
Targeting Innate Immunity in Cancer Therapy

Abstract
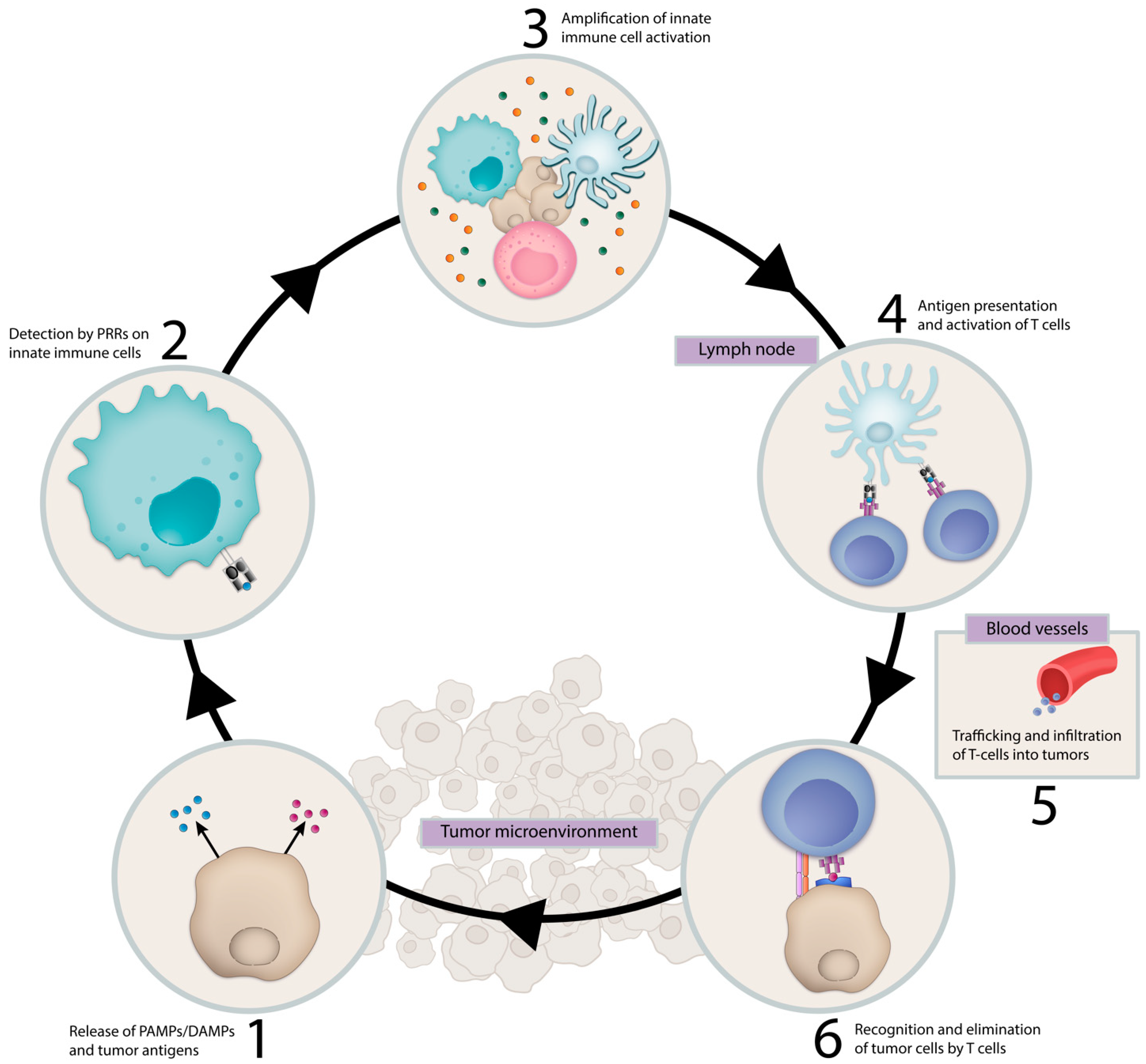
1. Introduction
2. Key Cellular Components of Innate Immunity
2.1. Dendritic Cells (DCs)
2.2. Macrophages
2.3. Neutrophils
2.4. Myeloid Derived Suppressor Cells (MDSCs)
2.5. Mast Cells
2.6. Natural Killer (NK) Cells
3. Activating the Innate Immune System in Cancer Therapy
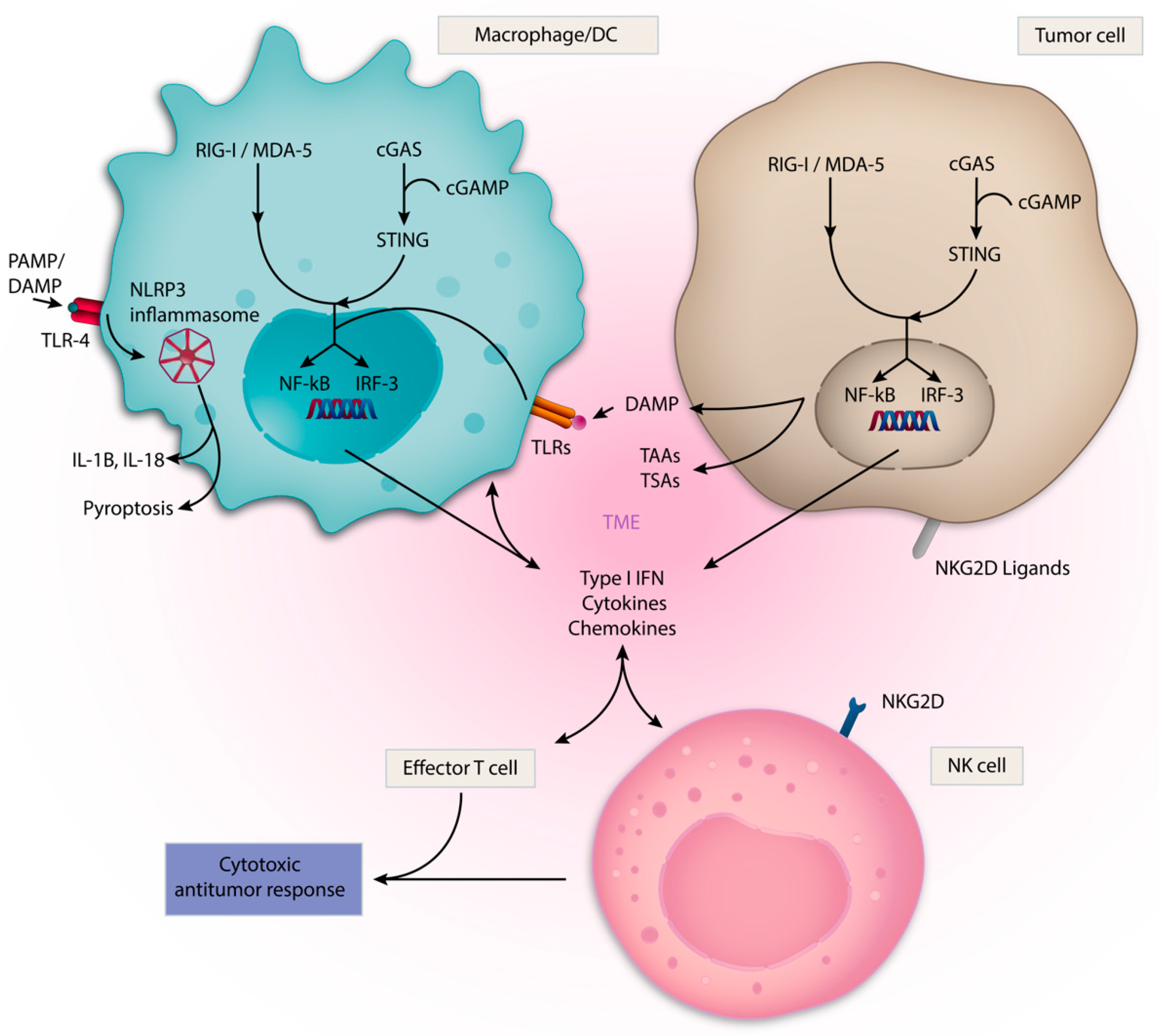
3.1. Toll-like Receptors (TLRs)
3.2. cGAS/STING Pathway
3.3. Retinoic Acid Inducible Gene-I-like Receptors (RLRs) and RIG-I
3.4. CD40
3.5. NLRP3-Inflammasome
3.6. Dendritic Cell Directed Strategies
3.7. Adoptive DC Strategies
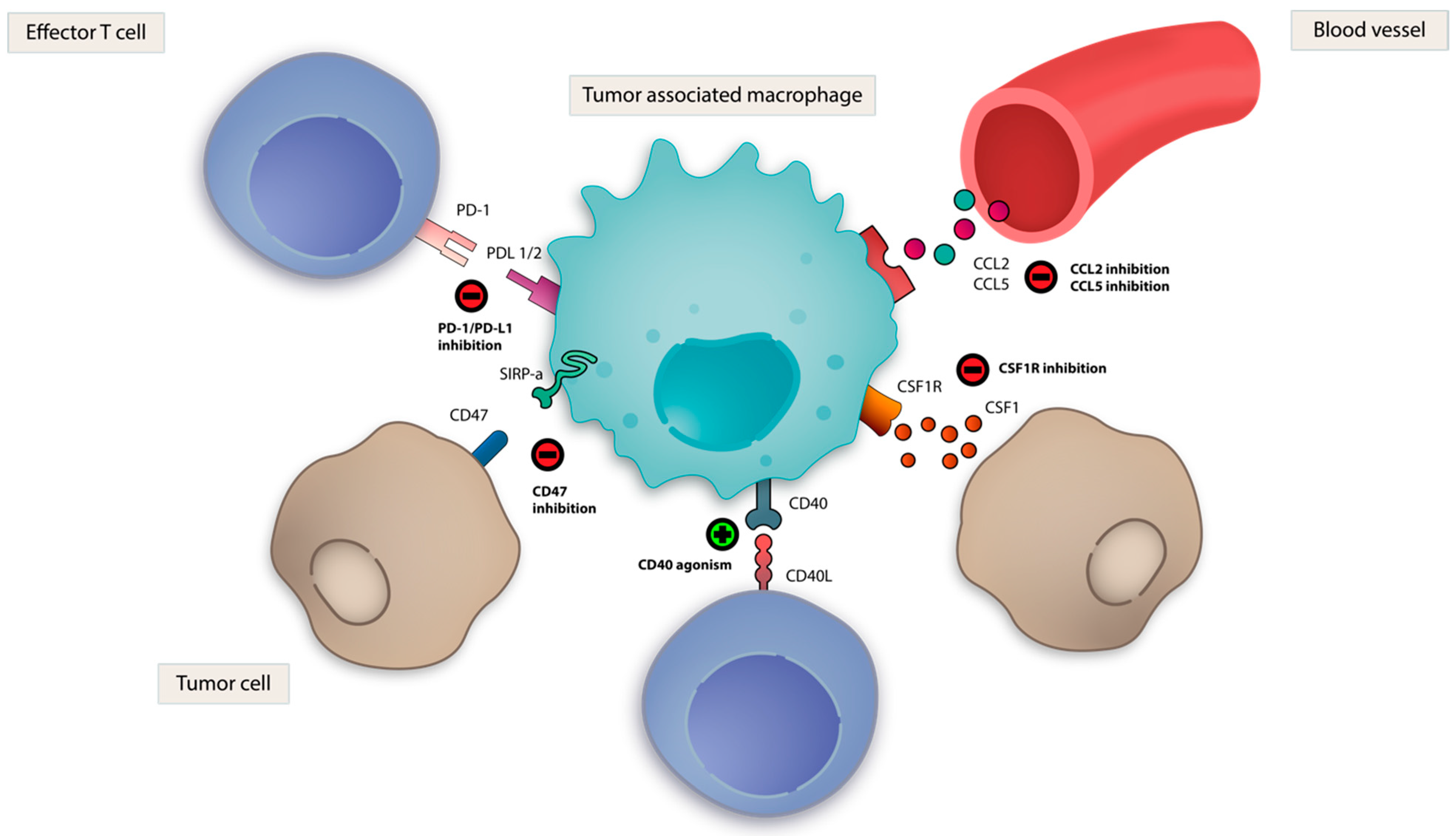
3.8. TAM Directed Strategies
3.8.1. Colony Stimulating Factor 1 (CSF-1)
3.8.2. PI3K-γ Inhibition
3.8.3. CD47- Signal-Recognition Protein Alpha (SIRPα)
3.8.4. Dendritic Cell-Specific Intercellular Adhesion Molecule-3-Grabbing Non-Integrin (DC-SIGN)
3.8.5. Other Macrophage-Directed Strategies
3.9. NK Cell Directed Strategies
3.9.1. Natural-Killer Group 2, Member D (NKG2D) Ligands
3.9.2. NK Cell Engagers (NKCEs)
3.9.3. NKG2A Inhibition
3.9.4. Adoptive NK Cell Strategies
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Kaplan, D.H.; Shankaran, V.; Dighe, A.S.; Stockert, E.; Aguet, M.; Old, L.J.; Schreiber, R.D. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA 1998, 95, 7556–7561. [Google Scholar] [CrossRef]
- Fuertes, M.B.; Kacha, A.K.; Kline, J.; Woo, S.R.; Kranz, D.M.; Murphy, K.M.; Gajewski, T.F. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J. Exp. Med. 2011, 208, 2005–2016. [Google Scholar] [CrossRef]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.e714. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Spranger, S.; Luke, J.J.; Bao, R.; Zha, Y.; Hernandez, K.M.; Li, Y.; Gajewski, A.P.; Andrade, J.; Gajewski, T.F. Density of immunogenic antigens does not explain the presence or absence of the T-cell–inflamed tumor microenvironment in melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, E7759–E7768. [Google Scholar] [CrossRef]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef] [PubMed]
- Ugel, S.; De Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Investig. 2015, 125, 3365–3376. [Google Scholar] [CrossRef] [PubMed]
- Barnes, T.A.; Amir, E. HYPE or HOPE: The prognostic value of infiltrating immune cells in cancer. Br. J. Cancer 2018, 118, e5. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Louahed, J.; Brichard, V.G. Gene signature in melanoma associated with clinical activity: A potential clue to unlock cancer immunotherapy. Cancer J. 2010, 16, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.-R.; Chasalow, S.D.; Wang, L.; Hamid, O.; Schmidt, H.; Cogswell, J.; Alaparthy, S.; Berman, D.; Jure-Kunkel, M.; Siemers, N.O.; et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. 2012, 61, 1019–1031. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Mildner, A.; Jung, S. Development and function of dendritic cell subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef]
- Sanchez-Paulete, A.R.; Cueto, F.J.; Martinez-Lopez, M.; Labiano, S.; Morales-Kastresana, A.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Azpilikueta, A.; Aznar, M.A.; Quetglas, J.I.; et al. Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti-PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer Discov. 2016, 6, 71–79. [Google Scholar] [CrossRef]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A.; et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4(+) T Cell Immunity. Cell 2019, 177, 556–571.e516. [Google Scholar] [CrossRef]
