CT
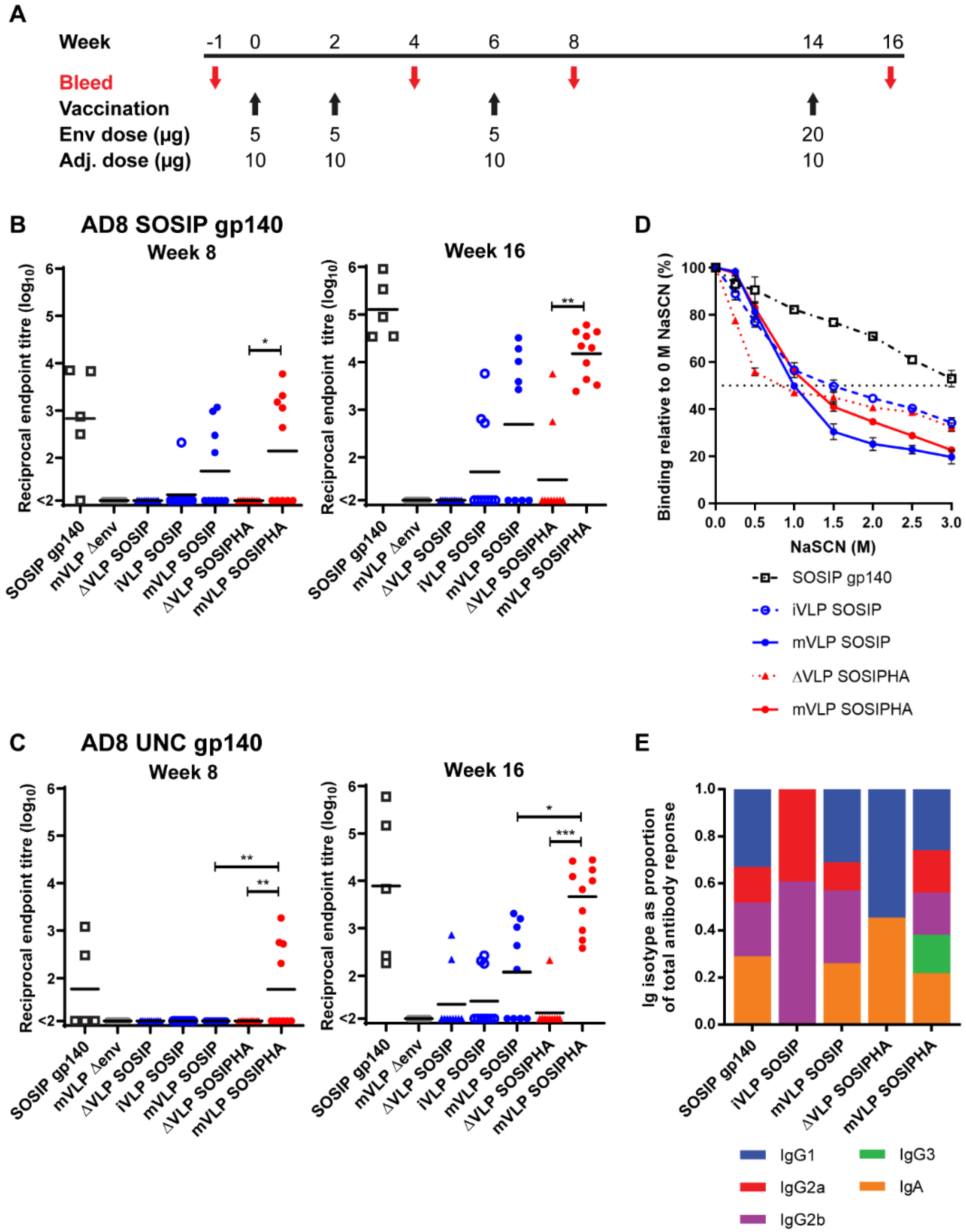
3.7. Vaccination with VLPs Bearing SOSIP-Stabilized Env Induces Lower Serum Titers of Env-Specific Antibodies Compared to Soluble SOSIP gp140
3.8. Env Presented on mVLPs Induces Broader Class Switching but Fails to Increase the Functional Avidity of the Antibody Response
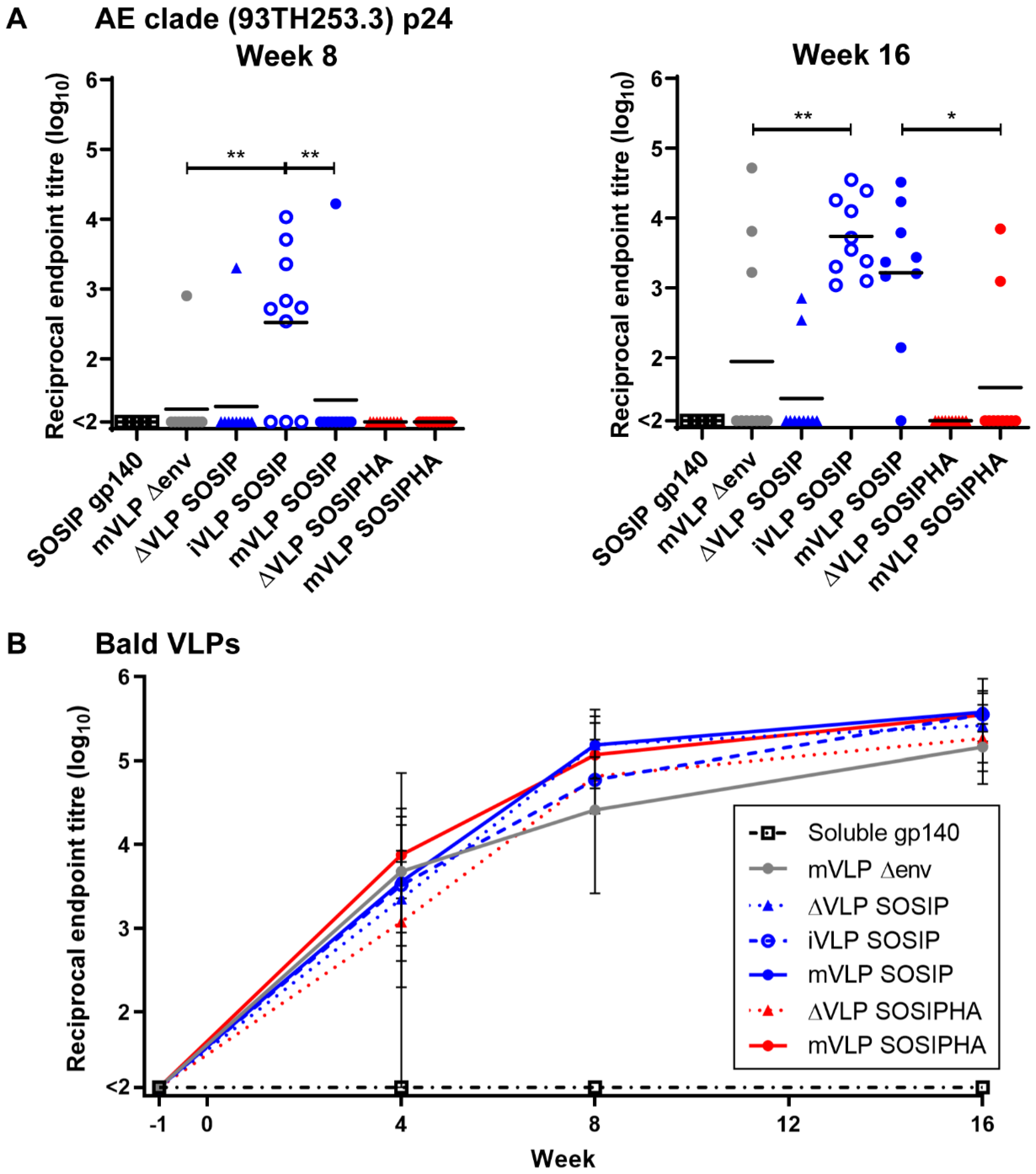
3.9. VLP Vaccinations Elicit Anti-Gag Antibody Responses That Do Not Negatively Impact Env-Specific Responses
3.10. Humoral Responses in Mice Following VLP Vaccination Are Largely Directed against Human Antigens Presented on the VLP Surface
3.11. Limited Neutralizing Antibody Responses Are Detected after mVLP SOSIPHA Vaccination of Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Escolano, A.; Dosenovic, P.; Nussenzweig, M.C. Progress toward active or passive HIV-1 vaccination. J. Exp. Med. 2017, 214, 3–16. [Google Scholar] [CrossRef]
- Kwong, P.D.; Mascola, J.R. HIV-1 Vaccines Based on Antibody Identification, B Cell Ontogeny, and Epitope Structure. Immunity 2018, 48, 855–871. [Google Scholar] [CrossRef]
- Reitter, J.N.; Means, R.E.; Desrosiers, R.C. A role for carbohydrates in immune evasion in AIDS. Nat. Med. 1998, 4, 679–684. [Google Scholar] [CrossRef]
- Wyatt, R.; Kwong, P.D.; Desjardins, E.; Sweet, R.W.; Robinson, J.; Hendrickson, W.A.; Sodroski, J.G. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 1998, 393, 705–711. [Google Scholar] [CrossRef]
- Wei, X.; Decker, J.M.; Wang, S.; Hui, H.; Kappes, J.C.; Wu, X.; Salazar-Gonzalez, J.F.; Salazar, M.G.; Kilby, J.M.; Saag, M.S.; et al. Antibody neutralization and escape by HIV-1. Nature 2003, 422, 307–312. [Google Scholar] [CrossRef]
- Zhou, T.; Doria-Rose, N.A.; Cheng, C.; Stewart-Jones, G.B.E.; Chuang, G.Y.; Chambers, M.; Druz, A.; Geng, H.; McKee, K.; Kwon, Y.D.; et al. Quantification of the Impact of the HIV-1-Glycan Shield on Antibody Elicitation. Cell Rep. 2017, 19, 719–732. [Google Scholar] [CrossRef]
- Ringe, R.P.; Pugach, P.; Cottrell, C.A.; LaBranche, C.C.; Seabright, G.E.; Ketas, T.J.; Ozorowski, G.; Kumar, S.; Schorcht, A.; van Gils, M.J.; et al. Closing and Opening Holes in the Glycan Shield of HIV-1 Envelope Glycoprotein SOSIP Trimers Can Redirect the Neutralizing Antibody Response to the Newly Unmasked Epitopes. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Poignard, P.; Moulard, M.; Golez, E.; Vivona, V.; Franti, M.; Venturini, S.; Wang, M.; Parren, P.W.; Burton, D.R. Heterogeneity of envelope molecules expressed on primary human immunodeficiency virus type 1 particles as probed by the binding of neutralizing and nonneutralizing antibodies. J. Virol. 2003, 77, 353–365. [Google Scholar] [CrossRef]
- Moore, P.L.; Crooks, E.T.; Porter, L.; Zhu, P.; Cayanan, C.S.; Grise, H.; Corcoran, P.; Zwick, M.B.; Franti, M.; Morris, L.; et al. Nature of nonfunctional envelope proteins on the surface of human immunodeficiency virus type 1. J. Virol. 2006, 80, 2515–2528. [Google Scholar] [CrossRef]
