
Development of Pandemic Vaccines: ERVEBO Case Study

 , , and
, , and
Abstract
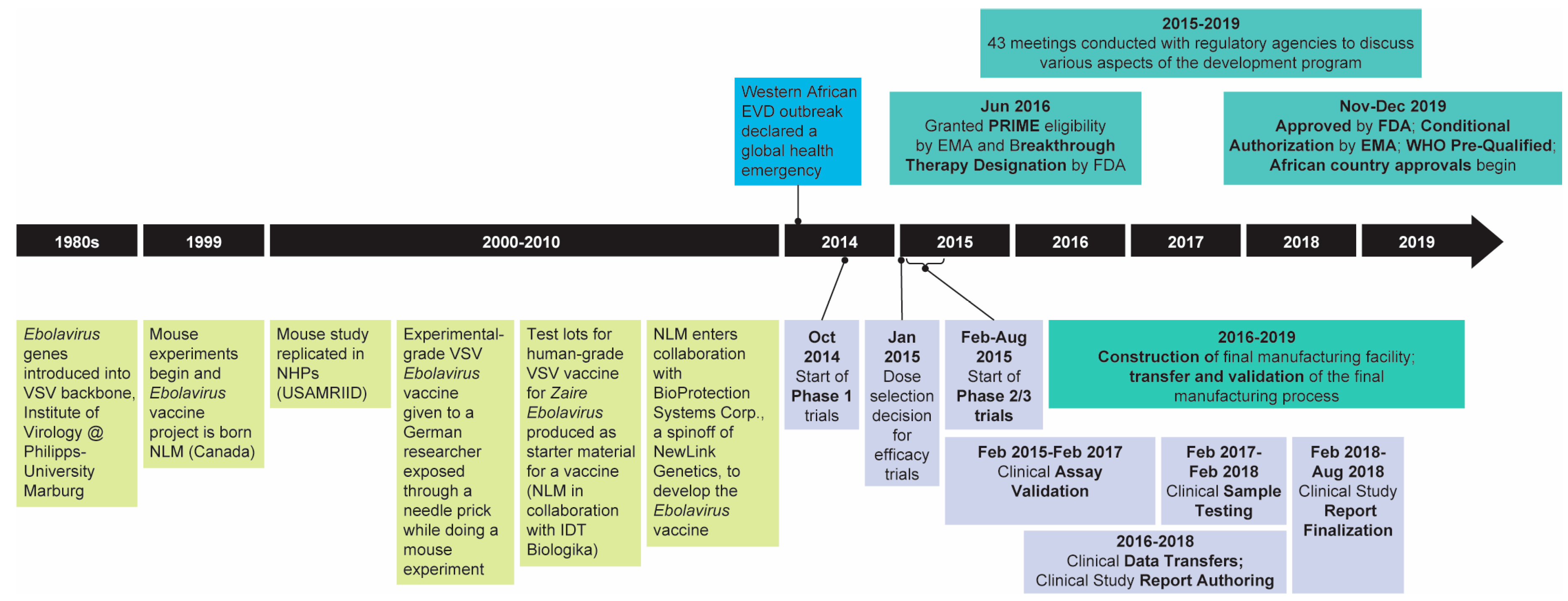
1. Introduction
2. Regulatory Considerations
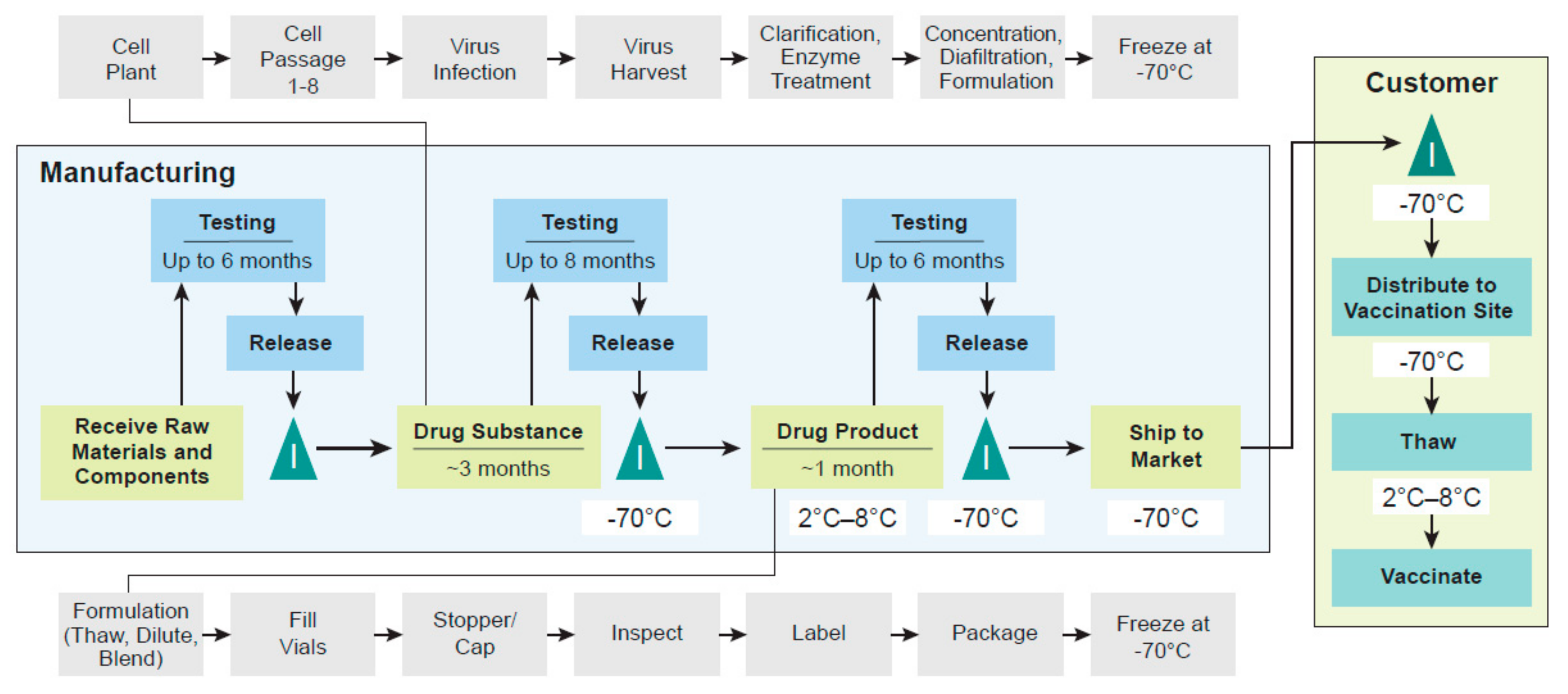
3. Manufacturing
4. Non-Clinical Overview
5. Clinical Development
6. Ongoing Work and Relevance to Vaccine Development for Other Emerging Infectious Diseases
- FLW trial: Part B of the Ebola Ҫa Suffit trial was an open-label safety and immunogenicity trial conducted in 2115 frontline workers in Guinea. The study was sponsored by the WHO and conducted by Médecins Sans Frontieres. The study was initiated at the same time as the Ebola Ҫa Suffit trial, but since efficacy was not assessed in this study, efforts were focused on the safety and efficacy trials for the initial license submissions. Data from the trial expanding our knowledge of the safety and immunogenicity of the vaccine have recently been published [53]. Importantly, given the conduct of this trial in the same country and at the same time as the efficacy trial, population-based approaches to evaluate potential correlates of protection for ERVEBOTM have been assessed using the data from the trial [57]. This evaluation suggests that the GP-ELISA assay, originally established by the FANG and subsequently validated at Q2 Solutions, correlates with protection for this vaccine, although a specific protective threshold was not defined. Evidence for a correlate of protection for rVSVΔG-ZEBOV-GP provides additional information for groups such as the WHO Strategic Advisory Group of Experts (SAGE) as they recommend the best way to use the vaccine to have the largest public health impact.