- Liu, Y.J. IPC: Professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 2005, 23, 275–306. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Allavena, P.; Garlanda, C.; Locati, M. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum. Immunol. 2009, 70, 325–330. [Google Scholar] [CrossRef]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- van Dalen, F.J.; van Stevendaal, M.; Fennemann, F.L.; Verdoes, M.; Ilina, O. Molecular Repolarisation of Tumour-Associated Macrophages. Molecules 2018, 24, 9. [Google Scholar] [CrossRef]
- Lin, E.Y.; Pollard, J.W. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. 2007, 67, 5064–5066. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef]
- Dehne, N.; Mora, J.; Namgaladze, D.; Weigert, A.; Brune, B. Cancer cell and macrophage cross-talk in the tumor microenvironment. Curr. Opin. Pharmacol. 2017, 35, 12–19. [Google Scholar] [CrossRef]
- Zaidi, N.E.; Shazali, N.A.H.; Chor, A.L.T.; Osman, M.A.; Ibrahim, K.; Jaoi-Edward, M.; Afizan Nik Abd Rahman, N.M. Time-Lapse 2D Imaging of Phagocytic Activity in M1 Macrophage-4T1 Mouse Mammary Carcinoma Cells in Co-cultures. J. Vis. Exp. 2019. [Google Scholar] [CrossRef]
- Chung, W.; Eum, H.H.; Lee, H.O.; Lee, K.M.; Lee, H.B.; Kim, K.T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [PubMed]