- Pancera, M.; Majeed, S.; Ban, Y.E.; Chen, L.; Huang, C.C.; Kong, L.; Kwon, Y.D.; Stuckey, J.; Zhou, T.; Robinson, J.E.; et al. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc. Natl. Acad. Sci. USA 2010, 107, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Munro, J.B.; Gorman, J.; Ma, X.; Zhou, Z.; Arthos, J.; Burton, D.R.; Koff, W.C.; Courter, J.R.; Smith, A.B., 3rd; Kwong, P.D.; et al. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science 2014, 346, 759–763. [Google Scholar] [CrossRef]
- Castillo-Menendez, L.R.; Witt, K.; Espy, N.; Princiotto, A.; Madani, N.; Pacheco, B.; Finzi, A.; Sodroski, J. Comparison of Uncleaved and Mature Human Immunodeficiency Virus Membrane Envelope Glycoprotein Trimers. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Thomson, M.M.; Pérez-Álvarez, L.; Nájera, R. Molecular epidemiology of HIV-1 genetic forms and its significance for vaccine development and therapy. Lancet Infect. Dis. 2002, 2, 461–471. [Google Scholar] [CrossRef]
- Korber, B.; Gaschen, B.; Yusim, K.; Thakallapally, R.; Kesmir, C.; Detours, V. Evolutionary and immunological implications of contemporary HIV-1 variation. Br. Med. Bull. 2001, 58, 19–42. [Google Scholar] [CrossRef]
- Lynch, R.M.; Shen, T.; Gnanakaran, S.; Derdeyn, C.A. Appreciating HIV type 1 diversity: Subtype differences in Env. AIDS Res. Hum. Retrovir. 2009, 25, 237–248. [Google Scholar] [CrossRef]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Rohrer, U.H.; Kündig, T.M.; Bürki, K.; Hengartner, H.; Zinkernagel, R.M. The influence of antigen organization on B cell responsiveness. Science 1993, 262, 1448–1451. [Google Scholar] [CrossRef]
- Thyagarajan, R.; Arunkumar, N.; Song, W. Polyvalent antigens stabilize B cell antigen receptor surface signaling microdomains. J. Immunol. 2003, 170, 6099–6106. [Google Scholar] [CrossRef]
- Tsunetsugu-Yokota, Y.; Morikawa, Y.; Isogai, M.; Kawana-Tachikawa, A.; Odawara, T.; Nakamura, T.; Grassi, F.; Autran, B.; Iwamoto, A. Yeast-derived human immunodeficiency virus type 1 p55(gag) virus-like particles activate dendritic cells (DCs) and induce perforin expression in Gag-specific CD8(+) T cells by cross-presentation of DCs. J. Virol. 2003, 77, 10250–10259. [Google Scholar] [CrossRef]
- GlaxoSmithKline Biologicals. ENGERIX-B [Hepatitis B Vaccine (Recombinant)]. Available online: https://www.fda.gov/vaccines-blood-biologics/vaccines/engerix-b (accessed on 1 March 2021).
- GlaxoSmithKline Biologicals. Cervarix [Human Papillomavirus Bivalent (Types 16 and 18) Vaccine, Recombinant]. Available online: https://www.fda.gov/vaccines-blood-biologics/vaccines/Cervarix (accessed on 1 March 2021).
- Merck & Co., Inc. RECOMBIVAX HB [Hepatitis B Vaccine (Recombinant)]. Available online: https://www.fda.gov/vaccines-blood-biologics/vaccines/recombivax-hb (accessed on 1 March 2021).
- Merck & Co., Inc. Gardasil [Human Papillomavirus Quadrivalent (Types 6, 11, 16, 18) Vaccine, Recombinant]. Available online: https://www.fda.gov/vaccines-blood-biologics/vaccines/gardasil (accessed on 1 March 2021).