- PREPARE (Pre-Exposure Prophylaxis in People at Potential Occupation Risk for Ebola Virus Exposure) trial: This ongoing National Institute of Allergy and Infectious Diseases (NIAID)-sponsored, randomized, and controlled trial being conducted in the US and Canada evaluates the durability of immune responses to rVSVΔG-ZEBOV-GP in individuals who are at occupational risk of exposure to Ebola (e.g., through working in BSL-4 laboratories, etc.) and includes randomization to assess the impact of a booster dose given at 18 months.
- Partnership for Research on Ebola Vaccination (PREVAC) Trial: This large randomized, placebo-controlled trial, which is evaluating three different vaccination strategies, including one or two doses of rVSVΔG-ZEBOV-GP, in adult and pediatric participants in Guinea, Liberia, Mali, and Sierra Leone is sponsored by the National Institute of Allergy and Infectious Diseases (NIAID), Institut National de la Santé et de la Recherche Médicale (INSERM), and the London School of Hygiene and Tropical Medicine (LSHTM). The trial, which is fully enrolled, includes children as young as 1 year of age. The data for the primary endpoint at one-year post-dose 1 is expected to be released soon. Additional follow-up of participants for five years post-dose 1 is planned. This study will provide critical data on the safety, tolerability, and immunogenicity of rVSVΔG-ZEBOV-GP in pediatric populations as young as 1 year of age and is expected to serve as the basis for expanding the indication, which is currently limited to adults, to include children.
- ACHIV (African-Canadian Study of HIV-Infected Adults and a Vaccine for Ebola) trial: This randomized, placebo-controlled trial is evaluating the safety and immunogenicity of rVSVΔG-ZEBOV-GP in HIV-positive adults and adolescents and includes the evaluation of one and two doses. This trial is sponsored by the University of Dalhousie and is being conducted in Burkina Faso, Canada, and Senegal with ongoing enrollment. Given that Ebola outbreaks may overlap with areas of high HIV prevalence, understanding the safety and immunogenicity of rVSVΔG-ZEBOV-GP in these populations is very important. It is noteworthy to say that this trial has been temporarily paused due to SARS-CoV-2, highlighting the complexity of conducting clinical trials in the context of outbreaks.
- Expanded access protocols related to the Ebola outbreaks in the DRC: Working with the Ministries of Health and local researchers, WHO has implemented expanded access protocols in the DRC, Uganda, Rwanda, South Sudan, and Burundi. These protocols are designed to provide access to individuals at potential risk of EVD by virtue of being a contact or contact of contacts for a case of EVD or being a healthcare provider or FLW in a region involved in an active outbreak or in a neighboring region that is at risk of spread. These protocols have been extensively used as part of the Ebola response to the recent outbreaks in the DRC (beginning with the Equateur Province outbreak in May 2018). Through these protocols, more than 300,000 individuals have been vaccinated in the DRC, including children as young as 6 months of age and women who are pregnant (post first trimester) or lactating [60]. In addition, more than 14,000 individuals have been vaccinated in the neighboring countries. Data from these expanded access efforts are expected to provide information on safety in these important populations and to provide information on the effectiveness of the vaccine, potentially including information on the durability of protection for rVSVΔG-ZEBOV-GP. The data generated through these expanded access efforts are much closer to “real world” data and will be an important complement to the data generated through randomized controlled trials.
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Ebola Strategy: Ebola and Marburg Virus Disease Epidemics: Preparedness, Alert, Control, and Evaluation; WHO: Geneva, Switzerland, 2014. [Google Scholar]
- Chertow, D.S. Understanding long-term effects of Ebola virus disease. Nat. Med. 2019, 25, 714–715. [Google Scholar] [CrossRef] [PubMed]
- Brolin Ribacke, K.J.; Saulnier, D.D.; Eriksson, A.; von Schreeb, J. Effects of the West Africa Ebola virus disease on health-care utilization—A systematic review. Front. Public Health 2016, 4, 222. [Google Scholar] [CrossRef] [PubMed]
- Gouglas, D.; Le, T.T.; Henderson, K.; Kaloudis, A.; Danielsen, T.; Hammersland, N.C.; Robinson, J.M.; Heaton, P.M.; Røttingen, J.-A. Estimating the cost of vaccine development against epidemic infectious diseases: A cost minimisation study. Lancet Glob. Health 2018, 6, e1386–e1396. [Google Scholar] [CrossRef]
- Hurford, P.; Davis, M.A. How much does it cost to research and develop a vaccine? Eff. Altruism Forum 2018. Available online: https://forum.effectivealtruism.org/posts/BjBmcfwg2awqPJLin/how-much-does-it-cost-to-research-and-develop-a-vaccine (accessed on 18 February 2021).
- Hurford, P.; Davis, M.A. How long does it take to research and develop a new vaccine? Eff. Altruism Forum 2017. Available online: https://forum.effectivealtruism.org/posts/8qMDseJTE3vCFiYec/how-long-does-it-take-to-research-and-develop-a-new-vaccine (accessed on 18 February 2021).