- Shiao, S.L.; Ruffell, B.; DeNardo, D.G.; Faddegon, B.A.; Park, C.C.; Coussens, L.M. TH2-Polarized CD4(+) T Cells and Macrophages Limit Efficacy of Radiotherapy. Cancer Immunol. Res. 2015, 3, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, M.; Karachaliou, N. Tumor immune microenvironment characterization and response to anti-PD-1 therapy. Cancer Biol. Med. 2015, 12, 74–78. [Google Scholar] [CrossRef]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef]
- Rao, H.L.; Chen, J.W.; Li, M.; Xiao, Y.B.; Fu, J.; Zeng, Y.X.; Cai, M.Y.; Xie, D. Increased intratumoral neutrophil in colorectal carcinomas correlates closely with malignant phenotype and predicts patients’ adverse prognosis. PLoS ONE 2012, 7, e30806. [Google Scholar] [CrossRef]
- Shen, M.; Hu, P.; Donskov, F.; Wang, G.; Liu, Q.; Du, J. Tumor-associated neutrophils as a new prognostic factor in cancer: A systematic review and meta-analysis. PLoS ONE 2014, 9, e98259. [Google Scholar] [CrossRef]
- Schmidt, H.; Bastholt, L.; Geertsen, P.; Christensen, I.J.; Larsen, S.; Gehl, J.; von der Maase, H. Elevated neutrophil and monocyte counts in peripheral blood are associated with poor survival in patients with metastatic melanoma: A prognostic model. Br. J. Cancer 2005, 93, 273–278. [Google Scholar] [CrossRef]
- Glodde, N.; Bald, T.; van den Boorn-Konijnenberg, D.; Nakamura, K.; O’Donnell, J.S.; Szczepanski, S.; Brandes, M.; Eickhoff, S.; Das, I.; Shridhar, N.; et al. Reactive Neutrophil Responses Dependent on the Receptor Tyrosine Kinase c-MET Limit Cancer Immunotherapy. Immunity 2017, 47, 789–802.e789. [Google Scholar] [CrossRef] [PubMed]