- Kowalski, M.; Potz, J.; Basiripour, L.; Dorfman, T.; Goh, W.C.; Terwilliger, E.; Dayton, A.; Rosen, C.; Haseltine, W.; Sodroski, J. Functional regions of the envelope glycoprotein of human immunodeficiency virus type 1. Science 1987, 237, 1351–1355. [Google Scholar] [CrossRef]
- Lu, M.; Blacklow, S.C.; Kim, P.S. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat. Struct. Biol. 1995, 2, 1075–1082. [Google Scholar] [CrossRef]
- Thali, M.; Furman, C.; Helseth, E.; Repke, H.; Sodroski, J. Lack of correlation between soluble CD4-induced shedding of the human immunodeficiency virus type 1 exterior envelope glycoprotein and subsequent membrane fusion events. J. Virol. 1992, 66, 5516–5524. [Google Scholar] [CrossRef]
- Fu, Y.K.; Hart, T.K.; Jonak, Z.L.; Bugelski, P.J. Physicochemical dissociation of CD4-mediated syncytium formation and shedding of human immunodeficiency virus type 1 gp120. J. Virol. 1993, 67, 3818–3825. [Google Scholar] [CrossRef]
- Chertova, E.; Bess, J.W., Jr.; Crise, B.J.; Sowder, I.R.; Schaden, T.M.; Hilburn, J.M.; Hoxie, J.A.; Benveniste, R.E.; Lifson, J.D.; Henderson, L.E.; et al. Envelope glycoprotein incorporation, not shedding of surface envelope glycoprotein (gp120/SU), Is the primary determinant of SU content of purified human immunodeficiency virus type 1 and simian immunodeficiency virus. J. Virol. 2002, 76, 5315–5325. [Google Scholar] [CrossRef]
- Zhu, P.; Chertova, E.; Bess, J., Jr.; Lifson, J.D.; Arthur, L.O.; Liu, J.; Taylor, K.A.; Roux, K.H. Electron tomography analysis of envelope glycoprotein trimers on HIV and simian immunodeficiency virus virions. Proc. Natl. Acad. Sci. USA 2003, 100, 15812–15817. [Google Scholar] [CrossRef]
- Zhu, P.; Liu, J.; Bess, J., Jr.; Chertova, E.; Lifson, J.D.; Grise, H.; Ofek, G.A.; Taylor, K.A.; Roux, K.H. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 2006, 441, 847–852. [Google Scholar] [CrossRef]
- Brandenberg, O.F.; Magnus, C.; Rusert, P.; Regoes, R.R.; Trkola, A. Different infectivity of HIV-1 strains is linked to number of envelope trimers required for entry. PLoS Pathog. 2015, 11, e1004595. [Google Scholar] [CrossRef]
- Klein, J.S.; Bjorkman, P.J. Few and far between: How HIV may be evading antibody avidity. PLoS Pathog. 2010, 6, e1000908. [Google Scholar] [CrossRef]
- Wang, B.Z.; Liu, W.; Kang, S.M.; Alam, M.; Huang, C.; Ye, L.; Sun, Y.; Li, Y.; Kothe, D.L.; Pushko, P.; et al. Incorporation of high levels of chimeric human immunodeficiency virus envelope glycoproteins into virus-like particles. J. Virol. 2007, 81, 10869–10878. [Google Scholar] [CrossRef]
- Cronin, J.; Zhang, X.Y.; Reiser, J. Altering the tropism of lentiviral vectors through pseudotyping. Curr. Gene Ther. 2005, 5, 387–398. [Google Scholar] [CrossRef]
- He, D.; Marles-Wright, J. Ferritin family proteins and their use in bionanotechnology. New Biotechnol. 2015, 32, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Sliepen, K.; Ozorowski, G.; Burger, J.A.; van Montfort, T.; Stunnenberg, M.; LaBranche, C.; Montefiori, D.C.; Moore, J.P.; Ward, A.B.; Sanders, R.W. Presenting native-like HIV-1 envelope trimers on ferritin nanoparticles improves their immunogenicity. Retrovirology 2015, 12, 82. [Google Scholar] [CrossRef]
- He, L.; de Val, N.; Morris, C.D.; Vora, N.; Thinnes, T.C.; Kong, L.; Azadnia, P.; Sok, D.; Zhou, B.; Burton, D.R.; et al. Presenting native-like trimeric HIV-1 antigens with self-assembling nanoparticles. Nat. Commun. 2016, 7, 12041. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, I.S.; Joyce, M.G.; Chen, R.E.; Leung, K.; McKee, K.; Druz, A.; Van Galen, J.G.; Kanekiyo, M.; Tsybovsky, Y.; Yang, E.S.; et al. Two-Component Ferritin Nanoparticles for Multimerization of Diverse Trimeric Antigens. ACS Infect. Dis. 2018, 4, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.D.; Azadnia, P.; de Val, N.; Vora, N.; Honda, A.; Giang, E.; Saye-Francisco, K.; Cheng, Y.; Lin, X.; Mann, C.J.; et al. Differential Antibody Responses to Conserved HIV-1 Neutralizing Epitopes in the Context of Multivalent Scaffolds and Native-Like gp140 Trimers. mBio 2017, 8. [Google Scholar] [CrossRef]
- Sliepen, K.; Han, B.W.; Bontjer, I.; Mooij, P.; Garces, F.; Behrens, A.J.; Rantalainen, K.; Kumar, S.; Sarkar, A.; Brouwer, P.J.M.; et al. Structure and immunogenicity of a stabilized HIV-1 envelope trimer based on a group-M consensus sequence. Nat. Commun. 2019, 10, 2355. [Google Scholar] [CrossRef]
- Tokatlian, T.; Read, B.J.; Jones, C.A.; Kulp, D.W.; Menis, S.; Chang, J.Y.H.; Steichen, J.M.; Kumari, S.; Allen, J.D.; Dane, E.L.; et al. Innate immune recognition of glycans targets HIV nanoparticle immunogens to germinal centers. Science 2019, 363, 649–654. [Google Scholar] [CrossRef]
- Ingale, J.; Stano, A.; Guenaga, J.; Sharma, S.K.; Nemazee, D.; Zwick, M.B.; Wyatt, R.T. High-Density Array of Well-Ordered HIV-1 Spikes on Synthetic Liposomal Nanoparticles Efficiently Activate B Cells. Cell Rep. 2016, 15, 1986–1999. [Google Scholar] [CrossRef]
- Bale, S.; Goebrecht, G.; Stano, A.; Wilson, R.; Ota, T.; Tran, K.; Ingale, J.; Zwick, M.B.; Wyatt, R.T. Covalent Linkage of HIV-1 Trimers to Synthetic Liposomes Elicits Improved B Cell and Antibody Responses. J. Virol. 2017, 91, e00443-00417. [Google Scholar] [CrossRef]
- Martinez-Murillo, P.; Tran, K.; Guenaga, J.; Lindgren, G.; Adori, M.; Feng, Y.; Phad, G.E.; Vazquez Bernat, N.; Bale, S.; Ingale, J.; et al. Particulate Array of Well-Ordered HIV Clade C Env Trimers Elicits Neutralizing Antibodies that Display a Unique V2 Cap Approach. Immunity 2017, 46, 804–817.e807. [Google Scholar] [CrossRef]
- Tokatlian, T.; Kulp, D.W.; Mutafyan, A.A.; Jones, C.A.; Menis, S.; Georgeson, E.; Kubitz, M.; Zhang, M.H.; Melo, M.B.; Silva, M.; et al. Enhancing Humoral Responses Against HIV Envelope Trimers via Nanoparticle Delivery with Stabilized Synthetic Liposomes. Sci. Rep. 2018, 8, 16527. [Google Scholar] [CrossRef]