- Wolf, J.; Bruno, S.; Eichberg, M.; Jannat, R.; Rudo, S.; VanRheenen, S.; Coller, B.-A. Applying lessons from the Ebola vaccine experience for SARS-CoV-2 and other epidemic pathogens. NPJ Vaccines 2020, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Eisele, P.; Irvine, S. ERVEBO® (Ebola Zaire Vaccine, Live) Now Registered in Four African Countries, Within 90 Days of Reference Country Approval and WHO Prequalification. Democratic Republic of the Congo One of the First African Countries to Register ERVEBO. 2020. Available online: https://www.merck.com/news/ervebo-ebola-zaire-vaccine-live-now-registered-in-four-african-countries-within-90-days-of-reference-country-approval-and-who-prequalification/ (accessed on 20 January 2021).
- European Medicine Agency. Ervebo: Ebola Zaire Vaccine (rVSV∆G-ZEBOV-GP, Live); EMA: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Jasarevic, T. WHO Prequalifies Ebola Vaccine, Paving the Way for Its Use in High-Risk Countries; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- United States Food & Drug Administration. ERVEBO, Ebola Zaire Vaccine, Live; USFDA: Silver Spring, MD, USA, 2020.
- RT Question More. Almost 100% Effective, No Side Effects: Russian Ebola Vaccine Presented to WHO. 2016. Available online: https://on.rt.com/74l0 (accessed on 20 January 2021).
- Liu, A. China Approves Domestic Ebola Vaccine Developed from Recent Outbreak. Fierce Pharma. 2017. Available online: https://www.fiercepharma.com/vaccines/china-approves-self-developed-ebola-vaccine-from-2014-outbreak-virus-type (accessed on 20 January 2021).
- Johnson & Johnson. Johnson & Johnson Announces European Commission Approval for Janssen’s Preventive Ebola Vaccine. 2020. Available online: https://www.jnj.com/johnson-johnson-announces-european-commission-approval-for-janssens-preventive-ebola-vaccine (accessed on 20 January 2021).
- World Health Organization. Essential Medicines and Health Products. WHO Publishes Roadmap for Introduction and Roll Out of a Licensed Ebola Vaccine. 2019. Available online: https://www.who.int/medicines/news/2019/roadmap_for_intro_roll_out_licensed_ebola_vaccine/en/ (accessed on 20 August 2020).
- World Health Organization. Lessons Learnt in Expediting Prequalification and Registration of Ebola Zaire Vaccine; Weekly Epidemiological Record; WHO: Geneva, Switzerland, 2020; Volume 95, pp. 369–380. [Google Scholar]
- Baylor, N.W. The regulatory evaluation of vaccines for human use. Methods Mol. Biol. 2016, 1404, 773–787. [Google Scholar] [CrossRef] [PubMed]
- Racine, T.; Kobinger, G.P.; Arts, E.J. Development of an HIV vaccine using a vesicular stomatitis virus vector expressing designer HIV-1 envelope glycoproteins to enhance humoral responses. AIDS Res. Ther. 2017, 14, 55. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Daddario-DiCaprio, K.M.; Lewis, M.G.; Geisbert, J.B.; Grolla, A.; Leung, A.; Paragas, J.; Matthias, L.; Smith, M.A.; Jones, S.M.; et al. Vesicular stomatitis virus-based ebola vaccine is well-tolerated and protects immunocompromised nonhuman primates. PLoS Pathog. 2008, 4, e1000225. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Geisbert, J.B.; Leung, A.; Daddario-DiCaprio, K.M.; Hensley, L.E.; Grolla, A.; Feldmann, H. Single-injection vaccine protects nonhuman primates against infection with marburg virus and three species of Ebola virus. J. Virol. 2009, 83, 7296–7304. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.M.; Feldmann, H.; Ströher, U.; Geisbert, J.B.; Fernando, L.; Grolla, A.; Klenk, H.-D.; Sullivan, N.J.; Volchkov, V.E.; Fritz, E.A.; et al. Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat. Med. 2005, 11, 786–790. [Google Scholar] [CrossRef] [PubMed]
- Marzi, A.; Engelmann, F.; Feldmann, F.; Haberthur, K.; Lesley Shupert, W.; Brining, D.; Scott, D.P.; Geisbert, T.W.; Kawaoka, Y.; Katze, M.G.; et al. Antibodies are necessary for rVSV/ZEBOV-GP-mediated protection against lethal Ebola virus challenge in nonhuman primates. Proc. Natl. Acad. Sci. USA 2013, 110, 1893–1898. [Google Scholar] [CrossRef]
- Qiu, X.; Fernando, L.; Alimonti, J.B.; Leno Melito, P.; Feldmann, F.; Dick, D.; Ströher, U.; Feldmann, H.; Jones, S.M. Mucosal immunization of cynomolgus macaques with the VSVDeltaG/ZEBOVGP vaccine stimulates strong ebola GP-specific immune responses. PLoS ONE 2009, 4, e5547. [Google Scholar] [CrossRef] [PubMed]
