
SARS-CoV-2 mRNA Vaccines: Immunological Mechanism and Beyond

{kind=link}
Abstract
1. Introduction
2. Design Strategies for SARS-CoV-2 mRNA Vaccines
2.1. Modifications to mRNA
2.2. Antigen Selection
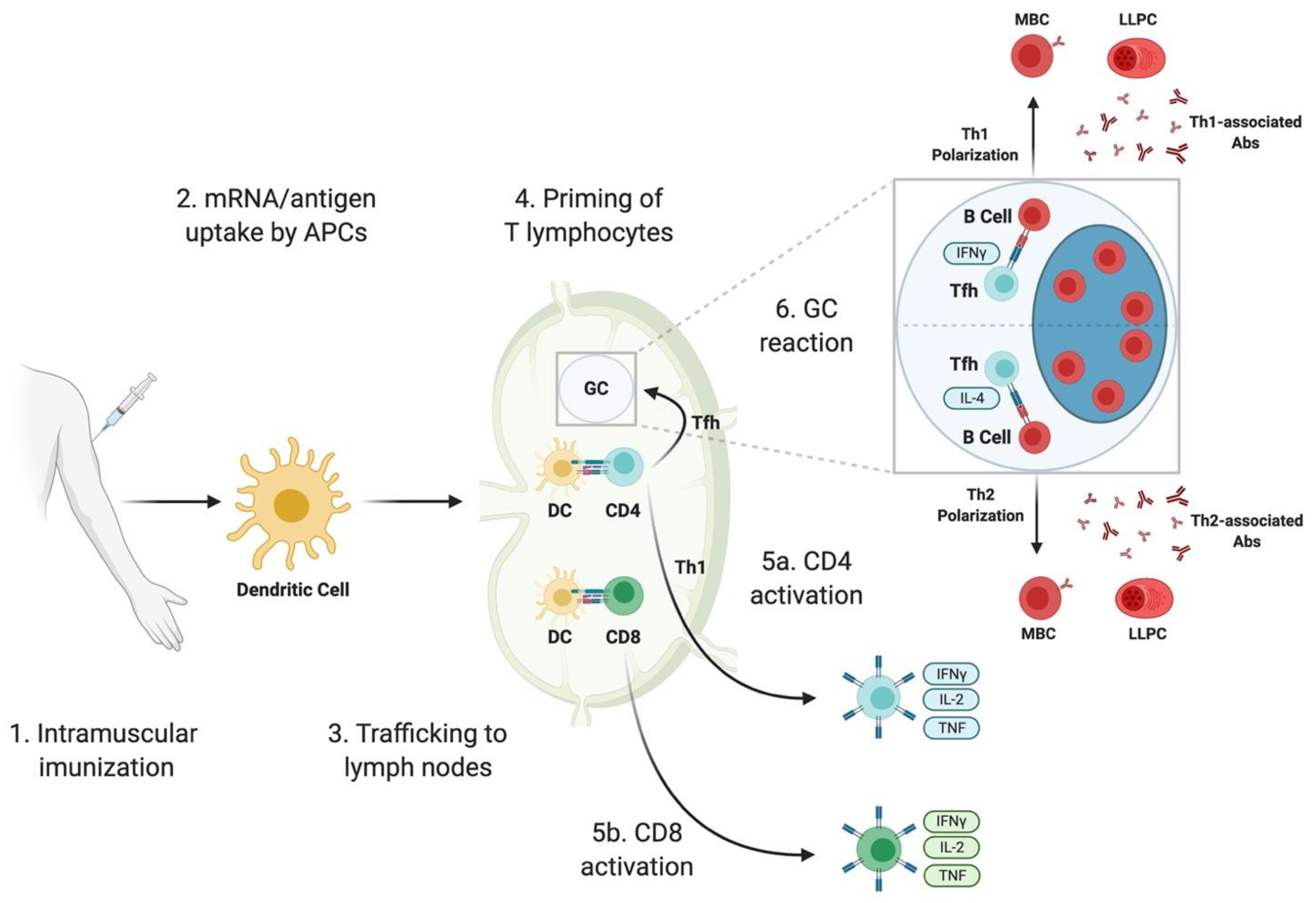
2.3. Delivery of mRNA
3. Immune Responses Elicited by SARS-CoV-2 mRNA Vaccines: Lessons from Animal Studies
3.1. B Cells and Antibody Responses
3.1.1. Induction of SARS-CoV-2 Binding and Neutralizing Abs
3.1.2. Germinal Center-Derived B Cell Response
3.2. T Cell Responses
3.2.1. T Follicular Helper Cell Response
3.2.2. Functional Polarization of CD4 T Cells
3.2.3. Cytotoxic T Cell Response
4. Results from Human Clinical Trial with SARS-CoV-2 mRNA Vaccines
4.1. Phase 1/2
4.1.1. Efficacy Profiles: Humoral Responses
4.1.2. Efficacy Profiles: Cellular Responses
4.1.3. Safety Profiles
4.2. Phase 3 Trials
5. Strengths and Limitations of SARS-CoV-2 mRNA Vaccine Candidates
5.1. Strengths
5.2. Challenges and Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Ab | Antibody |
| ACE2 | Angiotensin-converting enzyme 2 |
| APC | Antigen-presenting cell |
| COVID-19 | Coronavirus Disease 2019 |
| Δfurin | Full-length S lacking the furin cleavage site |
| DC ELISA | Dendritic cell Enzyme-linked immunosorbent assay |