- Sanders, R.W.; Derking, R.; Cupo, A.; Julien, J.P.; Yasmeen, A.; de Val, N.; Kim, H.J.; Blattner, C.; de la Pena, A.T.; Korzun, J.; et al. A next-generation cleaved, soluble HIV-1 Env trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog. 2013, 9, e1003618. [Google Scholar] [CrossRef]
- Julien, J.P.; Lee, J.H.; Ozorowski, G.; Hua, Y.; Torrents de la Pena, A.; de Taeye, S.W.; Nieusma, T.; Cupo, A.; Yasmeen, A.; Golabek, M.; et al. Design and structure of two HIV-1 clade C SOSIP.664 trimers that increase the arsenal of native-like Env immunogens. Proc. Natl. Acad. Sci. USA 2015, 112, 11947–11952. [Google Scholar] [CrossRef]
- Pugach, P.; Ozorowski, G.; Cupo, A.; Ringe, R.; Yasmeen, A.; de Val, N.; Derking, R.; Kim, H.J.; Korzun, J.; Golabek, M.; et al. A native-like SOSIP.664 trimer based on an HIV-1 subtype B env gene. J. Virol. 2015, 89, 3380–3395. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Madani, N.; Ding, H.; Elder, E.; Princiotto, A.; Gu, C.; Darby, P.; Alin, J.; Herschhorn, A.; Kappes, J.C.; et al. Evaluation of the contribution of the transmembrane region to the ectodomain conformation of the human immunodeficiency virus (HIV-1) envelope glycoprotein. Virol. J. 2017, 14, 33. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.; Turner, H.L.; Nogal, B.; Cottrell, C.A.; Oyen, D.; Pauthner, M.; Bastidas, R.; Nedellec, R.; McCoy, L.E.; Wilson, I.A.; et al. Electron-Microscopy-Based Epitope Mapping Defines Specificities of Polyclonal Antibodies Elicited during HIV-1 BG505 Envelope Trimer Immunization. Immunity 2018, 49, 288–300.e288. [Google Scholar] [CrossRef]
- Sanders, R.W.; van Gils, M.J.; Derking, R.; Sok, D.; Ketas, T.J.; Burger, J.A.; Ozorowski, G.; Cupo, A.; Simonich, C.; Goo, L.; et al. HIV-1 neutralizing antibodies induced by native-like envelope trimers. Science 2015, 349, aac4223. [Google Scholar] [CrossRef] [PubMed]
- Tong, T.; Crooks, E.T.; Osawa, K.; Binley, J.M. HIV-1 virus-like particles bearing pure env trimers expose neutralizing epitopes but occlude nonneutralizing epitopes. J. Virol. 2012, 86, 3574–3587. [Google Scholar] [CrossRef]
- Crooks, E.T.; Tong, T.; Osawa, K.; Binley, J.M. Enzyme digests eliminate nonfunctional Env from HIV-1 particle surfaces, leaving native Env trimers intact and viral infectivity unaffected. J. Virol. 2011, 85, 5825–5839. [Google Scholar] [CrossRef]
- Crooks, E.T.; Osawa, K.; Tong, T.; Grimley, S.L.; Dai, Y.D.; Whalen, R.G.; Kulp, D.W.; Menis, S.; Schief, W.R.; Binley, J.M. Effects of partially dismantling the CD4 binding site glycan fence of HIV-1 Envelope glycoprotein trimers on neutralizing antibody induction. Virology 2017, 505, 193–209. [Google Scholar] [CrossRef]
- Crooks, E.T.; Tong, T.; Chakrabarti, B.; Narayan, K.; Georgiev, I.S.; Menis, S.; Huang, X.; Kulp, D.; Osawa, K.; Muranaka, J.; et al. Vaccine-Elicited Tier 2 HIV-1 Neutralizing Antibodies Bind to Quaternary Epitopes Involving Glycan-Deficient Patches Proximal to the CD4 Binding Site. PLoS Pathog. 2015, 11, e1004932. [Google Scholar] [CrossRef]
- Effio, C.L.; Hubbuch, J. Next generation vaccines and vectors: Designing downstream processes for recombinant protein-based virus-like particles. Biotechnol. J. 2015, 10, 715–727. [Google Scholar] [CrossRef]
- Gonelli, C.A.; Khoury, G.; Center, R.J.; Purcell, D.F.J. HIV-1-based Virus-like Particles that Morphologically Resemble Mature, Infectious HIV-1 Virions. Viruses 2019, 11, 507. [Google Scholar] [CrossRef]
- Chojnacki, J.; Staudt, T.; Glass, B.; Bingen, P.; Engelhardt, J.; Anders, M.; Schneider, J.; Muller, B.; Hell, S.W.; Krausslich, H.G. Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science 2012, 338, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.; Chackerian, B. Why HIV virions have low numbers of envelope spikes: Implications for vaccine development. PLoS Pathog. 2014, 10, e1004254. [Google Scholar] [CrossRef]
- Thali, M.; Moore, J.P.; Furman, C.; Charles, M.; Ho, D.D.; Robinson, J.; Sodroski, J. Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120-CD4 binding. J. Virol. 1993, 67, 3978–3988. [Google Scholar] [CrossRef]
- Huang, J.; Kang, B.H.; Pancera, M.; Lee, J.H.; Tong, T.; Feng, Y.; Imamichi, H.; Georgiev, I.S.; Chuang, G.Y.; Druz, A.; et al. Broad and potent HIV-1 neutralization by a human antibody that binds the gp41-gp120 interface. Nature 2014, 515, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Gorny, M.K.; Conley, A.J.; Karwowska, S.; Buchbinder, A.; Xu, J.Y.; Emini, E.A.; Koenig, S.; Zolla-Pazner, S. Neutralization of diverse human immunodeficiency virus type 1 variants by an anti-V3 human monoclonal antibody. J. Virol. 1992, 66, 7538–7542. [Google Scholar] [CrossRef]
- Posner, M.R.; Cavacini, L.A.; Emes, C.L.; Power, J.; Byrn, R. Neutralization of HIV-1 by F105, a human monoclonal antibody to the CD4 binding site of gp120. J. Acquir. Immune Defic. Syndr. 1993, 6, 7–14. [Google Scholar]
- Cavacini, L.A.; Emes, C.L.; Wisnewski, A.V.; Power, J.; Lewis, G.; Montefiori, D.; Posner, M.R. Functional and molecular characterization of human monoclonal antibody reactive with the immunodominant region of HIV type 1 glycoprotein 41. AIDS Res. Hum. Retrovir. 1998, 14, 1271–1280. [Google Scholar] [CrossRef]
- Walker, L.M.; Huber, M.; Doores, K.J.; Falkowska, E.; Pejchal, R.; Julien, J.P.; Wang, S.K.; Ramos, A.; Chan-Hui, P.Y.; Moyle, M.; et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 2011, 477, 466–470. [Google Scholar] [CrossRef]
- Nelson, J.D.; Brunel, F.M.; Jensen, R.; Crooks, E.T.; Cardoso, R.M.; Wang, M.; Hessell, A.; Wilson, I.A.; Binley, J.M.; Dawson, P.E.; et al. An affinity-enhanced neutralizing antibody against the membrane-proximal external region of human immunodeficiency virus type 1 gp41 recognizes an epitope between those of 2F5 and 4E10. J. Virol. 2007, 81, 4033–4043. [Google Scholar] [CrossRef]