- Henao-Restrepo, A.M.; Camacho, A.; Longini, I.M.; Watson, C.H.; Edmunds, W.J.; Egger, M.; Carroll, M.W.; Dean, N.E.; Diatta, I.; Doumbia, M.; et al. Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease: Final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ca Suffit!). Lancet 2017, 389, 505–518. [Google Scholar] [CrossRef]
- Burns, D.L. Licensure of vaccines using the Animal Rule. Curr. Opin. Virol. 2012, 2, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Brindley, D.A. Conditional approval pathways: The “special” case of global regenerative medicine regulation. Rejuvenation Res. 2017, 20, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Garbutt, M.; Liebscher, R.; Wahl-Jensen, V.; Jones, S.; Möller, P.; Wagner, R.; Volchkov, V.; Klenk, H.-D.; Feldmann, H.; Ströher, U. Properties of replication-competent vesicular stomatitis virus vectors expressing glycoproteins of filoviruses and arenaviruses. J. Virol. 2004, 78, 5458–5465. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, E.; Saijo, M. Animal models for Ebola and Marburg virus infections. Front. Microbiol. 2013, 4, 267. [Google Scholar] [CrossRef] [PubMed]
- St. Claire, M.C.; Ragland, D.R.; Bollinger, L.; Jahrling, P.B. Animal models of ebolavirus infection. Comp. Med. 2017, 67, 253–262. [Google Scholar]
- Siragam, V.; Wong, G.; Qiu, X.G. Animal models for filovirus infections. Zool. Res. 2018, 39, 15–24. [Google Scholar] [CrossRef]
- Krause, P.R.; Bryant, P.R.; Clark, T.; Dempsey, W.; Henchal, E.; Michael, N.L.; Regules, J.A.; Gruber, M.F. Immunology of protection from Ebola virus infection. Sci. Transl. Med. 2015, 7, 286ps11. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Malherbe, D.C.; Bukreyev, A. Can Ebola virus vaccines have universal immune correlates of protection? Trends Microbiol. 2019, 27, 8–16. [Google Scholar] [CrossRef]
- Logue, J.; Tuznik, K.; Follmann, D.; Grandits, G.; Marchand, J.; Reilly, C.; Sarro, Y.D.S.; Pettitt, J.; Stavale, E.J.; Fallah, M.; et al. Use of the Filovirus Animal Non-Clinical Group (FANG) Ebola virus immuno-assay requires fewer study participants to power a study than the Alpha Diagnostic International assay. J. Virol. Methods 2018, 255, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Heppner, D.G., Jr.; Kemp, T.L.; Martin, B.K.; Ramsey, W.J.; Nichols, R.; Dasen, E.J.; Link, C.J.; Das, R.; Xu, Z.J.; Sheldon, E.A.; et al. Safety and immunogenicity of the rVSVΔG-ZEBOV-GP Ebola virus vaccine candidate in healthy adults: A phase 1b randomised, multicentre, double-blind, placebo-controlled, dose-response study. Lancet Infect. Dis. 2017, 17, 854–866. [Google Scholar] [CrossRef]
- Niemuth, N.A.; Rudge, T.L., Jr.; Sankovich, K.A.; Anderson, M.S.; Skomrock, N.D.; Badorrek, C.S.; Sabourin, C.L. Method feasibility for cross-species testing, qualification, and validation of the Filovirus Animal Nonclinical Group anti-Ebola virus glycoprotein immunoglobulin G enzyme-linked immunosorbent assay for non-human primate serum samples. PLoS ONE 2020, 15, e0241016. [Google Scholar] [CrossRef] [PubMed]
- Rudge, T.L.; Sankovich, K.A.; Niemuth, N.A.; Anderson, M.S.; Badorrek, C.S.; Skomrock, N.D.; Cirimotich, C.M.; Sabourin, C.L. Development, qualification, and validation of the Filovirus Animal Nonclinical Group anti-Ebola virus glycoprotein immunoglobulin G enzyme-linked immunosorbent assay for human serum samples. PLoS ONE 2019, 14, e0215457. [Google Scholar] [CrossRef] [PubMed]
- Monath, T.P.; Fast, P.E.; Modjarrad, K.; Clarke, D.K.; Martin, B.K.; Fusco, J.; Nichols, R.; Heppner, D.G.; Simon, J.K.; Dubey, S.; et al. rVSVΔG-ZEBOV-GP (also designated V920) recombinant vesicular stomatitis virus pseudotyped with Ebola Zaire Glycoprotein: Standardized template with key considerations for a risk/benefit assessment. Vaccine X 2019, 1, 100009. [Google Scholar] [CrossRef]
- Fuchs, J.D.; Frank, I.; Elizaga, M.L.; Allen, M.; Frahm, N.; Kochar, N.; Li, S.; Edupuganti, S.; Kalams, S.A.; Tomaras, G.D.; et al. First-in-human evaluation of the safety and immunogenicity of a recombinant vesicular stomatitis virus human immunodeficiency virus-1 gag vaccine (HVTN 090). Open Forum Infect. Dis. 2015, 2, ofv082. [Google Scholar] [CrossRef]
- Mire, C.E.; Miller, A.D.; Carville, A.; Westmoreland, S.V.; Geisbert, J.B.; Mansfield, K.G.; Feldmann, H.; Hensley, L.E.; Geisbert, T.W. Recombinant vesicular stomatitis virus vaccine vectors expressing filovirus glycoproteins lack neurovirulence in nonhuman primates. PLoS Negl. Trop. Dis. 2012, 6, e1567. [Google Scholar] [CrossRef]