| GC HA | Germinal center Hemagglutinin |
| Ig | Immunoglobulin |
| IL | Interleukin |
| LLPC | Long-lived plasma cell |
| LNP | Lipid nanoparticle |
| m1Ψ | N1-methyl-pseudouridine |
| MBC | Memory B cell |
| mRNA | Messenger RNA |
| nAb | Neutralizing antibody |
| RBD | Receptor binding domain (of SARS-CoV-2 S) |
| RIG-I | Retinoic acid inducible gene I |
| rRBD | Recombinant RBD |
| rRBD-AddaVax | rRBD given with the adjuvant AddaVax |
| S | Spike protein (of SARS-CoV-2) |
| S-2P | Spike 2-proline (SARS-CoV-2 S with proline substitutions at amino acids 986 and 987; stabilized in a prefusion conformation) |
| S1 | Subunit 1 (of SARS-CoV-2 S) |
| S2 | Subunit 2 (of SARS-CoV-2 S) |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus 2 |
| Tfh | T follicular helper cells |
| Th1 | T helper 1 |
| Th2 | T helper 2 |
| TLR | Toll-like receptor |
References
- COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU). Available online: https://Coronavirus.Jhu.Edu/Map.Html (accessed on 31 December 2020).
- Draft Landscape of COVID-19 Candidate Vaccines. Available online: https://www.Who.Int/Publications/m/Item/Draft-Landscape-of-Covid-19-Candidate-Vaccines (accessed on 29 December 2020).