- Huang, J.; Ofek, G.; Laub, L.; Louder, M.K.; Doria-Rose, N.A.; Longo, N.S.; Imamichi, H.; Bailer, R.T.; Chakrabarti, B.; Sharma, S.K.; et al. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 2012, 491, 406–412. [Google Scholar] [CrossRef]
- Blattner, C.; Lee, J.H.; Sliepen, K.; Derking, R.; Falkowska, E.; de la Pena, A.T.; Cupo, A.; Julien, J.P.; van Gils, M.; Lee, P.S.; et al. Structural delineation of a quaternary, cleavage-dependent epitope at the gp41-gp120 interface on intact HIV-1 Env trimers. Immunity 2014, 40, 669–680. [Google Scholar] [CrossRef]
- Doria-Rose, N.A.; Schramm, C.A.; Gorman, J.; Moore, P.L.; Bhiman, J.N.; DeKosky, B.J.; Ernandes, M.J.; Georgiev, I.S.; Kim, H.J.; Pancera, M.; et al. Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature 2014, 509, 55–62. [Google Scholar] [CrossRef]
- Wu, X.; Yang, Z.Y.; Li, Y.; Hogerkorp, C.M.; Schief, W.R.; Seaman, M.S.; Zhou, T.; Schmidt, S.D.; Wu, L.; Xu, L.; et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 2010, 329, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O.; Englund, G.; Martin, M.A. Role of the basic domain of human immunodeficiency virus type 1 matrix in macrophage infection. J. Virol. 1995, 69, 3949–3954. [Google Scholar] [CrossRef] [PubMed]
- Center, R.J.; Wheatley, A.K.; Campbell, S.M.; Gaeguta, A.J.; Peut, V.; Alcantara, S.; Siebentritt, C.; Kent, S.J.; Purcell, D.F. Induction of HIV-1 subtype B and AE-specific neutralizing antibodies in mice and macaques with DNA prime and recombinant gp140 protein boost regimens. Vaccine 2009, 27, 6605–6612. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Foley, B.T.; Kuiken, C.; Pillai, S.K.; Sodroski, J.G. Numbering positions in HIV relative to HXB2CG. In Human Retroviruses and AIDS 1998; Korber, C.K., Foley, B., Hahn, B., McCutchan, F., Mellors, J., Sodroski, J., Eds.; Theoretical Biology and Biophysics Group, Los Alamos National Laboratory: Los Alamos, NM, USA, 1998; pp. 102–111. [Google Scholar]
- Kramski, M.; Center, R.J.; Wheatley, A.K.; Jacobson, J.C.; Alexander, M.R.; Rawlin, G.; Purcell, D.F. Hyperimmune bovine colostrum as a low-cost, large-scale source of antibodies with broad neutralizing activity for HIV-1 envelope with potential use in microbicides. Antimicrob. Agents Chemother. 2012, 56, 4310–4319. [Google Scholar] [CrossRef]
- Anderson, J.L.; Johnson, A.T.; Howard, J.L.; Purcell, D.F. Both linear and discontinuous ribosome scanning are used for translation initiation from bicistronic human immunodeficiency virus type 1 env mRNAs. J. Virol. 2007, 81, 4664–4676. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Beroud, G.; Claustres, M.; Beroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Robertson, D.L.; Morrison, S.G.; Hui, H.; Craig, S.; Decker, J.; Fultz, P.N.; Girard, M.; Shaw, G.M.; Hahn, B.H.; et al. The heterosexual human immunodeficiency virus type 1 epidemic in Thailand is caused by an intersubtype (A/E) recombinant of African origin. J. Virol. 1996, 70, 7013–7029. [Google Scholar] [CrossRef]
- Dale, C.J.; De Rose, R.; Wilson, K.M.; Croom, H.A.; Thomson, S.; Coupar, B.E.; Ramsay, A.; Purcell, D.F.; Ffrench, R.; Law, M.; et al. Evaluation in macaques of HIV-1 DNA vaccines containing primate CpG motifs and fowlpoxvirus vaccines co-expressing IFNgamma or IL-12. Vaccine 2004, 23, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Center, R.J.; Earl, P.L.; Lebowitz, J.; Schuck, P.; Moss, B. The human immunodeficiency virus type 1 gp120 V2 domain mediates gp41-independent intersubunit contacts. J. Virol. 2000, 74, 4448–4455. [Google Scholar] [CrossRef] [PubMed]
- Binley, J.M.; Sanders, R.W.; Clas, B.; Schuelke, N.; Master, A.; Guo, Y.; Kajumo, F.; Anselma, D.J.; Maddon, P.J.; Olson, W.C.; et al. A recombinant human immunodeficiency virus type 1 envelope glycoprotein complex stabilized by an intermolecular disulfide bond between the gp120 and gp41 subunits is an antigenic mimic of the trimeric virion-associated structure. J. Virol. 2000, 74, 627–643. [Google Scholar] [CrossRef]
- Verkerke, H.P.; Williams, J.A.; Guttman, M.; Simonich, C.A.; Liang, Y.; Filipavicius, M.; Hu, S.L.; Overbaugh, J.; Lee, K.K. Epitope-Independent Purification of Native-Like Envelope Trimers from Diverse HIV-1 Isolates. J. Virol. 2016, 90, 9471–9482. [Google Scholar] [CrossRef] [PubMed]
- Ross, T.M.; Xu, Y.; Green, T.D.; Montefiori, D.C.; Robinson, H.L. Enhanced avidity maturation of antibody to human immunodeficiency virus envelope: DNA vaccination with gp120-C3d fusion proteins. AIDS Res. Hum. Retrovir. 2001, 17, 829–835. [Google Scholar] [CrossRef]
- Cavrois, M.; De Noronha, C.; Greene, W.C. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat. Biotechnol. 2002, 20, 1151–1154. [Google Scholar] [CrossRef]
- Landau, N.R.; Page, K.A.; Littman, D.R. Pseudotyping with human T-cell leukemia virus type I broadens the human immunodeficiency virus host range. J. Virol. 1991, 65, 162–169. [Google Scholar] [CrossRef]
- Sarzotti-Kelsoe, M.; Bailer, R.T.; Turk, E.; Lin, C.L.; Bilska, M.; Greene, K.M.; Gao, H.; Todd, C.A.; Ozaki, D.A.; Seaman, M.S.; et al. Optimization and validation of the TZM-bl assay for standardized assessments of neutralizing antibodies against HIV-1. J. Immunol. Methods 2014, 409, 131–146. [Google Scholar] [CrossRef]
- Byland, R.; Vance, P.J.; Hoxie, J.A.; Marsh, M. A conserved dileucine motif mediates clathrin and AP-2-dependent endocytosis of the HIV-1 envelope protein. Mol. Biol. Cell 2007, 18, 414–425. [Google Scholar] [CrossRef]
- Devitt, G.; Emerson, V.; Holtkotte, D.; Pfeiffer, T.; Pisch, T.; Bosch, V. Incorporation of chimeric HIV-SIV-Env and modified HIV-Env proteins into HIV pseudovirions. Virology 2007, 361, 465–471. [Google Scholar] [CrossRef]