- Sellers, R.S.; Nelson, K.; Bennet, B.; Wolf, J.; Tripathi, N.; Chamanza, R.; Lepage, M.-F.P.; Adkins, K.; Laurent, S.; Troth, S.P. Scientific and regulatory policy committee points to consider: Approaches to the conduct and interpretation of vaccine safety studies for clinical and anatomic pathologists. Toxicol. Pathol. 2019, 48, 257–276. [Google Scholar] [CrossRef]
- Bolay, F.K.; Grandits, G.; Lane, H.C.; Kennedy, S.B.; Johnson, M.P.; Fallah, M.P.; Wilson, B.; Njoh, W.S.; McNay, L.A.; Hensley, L.E.; et al. PREVAIL I cluster vaccination study with rVSVΔG-ZEBOV-GP as part of a public health response in Liberia. J. Infect. Dis. 2019, 219, 1634–1641. [Google Scholar] [CrossRef]
- Gsell, P.S.; Camacho, A.; Kucharski, A.J.; Watson, C.A.; Bagayoko, A.; Danmadji Nadlaou, S.; Dean, N.E.; Diallo, A.; Honora, D.A.; Doumbia, M.; et al. Ring vaccination with rVSV-ZEBOV under expanded access in response to an outbreak of Ebola virus disease in Guinea, 2016: An operational and vaccine safety report. Lancet Infect. Dis. 2017, 17, 1276–1284. [Google Scholar] [CrossRef]
- World Health Organization. Ebola Virus Disease: Democratic Republic of the Congo; External Situation Report 74; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Regules, J.A.; Beigel, J.H.; Paolino, K.M.; Voell, J.; Castellano, A.R.; Hu, Z.; Munoz, P.; Moon, J.E.; Ruck, R.C.; Bennett, J.W.; et al. A recombinant vesicular stomatitis virus Ebola vaccine. N. Engl. J. Med. 2017, 376, 330–341. [Google Scholar] [CrossRef]
- Elsherif, M.S.; Brown, C.; MacKinnon-Cameron, D.; Li, L.; Racine, T.; Alimonti, J.; Rudge, T.L.; Sabourin, C.; Silvera, P.; Hooper, J.W.; et al. Assessing the safety and immunogenicity of recombinant vesicular stomatitis virus Ebola vaccine in healthy adults: A randomized clinical trial. Can. Med. Assoc. J. 2017, 189, E819–E827. [Google Scholar] [CrossRef] [PubMed]
- Huttner, A.; Dayer, J.-A.; Yerly, S.; Combescure, C.; Auderset, F.; Desmeules, J.A.; Eickmann, M.; Finckh, A.; Goncalves, A.R.; Hooper, J.W.; et al. The effect of dose on the safety and immunogenicity of the VSV Ebola candidate vaccine: A randomised double-blind, placebo-controlled phase 1/2 trial. Lancet Infect. Dis. 2015, 15, 1156–1166. [Google Scholar] [CrossRef]
- Dahlke, C.; Kasonta, R.; Lunemann, S.; Krähling, V.; Zinser, M.E.; Biedenkopf, N.; Fehling, S.K.; Ly, M.L.; Rechtien, A.; Stubbe, H.C.; et al. Dose-dependent T-cell dynamics and cytokine cascade following rVSV-ZEBOV immunization. EBioMedicine 2017, 19, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Agnandji, S.T.; Fernandes, J.F.; Bache, E.B.; Obiang Mba, R.M.; Brosnahan, J.S.; Kabwende, L.; Pitzinger, P.; Staarink, P.; Massinga-Loembe, M.; Krähling, V.; et al. Safety and immunogenicity of rVSVΔG-ZEBOV-GP Ebola vaccine in adults and children in Lambarene, Gabon: A phase I randomised trial. PLoS Med. 2017, 14, e1002402. [Google Scholar] [CrossRef] [PubMed]
- Agnandji, S.T.; Huttner, A.; Zinser, M.E.; Njuguna, P.; Dahlke, C.; Fernandes, J.F.; Yerly, S.; Dayer, J.-A.; Kraehling, V.; Kasonta, R.; et al. Phase 1 trials of rVSV Ebola vaccine in Africa and Europe. N. Engl. J. Med. 2016, 374, 1647–1660. [Google Scholar] [CrossRef]
- Kennedy, S.B.; Bolay, F.; Kieh, M.; Grandits, G.; Badio, M.; Ballou, R.; Eckes, R.; Feinberg, M.; Follmann, D.; Grund, B.; et al. Phase 2 placebo-controlled trial of two vaccines to prevent Ebola in Liberia. N. Engl. J. Med. 2017, 377, 1438–1447. [Google Scholar] [CrossRef] [PubMed]
- Samai, M.; Seward, J.F.; Goldstein, S.T.; Mahon, B.E.; Radcliffe Lisk, D.; Widdowson, M.-A.; Jalloh, M.I.; Schrag, S.J.; Idriss, A.; Caster, R.J.; et al. The Sierra Leone trial to introduce a vaccine against Ebola: An evaluation of rVSVG-ZEBOV-GP vaccine tolerability and safety during the West Africa Ebola outbreak. J. Infect. Dis. 2018, 217, S6–S15. [Google Scholar] [CrossRef] [PubMed]
- Halperin, S.A.; Das, R.; Onorato, M.T.; Liu, K.; Martin, J.; Grant-Klein, R.J.; Nichols, R.; Coller, B.-A.; Helmond, F.A.; Simon, J.K.; et al. Immunogenicity, lot consistency, and extended safety of recombinant vesicular stomatitis virus–Zaire Ebola Virus envelope glycoprotein vaccine: A phase 3 randomized, double-blind, placebo-controlled study in healthy adults. J. Infect. Dis. 2019, 220, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Boum, Y.; Juan-Giner, A.; Hitchings, M.; Soumah, A.; Strecker, T.; Sadjo, M.; Cuthbertson, H.; Hayes, P.; Tchaton, M.; Jemmy, J.-P.; et al. Humoral and cellular immune response induced by rVSVΔG-ZEBOV-GP vaccine among frontline workers during the 2013–2016 West Africa Ebola outbreak in Guinea. Vaccine 2020, 38, 4877–4884. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. M4 Organization of the Common Technical Document for the Registration of Pharmaceuticals for Human Use: Guidance for Industry; US Department of Health and Human Services, USFDA: Silver Spring, MD, USA, 2017.
- Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. ERVEBO® (Ebola Zaire Vaccine, Live) Suspension for Intramuscular Injection Prescribing Information; Whitehouse Station: New Jersey, NJ, USA, 2019. [Google Scholar]
- Camacho, A.; Carroll, M.W.; Dean, N.E.; Doumbia, M.; Edmunds, W.J.; Egger, M.; Enwere, G.; Hall, Y.; Henao-Restrepo, A.M.; Hossmann, S.; et al. The ring vaccination trial: A novel cluster randomised controlled trial design to evaluate vaccine efficacy and effectiveness during outbreaks, with special reference to Ebola. BMJ 2015, 351, h3740. [Google Scholar] [CrossRef][Green Version]
- Grais, R.F.; Kennedy, S.B.; Mahon, B.E.; Dubey, S.A.; Grant-Klein, R.J.; Liu, K.; Hartzel, J.; Coller, B.-A.; Welebob, C.; Hanson, M.E.; et al. Estimation of the correlates of protection of the rVSVΔG-ZEBOV-GP Zaire ebolavirus vaccine: A post-hoc analysis of data from phase 2/3 clinical trials. Lancet Microbe 2021, 2, e70–e78. [Google Scholar] [CrossRef]
- Grant-Klein, R.J.; Antonello, J.; Nichols, R.; Dubey, S.; Simon, J. Effect of gamma irradiation on the antibody response measured in human serum from subjects vaccinated with recombinant vesicular stomatitis Virus–Zaire Ebola virus envelope glycoprotein vaccine. Am. J. Trop. Med. Hyg. 2019, 101, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Antonello, J.; Grant-Klein, R.J.; Nichols, R.; Kennedy, M.A.; Dubey, S.; Simon, J.K. Serostatus cutoff (SSCO) levels and fold increase to define seroresponse to recombinant vesicular stomatitis Virus–Zaire Ebola virus envelope glycoprotein vaccine (rVSVΔG-ZEBOV-GP): An evidence-based analysis. Vaccine 2020, 38, 4885–4891. [Google Scholar] [CrossRef]
- World Health Organization. Ebola Virus Disease: Democratic Republic of the Congo; External Situation Report 98; WHO: Geneva, Switzerland, 2020; pp. 1–9. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Manifestation | Immuno-Competent Mouse | Immuno-Compromised Mouse | Guinea Pig | Syrian Hamster | Ferret | NHP | Human |
|---|---|---|---|---|---|---|---|
| Lymphopenia | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Liver damage | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Thrombocytopenia | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Coagulopathy | No | Unknown | Yes | Yes | Yes | Yes | Yes |
| Cytokine Storm | Yes | Yes | Unknown | Yes | Unknown | Yes | Yes |
| Rash | No | No | No | Yes | Yes | Yes | Yes |
| Hemorrhage signs | No | Yes | Unknown | Yes | Yes | Yes | Yes |
| Protocol Number, Trial Name, Country Location, and Trial Registry Number | Trial Description | [Sponsor] External Trial Partner Organization/Funders, and Academic Partners | N Vaccinated with rVSVΔG-ZEBOV-GP | Dose Levels (pfu) | Subject Memory Aid Use (Y/N) | CSR MedDRA Version | AE Category | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Solicited Injection Site and Systemic AEs | Unsolicited AEs | SAEs | Viremia and Viral Shedding | Clinical Laboratory Safety Tests | |||||||
| Phase 1 * | |||||||||||
| V920-001 US NCT02269423 | Randomized single-center, double-blind, placebo-controlled, dose- escalation study [44] | [BPS/NLG] Walter Reed Army Institute of Research/DTRA, Imperial College of Science, Technology, and Medicine; University of Maryland School of Medicine, University of Texas-Austin | 30 | 3 × 106, 2 × 107, 1 × 108 (n = 10 each) or Placebo (n = 9) | Yes | 17.0 | Day 1–14 | Day 1–28 | Day 1–180 | Day 1, 3, 7, 14 | Day 0, 1, 3, 7, 28, 180 |
| V920-002 US NCT02280408 | Randomized double-blind, placebo-controlled, dose- escalation study [44] | [BPS/NLG] NIH/NIAID | 30 | 3 × 106, 2 × 107, 1 × 108 (n = 10 each), Placebo (n = 9) † Second identical dose at D28 | Yes | 17.0 | Day 1–14 | Day 1–28 | Day 1–365 | Day 0, 3, 7 following each dose | Day 0, 7, 28, 35, 56 following each dose |
| V920-003 Canada NCT02374385 | Randomized single-center, double-blind controlled, dose-ranging study [45] | [BPS/NLG] CIHR; PHAC, University of Ottawa, Dalhousie University | 30 | 1 × 105, 5 × 105, 3 × 106 (n = 10 each), Placebo (n = 10) | Yes | 17.0 | Day 1–14 | Day 1–28 | Day 1–180 | Day 1, 3, 7, 14 | Day 0, 1, 3, 7, 28, 180 |