- Krammer, F. SARS-CoV-2 Vaccines in Development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. MRNA Vaccines—A New Era in Vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of Pseudouridine Into MRNA Yields Superior Nonimmunogenic Vector With Increased Translational Capacity and Biological Stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An MRNA Vaccine against SARS-CoV-2—Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.S.; Flynn, B.; Foulds, K.E.; Francica, J.R.; Boyoglu-Barnum, S.; Werner, A.P.; Flach, B.; O’Connell, S.; Bock, K.W.; Minai, M.; et al. Evaluation of the MRNA-1273 Vaccine against SARS-CoV-2 in Nonhuman Primates. N. Engl. J. Med. 2020, 383, 1544–1555. [Google Scholar] [CrossRef]
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schäfer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 MRNA Vaccine Design Enabled by Prototype Pathogen Preparedness. Nature 2020, 586, 567–571. [Google Scholar] [CrossRef]
- Anderson, E.J.; Rouphael, N.G.; Widge, A.T.; Jackson, L.A.; Roberts, P.C.; Makhene, M.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; Pruijssers, A.J.; et al. Safety and Immunogenicity of SARS-CoV-2 MRNA-1273 Vaccine in Older Adults. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; Sahly, H.M.E.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the MRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Widge, A.T.; Rouphael, N.G.; Jackson, L.A.; Anderson, E.J.; Roberts, P.C.; Makhene, M.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; Pruijssers, A.J.; et al. Durability of Responses after SARS-CoV-2 MRNA-1273 Vaccination. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Vogel, A.B.; Kanevsky, I.; Che, Y.; Swanson, K.A.; Muik, A.; Vormehr, M.; Kranz, L.M.; Walzer, K.C.; Hein, S.; Güler, A.; et al. A Prefusion SARS-CoV-2 Spike RNA Vaccine Is Highly Immunogenic and Prevents Lung Infection in Non-Human Primates. Biorxiv 2020. [Google Scholar] [CrossRef]
- Sahin, U.; Muik, A.; Derhovanessian, E.; Vogler, I.; Kranz, L.M.; Vormehr, M.; Baum, A.; Pascal, K.; Quandt, J.; Maurus, D.; et al. COVID-19 Vaccine BNT162b1 Elicits Human Antibody and TH1 T Cell Responses. Nature 2020, 586, 594–599. [Google Scholar] [CrossRef]
- Mulligan, M.J.; Lyke, K.E.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Raabe, V.; Bailey, R.; Swanson, K.A.; et al. Phase I/II Study of COVID-19 RNA Vaccine BNT162b1 in Adults. Nature 2020, 586, 589–593. [Google Scholar] [CrossRef]
- Walsh, E.E.; Frenck, R.W., Jr.; Falsey, A.R.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Mulligan, M.J.; Bailey, R.; et al. Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates. N. Engl. J. Med. 2020, 383, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 MRNA Covid-19 Vaccine. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM Structure of the 2019-NCoV Spike in the Prefusion Conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional Assessment of Cell Entry and Receptor Usage for SARS-CoV-2 and Other Lineage B Betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Kirchdoerfer, R.N.; Cottrell, C.A.; Wang, N.; Pallesen, J.; Yassine, H.M.; Turner, H.L.; Corbett, K.S.; Graham, B.S.; McLellan, J.S.; Ward, A.B. Pre-Fusion Structure of a Human Coronavirus Spike Protein. Nature 2016, 531, 118–121. [Google Scholar] [CrossRef]