- Berman, P.W.; Gregory, T.J.; Riddle, L.; Nakamura, G.R.; Champe, M.A.; Porter, J.P.; Wurm, F.M.; Hershberg, R.D.; Cobb, E.K.; Eichberg, J.W. Protection of chimpanzees from infection by HIV-1 after vaccination with recombinant glycoprotein gp120 but not gp160. Nature 1990, 345, 622–625. [Google Scholar] [CrossRef] [PubMed]
- Clements, G.J.; Price-Jones, M.J.; Stephens, P.E.; Sutton, C.; Schulz, T.F.; Clapham, P.R.; McKeating, J.A.; McClure, M.O.; Thomson, S.; Marsh, M.; et al. The V3 loops of the HIV-1 and HIV-2 surface glycoproteins contain proteolytic cleavage sites: A possible function in viral fusion? AIDS Res. Hum. Retrovir. 1991, 7, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Du, S.X.; Xu, L.; Viswanathan, S.; Whalen, R.G. Inhibition of V3-specific cleavage of recombinant HIV-1 gp120 produced in Chinese hamster ovary cells. Protein Expr. Purif. 2008, 59, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Yuste, E.; Reeves, J.D.; Doms, R.W.; Desrosiers, R.C. Modulation of Env content in virions of simian immunodeficiency virus: Correlation with cell surface expression and virion infectivity. J. Virol. 2004, 78, 6775–6785. [Google Scholar] [CrossRef] [PubMed]
- Holtkotte, D.; Pfeiffer, T.; Pisch, T.; Bosch, V. Selection and characterization of a replication-competent human immunodeficiency virus type 1 variant encoding C-terminally truncated env. AIDS Res. Hum. Retrovir. 2006, 22, 57–65. [Google Scholar] [CrossRef]
- Chakrabarti, B.K.; Kong, W.P.; Wu, B.Y.; Yang, Z.Y.; Friborg, J.; Ling, X.; King, S.R.; Montefiori, D.C.; Nabel, G.J. Modifications of the human immunodeficiency virus envelope glycoprotein enhance immunogenicity for genetic immunization. J. Virol. 2002, 76, 5357–5368. [Google Scholar] [CrossRef]
- Georgiev, I.S.; Joyce, M.G.; Yang, Y.; Sastry, M.; Zhang, B.; Baxa, U.; Chen, R.E.; Druz, A.; Lees, C.R.; Narpala, S.; et al. Single-Chain Soluble BG505.SOSIP gp140 Trimers as Structural and Antigenic Mimics of Mature Closed HIV-1 Env. J. Virol. 2015, 89, 5318–5329. [Google Scholar] [CrossRef] [PubMed]
- Sanders, R.W.; Venturi, M.; Schiffner, L.; Kalyanaraman, R.; Katinger, H.; Lloyd, K.O.; Kwong, P.D.; Moore, J.P. The mannose-dependent epitope for neutralizing antibody 2G12 on human immunodeficiency virus type 1 glycoprotein gp120. J. Virol. 2002, 76, 7293–7305. [Google Scholar] [CrossRef]
- Kesavardhana, S.; Varadarajan, R. Stabilizing the native trimer of HIV-1 Env by destabilizing the heterodimeric interface of the gp41 postfusion six-helix bundle. J. Virol. 2014, 88, 9590–9604. [Google Scholar] [CrossRef]
- Abrahamyan, L.G.; Markosyan, R.M.; Moore, J.P.; Cohen, F.S.; Melikyan, G.B. Human immunodeficiency virus type 1 Env with an intersubunit disulfide bond engages coreceptors but requires bond reduction after engagement to induce fusion. J. Virol. 2003, 77, 5829–5836. [Google Scholar] [CrossRef]
- Binley, J.M.; Cayanan, C.S.; Wiley, C.; Schulke, N.; Olson, W.C.; Burton, D.R. Redox-triggered infection by disulfide-shackled human immunodeficiency virus type 1 pseudovirions. J. Virol. 2003, 77, 5678–5684. [Google Scholar] [CrossRef]
- Alsahafi, N.; Debbeche, O.; Sodroski, J.; Finzi, A. Effects of the I559P gp41 change on the conformation and function of the human immunodeficiency virus (HIV-1) membrane envelope glycoprotein trimer. PLoS ONE 2015, 10, e0122111. [Google Scholar] [CrossRef] [PubMed]
- Reuven, E.M.; Dadon, Y.; Viard, M.; Manukovsky, N.; Blumenthal, R.; Shai, Y. HIV-1 gp41 Transmembrane Domain Interacts with the Fusion Peptide: Implication in Lipid Mixing and Inhibition of Virus-Cell Fusion. Biochemistry 2012, 51, 2867–2878. [Google Scholar] [CrossRef]
- Crooks, E.T.; Moore, P.L.; Franti, M.; Cayanan, C.S.; Zhu, P.; Jiang, P.; de Vries, R.P.; Wiley, C.; Zharkikh, I.; Schulke, N.; et al. A comparative immunogenicity study of HIV-1 virus-like particles bearing various forms of envelope proteins, particles bearing no envelope and soluble monomeric gp120. Virology 2007, 366, 245–262. [Google Scholar] [CrossRef]
- Tong, T.; Crooks, E.T.; Osawa, K.; Robinson, J.E.; Barnes, M.; Apetrei, C.; Binley, J.M. Multi-parameter exploration of HIV-1 virus-like particles as neutralizing antibody immunogens in guinea pigs, rabbits and macaques. Virology 2014, 456–457, 55–69. [Google Scholar] [CrossRef]
- Gautam, R.; Nishimura, Y.; Lee, W.R.; Donau, O.; Buckler-White, A.; Shingai, M.; Sadjadpour, R.; Schmidt, S.D.; LaBranche, C.C.; Keele, B.F.; et al. Pathogenicity and mucosal transmissibility of the R5-tropic simian/human immunodeficiency virus SHIV(AD8) in rhesus macaques: Implications for use in vaccine studies. J. Virol. 2012, 86, 8516–8526. [Google Scholar] [CrossRef] [PubMed]
- Drummer, H.E.; Hill, M.K.; Maerz, A.L.; Wood, S.; Ramsland, P.A.; Mak, J.; Poumbourios, P. Allosteric modulation of the HIV-1 gp120-gp41 association site by adjacent gp120 variable region 1 (V1) N-glycans linked to neutralization sensitivity. PLoS Pathog. 2013, 9, e1003218. [Google Scholar] [CrossRef]
- Chapman, R.; van Diepen, M.; Galant, S.; Kruse, E.; Margolin, E.; Ximba, P.; Hermanus, T.; Moore, P.; Douglass, N.; Williamson, A.L.; et al. Immunogenicity of HIV-1 Vaccines Expressing Chimeric Envelope Glycoproteins on the Surface of Pr55 Gag Virus-Like Particles. Vaccines 2020, 8, 54. [Google Scholar] [CrossRef]
- Thery, C.; Boussac, M.; Veron, P.; Ricciardi-Castagnoli, P.; Raposo, G.; Garin, J.; Amigorena, S. Proteomic analysis of dendritic cell-derived exosomes: A secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 2001, 166, 7309–7318. [Google Scholar] [CrossRef] [PubMed]
- Cantin, R.; Diou, J.; Belanger, D.; Tremblay, A.M.; Gilbert, C. Discrimination between exosomes and HIV-1: Purification of both vesicles from cell-free supernatants. J. Immunol. Methods 2008, 338, 21–30. [Google Scholar] [CrossRef] [PubMed]