| V920-004 US NCT02314923 | Randomized multi-center, double-blind, placebo- controlled, dose-response study [34] | [BPS/NLG] BARDA | 418 | 3 × 103, 3 × 104, 3 × 105 (n = 64 each) 3 × 106 (n = 84) 9 × 106, 2 × 107 (n = 47 each) 1 × 108 (n = 48) Placebo (n = 94) | Yes | 17.0 | Cohort 1: Day 1–14 Cohort 2: Day 1–56 | Cohort 1: Day 1–14 Cohort 2: Day 1–56 | Day 1–360 | Day 0, 1, 2, 3, 4, 7, 14, 28 | Day 0, 7, 28 |
| V920-05 Switzerland NCT02287480 | Dose-finding, randomized, single-center, double-blind †, placebo- controlled study [46] | [University Hospitals of Geneva] WHO, Wellcome Trust, Innovative Medicines Initiative, University Hospitals of Geneva | 102 | 3 × 105 (n = 51) 1 × 107 (n = 35) 5 × 107 (n = 16) Placebo (n = 13) | Yes | 17.0 | Day 1–14 | Day 1–28 | Day 1–365 | Day 0, 1, 3, 7 | Day 0, 1, 3, 7, 14, 28, 365 (only blood count at Day 365) |
| V920-006 Germany NCT02283099 | Open-label, dose-escalation study [47] | [Universitätsklinikum Hamburg-Eppendorf] WHO; Wellcome Trust | 30 | 3 × 105, 3 × 106, 2 × 107 (n = 10 each) | Yes | 17.0 | Day 1–14 | Day 1–28 | Day 1–180 | Day 0, 1 to 7, 14, 28 | Day 0, 1, 3, 7, 14, 28, 180 |
| V920-007 Gabon PACTR201411000919191 | Randomized open-label, dose-escalation study [48] | [Universitätsklinikum Tübingen] WHO; Wellcome Trust, St. George’s University of London, Medical University Vienna, Austria | 115 ‡ | 3 × 103 (n = 20) 3 × 104 (n = 20) 3 × 105 (n = 20) 3 × 106 (n = 39) 2 × 107 (n = 16) | Yes | 17.0 | Day 1–14 | Day 1–28 | Day 1–365 | Day 0, 1, 2, and 7 | Day 0, 1, 2, 7, 28, 84, 180, 365 |
| V920-008 Kenya NCT02296983 | Open-label, dose-escalation study [49] | [University of Oxford] WHO; Wellcome Trust | 40 | 3 × 106 2 × 107 (n = 20 each) | Yes | 17.0 | Day 1–14 | Day 1–28 | Day 1–365 | Day 1, 3, 7 | Day 0, 7, 30 |
| V920-009 PREVAIL Liberia NCT02344407 | Randomized double-blind, placebo-controlled, 3-arm trial [50] | [NIH/NIAID] Liberian Ministry of Health and Social Welfare, BARDA, GlaxoSmithKline, University of Minnesota | 500 | 2 × 107 (n = 500) GSK (n = 500) Placebo (n = 500) | No | 20.0 | Week 1, Week 2, Month 1 ₶ | Week 1 and Month 1 | Wk 1, Month 1 and 2, every 2 Months to trial end | Not collected | At Week 1 and Month 1 |
| V920-014 Lambaréné, Gabon Not registered | Randomized, open-label, controlled | Centre de Recherches Médicales de Lambaréné (CERMEL), | Planned: 40 | 2 × 107 n = 40 Varicella vaccine N = 20 | No | N/A | Days 1–28 | Days 1–28 | Days 1–365 | Days 1–56 | Screening, D7, 28, 84, 180, 365 |
| Phase 2/3 | |||||||||||
| V920-010 Ebola Ҫa Suffit Guinea PACTR201503001057193 | Open-label, cluster-randomized ring vaccination trial [24] | [WHO] Norwegian Research Council; MSF; Wellcome Trust; PHAC, Guinea Ministry of Health and Public Hygiene | 5837 | 2 × 107 (n = 5837) | No | N/A § | Minute 30, Day 3, and Day 14 | Day 1–14 | Day 1–84 | Not collected | Not collected |
| V920-011 STRIVE Sierra Leone NCT02378753 | Randomized unblinded trial design [51] | [US CDC] BARDA, Sierra Leone Ministry of Health and Sanitation, College of Medicine and Allied Health Sciences—Sierra Leone | 7998 | 2 × 107 (n = 7998) | Yes | 19.0 | Safety sub study participants: Day 0–28 | Overall pop: Day 0–28 | Overall pop: Day 0–180 | Not collected | Not collected |
| V920-012 Lot Consistency US, Canada, Spain NCT02503202 | Randomized placebo-controlled, safety and lot consistency immunogenicity study [52] | [MSD, a subsidiary of Merck & Co., Inc, Kenilworth, NJ, USA] BARDA; Dalhousie University | 1061 | 2 × 107 (n = 797) 1 × 108 (n = 264) Placebo (n = 133) | Yes | 19.1 | Day 1–42 ¥ | Day 1 to 42 | Day 1 to Month 24 | Not collected | As needed for arthralgia, arthritis, rash or vesicles— follow-up only |
| V920-013 PREPARE US, Canada NCT02788227 | Randomized open-label, booster or no booster at 18 months in individuals at potential occupational risk | [NIH/NIAID] BARDA, University of Texas, Galveston, TX, Emory University, PHAC, Winnipeg; CIRN, University of Nebraska Medical Center, Boston Medical Center/Boston University, Universitätsklinikum Hamburg-Eppendorf | Planned enrollment N up to 1000 | 2 × 107 | Yes | N/A | Day 1–14, Month 1, Month 18 (booster), Month 19 (post-booster) | Day 1–42 | Day 1 to Year 3 | Not collected | Not collected |
| V920-015 ACHIV Canada, Burkina Faso, Senegal NCT03031912 | Randomized double-blind, placebo- controlled, one or two doses of rVSVΔG-ZEBOV-GP | [Dalhousie University] BARDA, CIRN | Planned enrollment ~250 | 2 × 107 pfu/mL (n~200) Placebo (n~50) | Yes | N/A | Day 1, 3, 7, 14, 28, 42 | Day 1, 3, 7, 14, 28, 42 | Day 0–365 | Day 3, 7, 14, 28, 42 | Only as clinically needed |