- Pallesen, J.; Wang, N.; Corbett, K.S.; Wrapp, D.; Kirchdoerfer, R.N.; Turner, H.L.; Cottrell, C.A.; Becker, M.M.; Wang, L.; Shi, W.; et al. Immunogenicity and Structures of a Rationally Designed Prefusion MERS-CoV Spike Antigen. Proc. Natl. Acad. Sci. USA 2017, 114, E7348–E7357. [Google Scholar] [CrossRef]
- Graham, B.S.; Gilman, M.S.A.; McLellan, J.S. Structure-Based Vaccine Antigen Design. Annu. Rev. Med. 2019, 70, 91–104. [Google Scholar] [CrossRef]
- Laczkó, D.; Hogan, M.J.; Toulmin, S.A.; Hicks, P.; Lederer, K.; Gaudette, B.T.; Castaño, D.; Amanat, F.; Muramatsu, H.; Oguin, T.H.; et al. A Single Immunization with Nucleoside-Modified MRNA Vaccines Elicits Strong Cellular and Humoral Immune Responses against SARS-CoV-2 in Mice. Immunity 2020, 53, 724–732.e7. [Google Scholar] [CrossRef]
- Lederer, K.; Castaño, D.; Atria, D.G.; Oguin, T.H.; Wang, S.; Manzoni, T.B.; Muramatsu, H.; Hogan, M.J.; Amanat, F.; Cherubin, P.; et al. SARS-CoV-2 MRNA Vaccines Foster Potent Antigen-Specific Germinal Center Responses Associated with Neutralizing Antibody Generation. Immunity 2020, 53, 1281–1295.e5. [Google Scholar] [CrossRef]
- Zost, S.J.; Gilchuk, P.; Case, J.B.; Binshtein, E.; Chen, R.E.; Nkolola, J.P.; Schäfer, A.; Reidy, J.X.; Trivette, A.; Nargi, R.S.; et al. Potently Neutralizing and Protective Human Antibodies against SARS-CoV-2. Nature 2020, 584, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Zost, S.J.; Gilchuk, P.; Chen, R.E.; Case, J.B.; Reidy, J.X.; Trivette, A.; Nargi, R.S.; Sutton, R.E.; Suryadevara, N.; Chen, E.C.; et al. Rapid Isolation and Profiling of a Diverse Panel of Human Monoclonal Antibodies Targeting the SARS-CoV-2 Spike Protein. Nat. Med. 2020, 26, 1422–1427. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, P.J.M.; Caniels, T.G.; van der Straten, K.; Snitselaar, J.L.; Aldon, Y.; Bangaru, S.; Torres, J.L.; Okba, N.M.A.; Claireaux, M.; Kerster, G.; et al. Potent Neutralizing Antibodies from COVID-19 Patients Define Multiple Targets of Vulnerability. Science 2020, 369, 643–650. [Google Scholar] [CrossRef]
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.-W.; Sahi, V.; Figueroa, A.; et al. Potent Neutralizing Antibodies against Multiple Epitopes on SARS-CoV-2 Spike. Nature 2020, 584, 450–456. [Google Scholar] [CrossRef]
- Robbiani, D.F.; Gaebler, C.; Muecksch, F.; Lorenzi, J.C.C.; Wang, Z.; Cho, A.; Agudelo, M.; Barnes, C.O.; Gazumyan, A.; Finkin, S.; et al. Convergent Antibody Responses to SARS-CoV-2 in Convalescent Individuals. Nature 2020, 584, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Tuyishime, S.; Muramatsu, H.; Kariko, K.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Weissman, D. Expression Kinetics of Nucleoside-Modified MRNA Delivered in Lipid Nanoparticles to Mice by Various Routes. J. Control. Release 2015, 217, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Naradikian, M.S.; Parkhouse, K.; Cain, D.W.; Jones, L.; Moody, M.A.; Verkerke, H.P.; Myles, A.; Willis, E.; et al. Nucleoside-Modified MRNA Vaccines Induce Potent T Follicular Helper and Germinal Center B Cell Responses. J. Exp. Med. 2018, 215, 1571–1588. [Google Scholar] [CrossRef]
- Liang, F.; Lindgren, G.; Lin, A.; Thompson, E.A.; Ols, S.; Röhss, J.; John, S.; Hassett, K.; Yuzhakov, O.; Bahl, K.; et al. Efficient Targeting and Activation of Antigen-Presenting Cells In Vivo after Modified MRNA Vaccine Administration in Rhesus Macaques. Mol. Ther. 2017, 25, 2635–2647. [Google Scholar] [CrossRef]