- El Andaloussi, S.; Mager, I.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef]
- Booth, A.M.; Fang, Y.; Fallon, J.K.; Yang, J.M.; Hildreth, J.E.; Gould, S.J. Exosomes and HIV Gag bud from endosome-like domains of the T cell plasma membrane. J. Cell Biol. 2006, 172, 923–935. [Google Scholar] [CrossRef]
- Lavado-García, J.; González-Domínguez, I.; Cervera, L.; Jorge, I.; Vázquez, J.; Gòdia, F. Molecular Characterization of the Coproduced Extracellular Vesicles in HEK293 during Virus-Like Particle Production. J. Proteome Res. 2020, 19, 4516–4532. [Google Scholar] [CrossRef]
- Trubey, C.M.; Chertova, E.; Coren, L.V.; Hilburn, J.M.; Hixson, C.V.; Nagashima, K.; Lifson, J.D.; Ott, D.E. Quantitation of HLA class II protein incorporated into human immunodeficiency type 1 virions purified by anti-CD45 immunoaffinity depletion of microvesicles. J. Virol. 2003, 77, 12699–12709. [Google Scholar] [CrossRef]
- Ott, D.E. Purification of HIV-1 virions by subtilisin digestion or CD45 immunoaffinity depletion for biochemical studies. Methods Mol. Biol. 2009, 485, 15–25. [Google Scholar] [CrossRef]
- Esser, M.T.; Graham, D.R.; Coren, L.V.; Trubey, C.M.; Bess, J.W., Jr.; Arthur, L.O.; Ott, D.E.; Lifson, J.D. Differential incorporation of CD45, CD80 (B7-1), CD86 (B7-2), and major histocompatibility complex class I and II molecules into human immunodeficiency virus type 1 virions and microvesicles: Implications for viral pathogenesis and immune regulation. J. Virol. 2001, 75, 6173–6182. [Google Scholar] [CrossRef]
- Wubbolts, R.; Leckie, R.S.; Veenhuizen, P.T.; Schwarzmann, G.; Mobius, W.; Hoernschemeyer, J.; Slot, J.W.; Geuze, H.J.; Stoorvogel, W. Proteomic and biochemical analyses of human B cell-derived exosomes. Potential implications for their function and multivesicular body formation. J. Biol. Chem. 2003, 278, 10963–10972. [Google Scholar] [CrossRef]
- Miguet, L.; Pacaud, K.; Felden, C.; Hugel, B.; Martinez, M.C.; Freyssinet, J.M.; Herbrecht, R.; Potier, N.; van Dorsselaer, A.; Mauvieux, L. Proteomic analysis of malignant lymphocyte membrane microparticles using double ionization coverage optimization. Proteomics 2006, 6, 153–171. [Google Scholar] [CrossRef]
- Steppert, P.; Burgstaller, D.; Klausberger, M.; Berger, E.; Aguilar, P.P.; Schneider, T.A.; Kramberger, P.; Tover, A.; Nobauer, K.; Razzazi-Fazeli, E.; et al. Purification of HIV-1 gag virus-like particles and separation of other extracellular particles. J. Chromatogr. A 2016, 1455, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Steppert, P.; Burgstaller, D.; Klausberger, M.; Kramberger, P.; Tover, A.; Berger, E.; Nobauer, K.; Razzazi-Fazeli, E.; Jungbauer, A. Separation of HIV-1 gag virus-like particles from vesicular particles impurities by hydroxyl-functionalized monoliths. J. Sep. Sci. 2017, 40, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Pereira Aguilar, P.; Reiter, K.; Wetter, V.; Steppert, P.; Maresch, D.; Ling, W.L.; Satzer, P.; Jungbauer, A. Capture and purification of Human Immunodeficiency Virus-1 virus-like particles: Convective media vs porous beads. J. Chromatogr. A 2020, 1627, 461378. [Google Scholar] [CrossRef]
- Hammonds, J.; Chen, X.; Fouts, T.; DeVico, A.; Montefiori, D.; Spearman, P. Induction of neutralizing antibodies against human immunodeficiency virus type 1 primary isolates by Gag-Env pseudovirion immunization. J. Virol. 2005, 79, 14804–14814. [Google Scholar] [CrossRef] [PubMed]
- Benen, T.D.; Tonks, P.; Kliche, A.; Kapzan, R.; Heeney, J.L.; Wagner, R. Development and immunological assessment of VLP-based immunogens exposing the membrane-proximal region of the HIV-1 gp41 protein. J. Biomed. Sci. 2014, 21, 79. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Linde, M.E.; Colquhoun, D.R.; Ubaida Mohien, C.; Kole, T.; Aquino, V.; Cotter, R.; Edwards, N.; Hildreth, J.E.; Graham, D.R. The conserved set of host proteins incorporated into HIV-1 virions suggests a common egress pathway in multiple cell types. J. Proteome Res. 2013, 12, 2045–2054. [Google Scholar] [CrossRef]
- Trono, D.; Baltimore, D. A human cell factor is essential for HIV-1 Rev action. EMBO J. 1990, 9, 4155–4160. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Kindt, T.J.; Zhao, T.M.; Sawasdikosol, S.; Hague, B.F. Replication of HIV type 1 in rabbit cell lines is not limited by deficiencies in tat, rev, or long terminal repeat function. AIDS Res. Hum. Retrovir. 1995, 11, 1487–1493. [Google Scholar] [CrossRef]
- McCoy, L.E.; van Gils, M.J.; Ozorowski, G.; Messmer, T.; Briney, B.; Voss, J.E.; Kulp, D.W.; Macauley, M.S.; Sok, D.; Pauthner, M.; et al. Holes in the Glycan Shield of the Native HIV Envelope Are a Target of Trimer-Elicited Neutralizing Antibodies. Cell Rep. 2016, 16, 2327–2338. [Google Scholar] [CrossRef]
- Popkov, M.; Mage, R.G.; Alexander, C.B.; Thundivalappil, S.; Barbas, C.F., 3rd; Rader, C. Rabbit immune repertoires as sources for therapeutic monoclonal antibodies: The impact of kappa allotype-correlated variation in cysteine content on antibody libraries selected by phage display. J. Mol. Biol. 2003, 325, 325–335. [Google Scholar] [CrossRef]
- Walker, L.M.; Phogat, S.K.; Chan-Hui, P.Y.; Wagner, D.; Phung, P.; Goss, J.L.; Wrin, T.; Simek, M.D.; Fling, S.; Mitcham, J.L.; et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 2009, 326, 285–289. [Google Scholar] [CrossRef]
- Walker, L.M.; Sok, D.; Nishimura, Y.; Donau, O.; Sadjadpour, R.; Gautam, R.; Shingai, M.; Pejchal, R.; Ramos, A.; Simek, M.D.; et al. Rapid development of glycan-specific, broad, and potent anti-HIV-1 gp120 neutralizing antibodies in an R5 SIV/HIV chimeric virus infected macaque. Proc. Natl. Acad. Sci. USA 2011, 108, 20125–20129. [Google Scholar] [CrossRef]