| V920-016 PREVAC Guinea, Liberia, Mali, Sierra Leone NCT02876328 | Randomized double-blind, placebo- controlled trial of three vaccine strategies (Ad26.ZEBOV/MVA-BN-Filo vaccine-Janssen, rVSVΔG-ZEBOV-GP vaccine-MSD with or without boost at 56 days) in adults and children ≥1 year | [Office of Clinical Research Operations and Regulatory Compliance Division of Clinical Research NIAID, NIH; Institut National de la Santé et de la Recherche Médicale; LSHTM] BARDA, MSD, Janssen, University of Minnesota | ~1822 | 2 × 107 pfu/mL, (N~1822) Also includes Janssen vaccine and placebo | No | N/A | Adults: Day 0, 7, 14, 28, 56, 63, Month 3 Children: Day 0, daily contacts, Day 1–6, Day 7, 14, 28, 56, 63, Month 3 | Grade 3 and 4 unsolicited AEs only. Adults: Day 0, 7, 14, 28, 56, 63, Month 3 Children: Day 0, daily contacts Day 1–6, Day 7, 14, 28, 56, 63, Month 3 | Day 0-Month 12 | Subset of children: Day 0, 7, 14, 28, 56, 63, Month 3 | Adults: Day 0 Children: Day 0, 7, 63 |
| V920-018 Front-Line Workers (FLW) Guinea PACTR201503001057193 | Open-label, cluster-randomized ring vaccination trial [53] | [WHO] University of Maryland, University of Bern, LSHTM, University of Florida | 2016 | 2 × 107(N = 2016) | No | N/A | Not collected | Day 3, 14 | Day 0–84 | Not collected | Not collected |
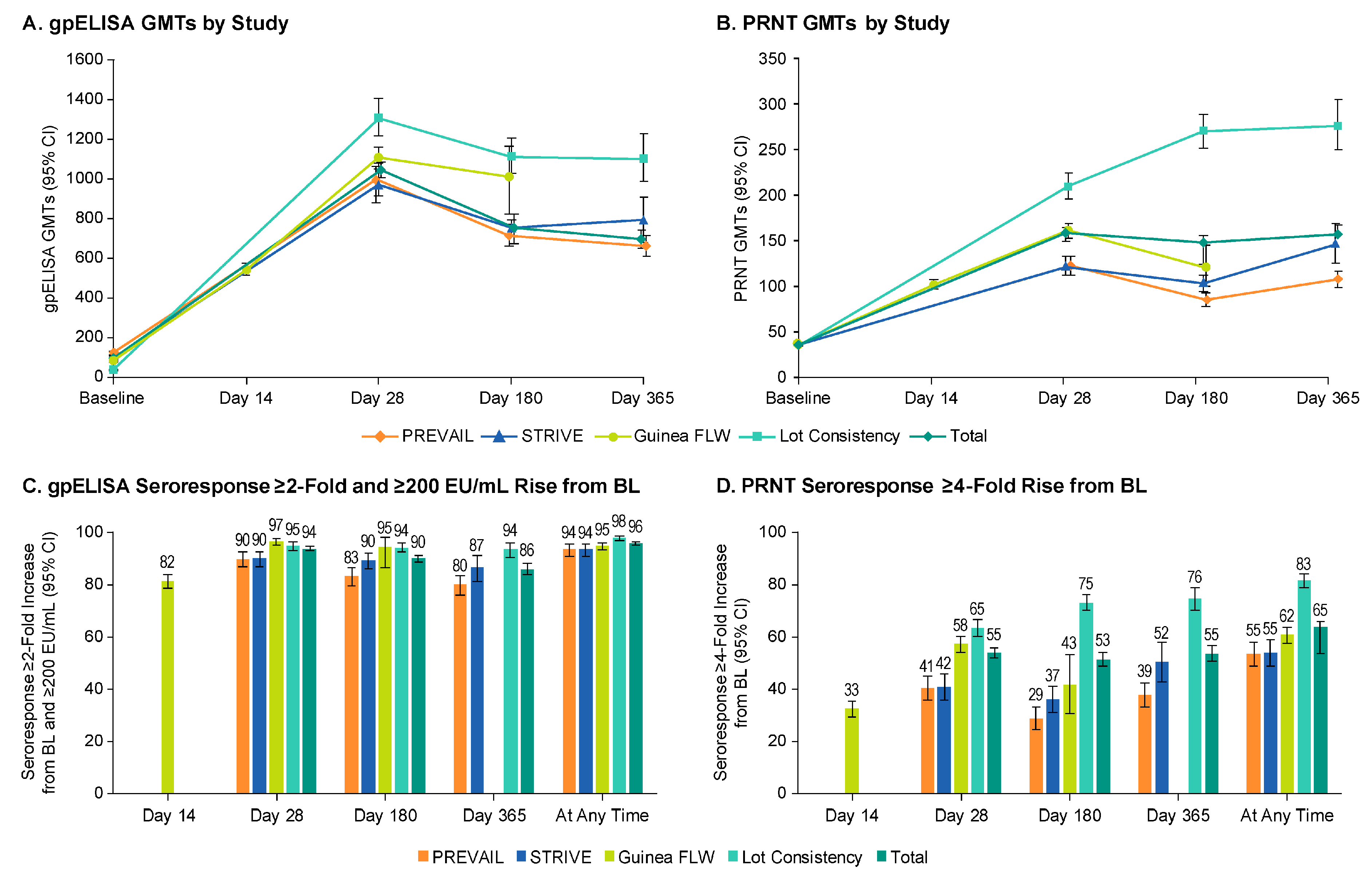
| Baseline | Day 14 | Day 28 | Day 180 | Day 365 | Day 14 | Day 28 | Day 180 | Day 365 | At Any Time | |
|---|---|---|---|---|---|---|---|---|---|---|
| GP-ELISA GMTs (95% CI) | GP-ELISA Seroresponse two-fold and 200 EU/mL Increase from Baseline | |||||||||
| FLW | 82.8 (79.4, 86.3) | 543.2 (512.5, 575.8) | 1106.5 (1053.4, 1162.2) | 1008.8 (849.8, 1197.6) | -- | 81.5 (78.1, 83.9) | 96.7 (95.3, 97.7) | 94.6 (86.7, 98.5) | -- | 94.9 (93.4, 96.1) |
| PREVAIL | 121.8 (112.1, 132.4) | -- | 994.7 (915.0, 1081.3) | 712.2 (659.4, 769.3) | 661.4 (613.2, 713.4) | -- | 90.0 (86.9, 92.6) | 83.2 (79.5, 86.5) | 80.1 (76.2, 83.7) | 93.8 (91.1, 95.8) |
| STRIVE | 97.1 (89.7, 105.0) | -- | 969.9 (885.3, 1062.4) | 755.8 (695.7, 821.2) | 795.0 (697.9, 905.7) | -- | 90.1 (87.0, 92.7) | 89.5 (86.0, 92.3) | 87.0 (81.4, 91.4) | 93.6 (91.0, 95.7) |
| Lot Consistency | 36.1 (36.1, 36.1) | -- | 1307.3 (1214.8, 1406.7) | 1113.4 (1029.5, 1204.1) | 1101.1 (986.3, 1229.3) | -- | 95.0 (93.2, 96.4) | 94.4 (92.5, 96.0) | 93.6 (90.5, 96.0) | 98.1 (96.9, 98.9) |
| Total | 93.7 (90.5, 97.1) | -- | 1045.6 (1005.6, 1087.1) | 752.0 (712.6, 793.6) | 697.4 (653.0, 744.8) | -- | 93.9 (92.9, 94.7) | 90.1 (88.6, 91.5) | 86.1 (83.8, 88.2) | 96.1 (95.2, 96.8) |
| PRNT GMTs (95% CI) | PRNT Seroresponse four-fold Increase from Baseline | |||||||||
| FLW | 35.2 (34.9, 35.5) | 102.9 (97.8, 108.3) | 162.2 (153.9, 170.9) | 121.4 (101.3, 145.6) | -- | 33.2 (30.3, 36.3) | 58.5 (55.2, 61.7) | 42.7 (31.3, 54.6) | -- | 62.1 (59.1, 65.0) |
| PREVAIL | 36.5 (35.0, 38.1) | -- | 123.1 (112.5, 134.7) | 85.3 (78.5, 92.7) | 107.8 (99.1, 117.3) | -- | 41.4 (36.6, 46.2) | 29.4 (25.2, 34.0) | 38.6 (34.0, 43.4) | 54.7 (49.8, 59.5) |
| STRIVE | 35.6 (35.0, 36.3) | -- | 122.8 (112.89, 133.6) | 103.7 (95.2, 113.0) | 147.3 (127.0, 170.8) | -- | 41.6 (36.6, 46.7) | 36.8 (31.6, 42.2) | 51.5 (43.6, 59.4) | 55.2 (50.2, 60.1) |
| Lot Consistency | 35.5 (35.5, 36.0) | -- | 211.7 (198.1, 226.3) | 271.8 (253.4, 291.5) | 278.3 (251.8, 307.5) | -- | 64.9 (61.4, 68.3) | 74.8 (71.5, 78.0) | 76.3 (71.5, 80.7) | 83.3 (80.5, 85.9) |
| Total | 35.6 (35.3, 35.9) | -- | 159.3 (153.8, 165.1) | 149.7 (142.4, 157.3) | 158.1 (148.4, 168.5) | -- | 54.9 (53.0, 56.9) | 52.6 (50.1, 55.1) | 54.8 (51.6, 58.0) | 65.4 (54.7, 67.2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wolf, J.; Jannat, R.; Dubey, S.; Troth, S.; Onorato, M.T.; Coller, B.-A.; Hanson, M.E.; Simon, J.K. Development of Pandemic Vaccines: ERVEBO Case Study. Vaccines 2021, 9, 190. https://doi.org/10.3390/vaccines9030190
Wolf J, Jannat R, Dubey S, Troth S, Onorato MT, Coller B-A, Hanson ME, Simon JK. Development of Pandemic Vaccines: ERVEBO Case Study. Vaccines. 2021; 9(3):190. https://doi.org/10.3390/vaccines9030190
Chicago/Turabian StyleWolf, Jayanthi, Risat Jannat, Sheri Dubey, Sean Troth, Matthew T. Onorato, Beth-Ann Coller, Mary E. Hanson, and Jakub K. Simon. 2021. "Development of Pandemic Vaccines: ERVEBO Case Study" Vaccines 9, no. 3: 190. https://doi.org/10.3390/vaccines9030190
APA StyleWolf, J., Jannat, R., Dubey, S., Troth, S., Onorato, M. T., Coller, B.-A., Hanson, M. E., & Simon, J. K. (2021). Development of Pandemic Vaccines: ERVEBO Case Study. Vaccines, 9(3), 190. https://doi.org/10.3390/vaccines9030190