- Maier, M.A.; Jayaraman, M.; Matsuda, S.; Liu, J.; Barros, S.; Querbes, W.; Tam, Y.K.; Ansell, S.M.; Kumar, V.; Qin, J.; et al. Biodegradable Lipids Enabling Rapidly Eliminated Lipid Nanoparticles for Systemic Delivery of RNAi Therapeutics. Mol. Ther. 2013, 21, 1570–1578. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.J.; Webber, M.J.; Anderson, D.G. Materials for Non-Viral Intracellular Delivery of Messenger RNA Therapeutics. J. Control. Release 2016, 240, 227–234. [Google Scholar] [CrossRef]
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. 2017, 25, 1467–1475. [Google Scholar] [CrossRef]
- Cagigi, A.; Loré, K. Immune Responses Induced by MRNA Vaccination in Mice, Monkeys and Humans. Nato Adv. Sci. Inst. Se 2021, 9, 61. [Google Scholar] [CrossRef]
- Plotkin, S.A. Correlates of Protection Induced by Vaccination. Clin. Vaccine Immunol. 2010, 17, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Mesin, L.; Ersching, J.; Victora, G.D. Germinal Center B Cell Dynamics. Immunity 2016, 45, 471–482. [Google Scholar] [CrossRef]
- Sallusto, F.; Lanzavecchia, A.; Araki, K.; Ahmed, R. From Vaccines to Memory and Back. Immunity 2010, 33, 451–463. [Google Scholar] [CrossRef]
- Amanna, I.J.; Carlson, N.E.; Slifka, M.K. Duration of Humoral Immunity to Common Viral and Vaccine Antigens. N. Engl. J. Med. 2007, 357, 1903–1915. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Shan, C.; Duan, X.; Chen, Z.; Liu, P.; Song, J.; Song, T.; Bi, X.; Han, C.; Wu, L.; et al. A Human Neutralizing Antibody Targets the Receptor-Binding Site of SARS-CoV-2. Nature 2020, 584, 120–124. [Google Scholar] [CrossRef]
- Rauch, S.; Roth, N.; Schwendt, K.; Fotin-Mleczek, M.; Mueller, S.O.; Petsch, B. MRNA Based SARS-CoV-2 Vaccine Candidate CVnCoV Induces High Levels of Virus Neutralizing Antibodies and Mediates Protection in Rodents. Biorxiv 2020. [Google Scholar] [CrossRef]
- Zhang, N.-N.; Li, X.-F.; Deng, Y.-Q.; Zhao, H.; Huang, Y.-J.; Yang, G.; Huang, W.-J.; Gao, P.; Zhou, C.; Zhang, R.-R.; et al. A Thermostable MRNA Vaccine against COVID-19. Cell 2020, 182, 1271–1283.e16. [Google Scholar] [CrossRef]
- Lu, J.; Lu, G.; Tan, S.; Xia, J.; Xiong, H.; Yu, X.; Qi, Q.; Yu, X.; Li, L.; Yu, H.; et al. A COVID-19 MRNA Vaccine Encoding SARS-CoV-2 Virus-like Particles Induces a Strong Antiviral-like Immune Response in Mice. Cell Res. 2020, 30, 936–939. [Google Scholar] [CrossRef]
- Tai, W.; Zhang, X.; Drelich, A.; Shi, J.; Hsu, J.C.; Luchsinger, L.; Hillyer, C.D.; Tseng, C.-T.K.; Jiang, S.; Du, L. A Novel Receptor-Binding Domain (RBD)-Based MRNA Vaccine against SARS-CoV-2. Cell Res. 2020, 30, 932–935. [Google Scholar] [CrossRef]
- Muñoz-Fontela, C.; Dowling, W.E.; Funnell, S.G.P.; Gsell, P.-S.; Riveros-Balta, A.X.; Albrecht, R.A.; Andersen, H.; Baric, R.S.; Carroll, M.W.; Cavaleri, M.; et al. Animal Models for COVID-19. Nature 2020, 586, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Rauch, S.; Gooch, K.; Hall, Y.; Salguero, F.J.; Dennis, M.J.; Gleeson, F.V.; Harris, D.; Ho, C.; Humphries, H.E.; Longet, S.; et al. MRNA Vaccine CVnCoV Protects Non-Human Primates from SARS-CoV-2 Challenge Infection. Biorxiv 2020. [Google Scholar] [CrossRef]
- Graham, B.S. Rapid COVID-19 Vaccine Development. Science 2020, 368, 945–946. [Google Scholar] [CrossRef]