- Stavnezer, J.; Schrader, C.E. IgH chain class switch recombination: Mechanism and regulation. J. Immunol. 2014, 193, 5370–5378. [Google Scholar] [CrossRef]
- Pulendran, B.; Kannourakis, G.; Nouri, S.; Smith, K.G.; Nossal, G.J. Soluble antigen can cause enhanced apoptosis of germinal-centre B cells. Nature 1995, 375, 331–334. [Google Scholar] [CrossRef]
- Kim, Y.M.; Pan, J.Y.; Korbel, G.A.; Peperzak, V.; Boes, M.; Ploegh, H.L. Monovalent ligation of the B cell receptor induces receptor activation but fails to promote antigen presentation. Proc. Natl. Acad. Sci. USA 2006, 103, 3327–3332. [Google Scholar] [CrossRef]
- Minguet, S.; Dopfer, E.P.; Schamel, W.W. Low-valency, but not monovalent, antigens trigger the B-cell antigen receptor (BCR). Int. Immunol. 2010, 22, 205–212. [Google Scholar] [CrossRef]
- Finkelman, F.D.; Katona, I.M.; Mosmann, T.R.; Coffman, R.L. IFN-gamma regulates the isotypes of Ig secreted during in vivo humoral immune responses. J. Immunol. 1988, 140, 1022–1027. [Google Scholar]
- Snapper, C.M.; McIntyre, T.M.; Mandler, R.; Pecanha, L.M.; Finkelman, F.D.; Lees, A.; Mond, J.J. Induction of IgG3 secretion by interferon gamma: A model for T cell-independent class switching in response to T cell-independent type 2 antigens. J. Exp. Med. 1992, 175, 1367–1371. [Google Scholar] [CrossRef]
- Huang, Y.; Kong, W.P.; Nabel, G.J. Human immunodeficiency virus type 1-specific immunity after genetic immunization is enhanced by modification of Gag and Pol expression. J. Virol. 2001, 75, 4947–4951. [Google Scholar] [CrossRef]
- Garnier, L.; Ratner, L.; Rovinski, B.; Cao, S.X.; Wills, J.W. Particle size determinants in the human immunodeficiency virus type 1 Gag protein. J. Virol. 1998, 72, 4667–4677. [Google Scholar] [CrossRef]
- Chung, A.W.; Ghebremichael, M.; Robinson, H.; Brown, E.; Choi, I.; Lane, S.; Dugast, A.S.; Schoen, M.K.; Rolland, M.; Suscovich, T.J.; et al. Polyfunctional Fc-effector profiles mediated by IgG subclass selection distinguish RV144 and VAX003 vaccines. Sci. Transl. Med. 2014, 6, 228ra238. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mAb | PBS Only | Env Only | Bald | WT | WT SOS | WT SOSIP | L0 | L0 SOS | L0 SOSIP | L10 | L10 SOS | L10 SOSIP | L15 | L15 SOS | L15 SOSIP |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2G12 | 0.03 | 0.31 | 0.04 | 1 | 1.00 | 1.12 | 0.90 | 1.10 | 1.11 | 0.92 | 1.04 | 1.12 | 0.86 | 1.07 | 1.13 |
| 2F5 | 0.06 | 0.24 | 0.06 | 1 | 1.96 | 1.88 | 1.02 | 2.12 | 2.13 | 0.90 | 1.87 | 2.46 | 0.97 | 2.00 | 2.42 |
| VRC01 | 0.02 | 0.33 | 0.02 | 1 | 1.09 | 1.21 | 0.97 | 1.05 | 1.23 | 0.73 | 0.96 | 1.20 | 0.72 | 0.96 | 1.15 |
| VRC26.06 | 0.20 | 0.41 | 0.22 | 1 | 1.16 | 3.62 | 0.64 | 0.71 | 2.96 | 0.66 | 0.72 | 1.74 | 0.69 | 0.64 | 1.47 |
| PGT121 | 0.02 | 0.30 | 0.02 | 1 | 1.11 | 1.48 | 0.91 | 0.97 | 1.47 | 0.63 | 0.80 | 1.33 | 0.55 | 0.78 | 1.21 |
| 35O22 | 0.11 | 0.20 | 0.12 | 1 | 1.87 | 4.84 | 0.57 | 1.78 | 4.38 | 0.59 | 1.66 | 4.42 | 0.58 | 1.61 | 3.78 |
| PGT151 | 0.11 | 0.21 | 0.11 | 1 | 1.79 | 1.33 | 0.24 | 0.26 | 0.40 | 0.25 | 0.24 | 0.67 | 0.24 | 0.22 | 0.67 |
| 10E8 | 0.02 | 0.05 | 0.02 | 1 | 0.86 | 0.84 | 0.73 | 0.99 | 0.89 | 0.62 | 0.86 | 1.12 | 0.75 | 0.93 | 1.09 |
| F105 | 0.02 | 0.21 | 0.02 | 1 | 1.11 | 0.86 | 1.12 | 1.26 | 0.93 | 0.91 | 1.11 | 1.09 | 0.80 | 1.17 | 1.15 |
| 17b | 0.04 | 0.26 | 0.04 | 1 | 2.28 | 2.13 | 1.23 | 1.00 | 2.27 | 0.92 | 0.91 | 3.35 | 0.84 | 0.88 | 3.46 |
| 447-52D | 0.01 | 0.41 | 0.01 | 1 | 1.01 | 0.91 | 0.96 | 0.98 | 0.93 | 0.74 | 0.85 | 0.88 | 0.66 | 0.82 | 0.88 |
| F240 | 0.01 | 0.29 | 0.01 | 1 | 0.16 | 0.04 | 0.81 | 0.20 | 0.04 | 0.65 | 0.16 | 0.04 | 0.68 | 0.15 | 0.03 |
| bNAb | ΔVLP SOSIP vs. mVLP SOSIP | ΔVLP SOSIPHA vs. mVLP SOSIP |
|---|---|---|
| VRC01 | 2.0 | 1.5 |
| PG9 | 3.7 | 2.2 |
| PGT145 | 3.3 | 1.7 |
| 2G12 | 1.9 | 1.5 |
| PGT121 | 3.5 | 1.7 |
| 35O22 | 5.4 | 1.7 |
| PGT151 | 14.6 | 2.8 |
| 10E8 | 3.4 | 1.5 |
| Immunogen | Normalization |
|---|---|
| Adjuvant only | N/A |
| SOSIP gp140 | Env |
| mVLP Δenv | Gag normalized to mVLP SOSIP |
| ΔVLP SOSIP | Volume of iVLP/mVLP SOSIP (largest volume) |
| iVLP SOSIP | Env |
| mVLP SOSIP | Env |
| ΔVLP SOSIPHA | Volume of mVLP SOSIPHA |
| mVLP SOSIPHA | Env |
| Reciprocal ID50 Serum Titers a | |||
|---|---|---|---|
| Immunogen | MN | SF162 | AD8 |
| Adjuvant only | <25 | <25 | <25 |
| SOSIP gp140 | 8625 | 59.2 | <25 |
| mVLP Δenv | <25 | <25 | <25 |
| ΔVLP SOSIP | <25 | <25 | <25 |
| iVLP SOSIP | <25 | <25 | <25 |
| mVLP SOSIP | <25 | <25 | <25 |
| ΔVLP SOSIPHA | <25 | <25 | <25 |
| mVLP SOSIPHA | 81.7 | <25 | <25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonelli, C.A.; King, H.A.D.; Mackenzie, C.; Sonza, S.; Center, R.J.; Purcell, D.F.J. Immunogenicity of HIV-1-Based Virus-Like Particles with Increased Incorporation and Stability of Membrane-Bound Env. Vaccines 2021, 9, 239. https://doi.org/10.3390/vaccines9030239
Gonelli CA, King HAD, Mackenzie C, Sonza S, Center RJ, Purcell DFJ. Immunogenicity of HIV-1-Based Virus-Like Particles with Increased Incorporation and Stability of Membrane-Bound Env. Vaccines. 2021; 9(3):239. https://doi.org/10.3390/vaccines9030239
Chicago/Turabian StyleGonelli, Christopher A., Hannah A. D. King, Charlene Mackenzie, Secondo Sonza, Rob J. Center, and Damian F. J. Purcell. 2021. "Immunogenicity of HIV-1-Based Virus-Like Particles with Increased Incorporation and Stability of Membrane-Bound Env" Vaccines 9, no. 3: 239. https://doi.org/10.3390/vaccines9030239
APA StyleGonelli, C. A., King, H. A. D., Mackenzie, C., Sonza, S., Center, R. J., & Purcell, D. F. J. (2021). Immunogenicity of HIV-1-Based Virus-Like Particles with Increased Incorporation and Stability of Membrane-Bound Env. Vaccines, 9(3), 239. https://doi.org/10.3390/vaccines9030239