- Tam, H.H.; Melo, M.B.; Kang, M.; Pelet, J.M.; Ruda, V.M.; Foley, M.H.; Hu, J.K.; Kumari, S.; Crampton, J.; Baldeon, A.D.; et al. Sustained Antigen Availability during Germinal Center Initiation Enhances Antibody Responses to Vaccination. Proc. Natl. Acad. Sci. USA 2016, 113, E6639–E6648. [Google Scholar] [CrossRef]
- Schultheiß, C.; Paschold, L.; Simnica, D.; Mohme, M.; Willscher, E.; von Wenserski, L.; Scholz, R.; Wieters, I.; Dahlke, C.; Tolosa, E.; et al. Next Generation Sequencing of T and B Cell Receptor Repertoires from COVID-19 Patients Showed Signatures Associated with Severity of Disease. Immunity 2020, 53, 442–455.e4. [Google Scholar] [CrossRef] [PubMed]
- Kreer, C.; Zehner, M.; Weber, T.; Ercanoglu, M.S.; Gieselmann, L.; Rohde, C.; Halwe, S.; Korenkov, M.; Schommers, P.; Vanshylla, K.; et al. Longitudinal Isolation of Potent Near-Germline SARS-CoV-2-Neutralizing Antibodies from COVID-19 Patients. Cell 2020, 182, 843–854.e12. [Google Scholar] [CrossRef] [PubMed]
- Seydoux, E.; Homad, L.J.; MacCamy, A.J.; Parks, K.R.; Hurlburt, N.K.; Jennewein, M.F.; Akins, N.R.; Stuart, A.B.; Wan, Y.-H.; Feng, J.; et al. Analysis of a SARS-CoV-2-Infected Individual Reveals Development of Potent Neutralizing Antibodies with Limited Somatic Mutation. Immunity 2020, 53, 98–105.e5. [Google Scholar] [CrossRef]
- Wajnberg, A.; Amanat, F.; Firpo, A.; Altman, D.R.; Bailey, M.J.; Mansour, M.; McMahon, M.; Meade, P.; Mendu, D.R.; Muellers, K.; et al. Robust Neutralizing Antibodies to SARS-CoV-2 Infection Persist for Months. Science 2020, 370, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Dan, J.M.; Mateus, J.; Kato, Y.; Hastie, K.M.; Yu, E.D.; Faliti, C.E.; Grifoni, A.; Ramirez, S.I.; Haupt, S.; Frazier, A.; et al. Immunological Memory to SARS-CoV-2 Assessed for up to Eight Months after Infection. Biorxiv 2020. [Google Scholar] [CrossRef]
- Rodda, L.B.; Netland, J.; Shehata, L.; Pruner, K.B.; Morawski, P.A.; Thouvenel, C.D.; Takehara, K.K.; Eggenberger, J.; Hemann, E.A.; Waterman, H.R.; et al. Functional SARS-CoV-2-Specific Immune Memory Persists after Mild COVID-19. Cell 2020. [Google Scholar] [CrossRef]
- Seow, J.; Graham, C.; Merrick, B.; Acors, S.; Pickering, S.; Steel, K.J.A.; Hemmings, O.; O’Byrne, A.; Kouphou, N.; Galao, R.P.; et al. Longitudinal Observation and Decline of Neutralizing Antibody Responses in the Three Months Following SARS-CoV-2 Infection in Humans. Nat. Microbiol. 2020, 5, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin-Bussières, G.; Laumaea, A.; Anand, S.P.; Prévost, J.; Gasser, R.; Goyette, G.; Medjahed, H.; Perreault, J.; Tremblay, T.; Lewin, A.; et al. Decline of Humoral Responses against SARS-CoV-2 Spike in Convalescent Individuals. Mbio 2020, 11. [Google Scholar] [CrossRef]
- Kaneko, N.; Kuo, H.-H.; Boucau, J.; Farmer, J.R.; Allard-Chamard, H.; Mahajan, V.S.; Piechocka-Trocha, A.; Lefteri, K.; Osborn, M.; Bals, J.; et al. Loss of Bcl-6-Expressing T Follicular Helper Cells and Germinal Centers in COVID-19. Cell 2020, 183. [Google Scholar] [CrossRef]
- Crotty, S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 2019, 50, 1132–1148. [Google Scholar] [CrossRef]
- Vinuesa, C.G.; Linterman, M.A.; Yu, D.; MacLennan, I.C.M. Follicular Helper T Cells. Annu. Rev. Immunol. 2015, 34, 1–34. [Google Scholar] [CrossRef]
- Soresina, A.; Moratto, D.; Chiarini, M.; Paolillo, C.; Baresi, G.; Focà, E.; Bezzi, M.; Baronio, B.; Giacomelli, M.; Badolato, R. Two X-linked Agammaglobulinemia Patients Develop Pneumonia as COVID-19 Manifestation but Recover. Pediatr. Allergy Immu. 2020, 31, 565–569. [Google Scholar] [CrossRef]
- Quinti, I.; Lougaris, V.; Milito, C.; Cinetto, F.; Pecoraro, A.; Mezzaroma, I.; Mastroianni, C.M.; Turriziani, O.; Bondioni, M.P.; Filippini, M.; et al. A Possible Role for B Cells in COVID-19?: Lesson from Patients with Agammaglobulinemia. J. Allergy Clin. Immun 2020, 146, 211–213.e4. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, G.; Ols, S.; Liang, F.; Thompson, E.A.; Lin, A.; Hellgren, F.; Bahl, K.; John, S.; Yuzhakov, O.; Hassett, K.J.; et al. Induction of Robust B Cell Responses after Influenza MRNA Vaccination Is Accompanied by Circulating Hemagglutinin-Specific ICOS+ PD-1+ CXCR3+ T Follicular Helper Cells. Front. Immunol. 2017, 8, 1539. [Google Scholar] [CrossRef]
- Crotty, S. A Brief History of T Cell Help to B Cells. Nat. Rev. Immunol. 2015, 15, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, R.L.; Liang, H.-E.; Locksley, R.M. Cytokine-Secreting Follicular T Cells Shape the Antibody Repertoire. Nat. Immunol. 2009, 10, 385–393. [Google Scholar] [CrossRef]
- Stevens, T.L.; Bossie, A.; Sanders, V.M.; Fernandez-Botran, R.; Coffman, R.L.; Mosmann, T.R.; Vitetta, E.S. Regulation of Antibody Isotype Secretion by Subsets of Antigen-Specific Helper T Cells. Nature 1988, 334, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Snapper, C.; Paul, W. Interferon-Gamma and B Cell Stimulatory Factor-1 Reciprocally Regulate Ig Isotype Production. Science 1987, 236, 944–947. [Google Scholar] [CrossRef] [PubMed]
- Bolles, M.; Deming, D.; Long, K.; Agnihothram, S.; Whitmore, A.; Ferris, M.; Funkhouser, W.; Gralinski, L.; Totura, A.; Heise, M.; et al. A Double-Inactivated Severe Acute Respiratory Syndrome Coronavirus Vaccine Provides Incomplete Protection in Mice and Induces Increased Eosinophilic Proinflammatory Pulmonary Response upon Challenge. J. Virol. 2011, 85, 12201–12215. [Google Scholar] [CrossRef]
- Juno, J.A.; Tan, H.-X.; Lee, W.S.; Reynaldi, A.; Kelly, H.G.; Wragg, K.; Esterbauer, R.; Kent, H.E.; Batten, C.J.; Mordant, F.L.; et al. Humoral and Circulating Follicular Helper T Cell Responses in Recovered Patients with COVID-19. Nat. Med. 2020, 26, 1428–1434. [Google Scholar] [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e15. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, S.H.; Mansatta, K.; Mallett, G.; Harris, V.; Emary, K.R.W.; Pollard, A.J. What Defines an Efficacious COVID-19 Vaccine? A Review of the Challenges Assessing the Clinical Efficacy of Vaccines against SARS-CoV-2. Lancet Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Lang, P.O.; Govind, S.; Michel, J.P.; Aspinall, R.; Mitchell, W.A. Immunosenescence: Implications for Vaccination Programmes in Adults. Maturitas 2011, 68, 322–330. [Google Scholar] [CrossRef]
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors Associated with COVID-19-Related Death Using OpenSAFELY. Nature 2020, 584, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Safety and Immunogenicity Study of 2019-nCoV Vaccine (mRNA-1273) for Prophylaxis of SARS-895 CoV-2 Infection (COVID-19). Available online: https://clinicaltrials.gov/ct2/show/NCT04283461 (accessed on 30 December 2020).
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- A Trial Investigating the Safety and Effects of Four BNT162 Vaccines Against COVID-2019 in Healthy 901 Adults. Available online: https://clinicaltrials.gov/ct2/show/NCT04380701 (accessed on 30 December 2020).
- Study to Describe the Safety, Tolerability, Immunogenicity, and Efficacy of RNA Vaccine Candidates 905 Against COVID-19 in Healthy Individuals. Available online: https://clinicaltrials.gov/ct2/show/NCT04368728?term=BNT162b1&phase=01&draw=2&rank=3 (accessed on 30 December 2020).
- Development and Licensure of Vaccines to Prevent COVID-19. Available online: https://Www.Fda.Gov/Media/139638/Download (accessed on 30 December 2020).
- Voysey, M.; Clemens, S.A.C.; Madhi, S.A.; Weckx, L.Y.; Folegatti, P.M.; Aley, P.K.; Angus, B.; Baillie, V.L.; Barnabas, S.L.; Bhorat, Q.E.; et al. Safety and Efficacy of the ChAdOx1 NCoV-19 Vaccine (AZD1222) against SARS-CoV-2: An Interim Analysis of Four Randomised Controlled Trials in Brazil, South Africa, and the UK. Lancet 2020. [Google Scholar] [CrossRef]
- Zhu, F.-C.; Li, Y.-H.; Guan, X.-H.; Hou, L.-H.; Wang, W.-J.; Li, J.-X.; Wu, S.-P.; Wang, B.-S.; Wang, Z.; Wang, L.; et al. Safety, Tolerability, and Immunogenicity of a Recombinant Adenovirus Type-5 Vectored COVID-19 Vaccine: A Dose-Escalation, Open-Label, Non-Randomised, First-in-Human Trial. Lancet 2020, 395, 1845–1854. [Google Scholar] [CrossRef]
- COVID-19 Vaccines and Allergic Reactions. Available online: https://www.Cdc.Gov/Coronavirus/2019-Ncov/Vaccines/Safety/Allergic-Reaction.Html (accessed on 30 December 2020).
- mRNA Vaccines against COVID-19: Pfizer-BioNTech COVID-19 vaccineBNT162b2. Available online: https://www.who.int/publications/i/item/mrna-vaccines-against-covid-19-pfizer-biontech-covid-19-vaccinebnt162b2 (accessed on 30 December 2020).
- Fact Sheet for Healthcare Providers Administering Vaccine. Available online: https://www.Fda.Gov/Media/144637/Download (accessed on 30 December 2020).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bettini, E.; Locci, M. SARS-CoV-2 mRNA Vaccines: Immunological Mechanism and Beyond. Vaccines 2021, 9, 147. https://doi.org/10.3390/vaccines9020147
Bettini E, Locci M. SARS-CoV-2 mRNA Vaccines: Immunological Mechanism and Beyond. Vaccines. 2021; 9(2):147. https://doi.org/10.3390/vaccines9020147
Chicago/Turabian StyleBettini, Emily, and Michela Locci. 2021. "SARS-CoV-2 mRNA Vaccines: Immunological Mechanism and Beyond" Vaccines 9, no. 2: 147. https://doi.org/10.3390/vaccines9020147
APA StyleBettini, E., & Locci, M. (2021). SARS-CoV-2 mRNA Vaccines: Immunological Mechanism and Beyond. Vaccines, 9(2), 147. https://doi.org/10.3390/vaccines9020147