- Benevides, L.; da Fonseca, D.M.; Donate, P.B.; Tiezzi, D.G.; De Carvalho, D.D.; de Andrade, J.M.; Martins, G.A.; Silva, J.S. IL17 Promotes Mammary Tumor Progression by Changing the Behavior of Tumor Cells and Eliciting Tumorigenic Neutrophils Recruitment. Cancer Res. 2015, 75, 3788–3799. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.S.; Verstegen, N.J.M.; Ciampricotti, M.; Hawinkels, L.; Jonkers, J.; et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015, 522, 345–348. [Google Scholar] [CrossRef]
- Gerrard, T.L.; Cohen, D.J.; Kaplan, A.M. Human neutrophil-mediated cytotoxicity to tumor cells. J. Natl. Cancer Inst. 1981, 66, 483–488. [Google Scholar]
- Granot, Z.; Henke, E.; Comen, E.A.; King, T.A.; Norton, L.; Benezra, R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell 2011, 20, 300–314. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Mishalian, I.; Singhal, S.; Cheng, G.; Kapoor, V.; Horng, W.; Fridlender, G.; Bayuh, R.; Worthen, G.S.; et al. Transcriptomic analysis comparing tumor-associated neutrophils with granulocytic myeloid-derived suppressor cells and normal neutrophils. PLoS ONE 2012, 7, e31524. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Bird, L. Controlling neutrophil plasticity. Nat. Rev. Immunol. 2010, 10, 752. [Google Scholar] [CrossRef] [PubMed]
- Sagiv, J.Y.; Michaeli, J.; Assi, S.; Mishalian, I.; Kisos, H.; Levy, L.; Damti, P.; Lumbroso, D.; Polyansky, L.; Sionov, R.V.; et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 2015, 10, 562–573. [Google Scholar] [CrossRef]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef]
- Pawelec, G.; Verschoor, C.P.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cells: Not Only in Tumor Immunity. Front. Immunol. 2019, 10, 1099. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Wang, K.; Huang, X.J. Myeloid-derived suppressor cells in hematological malignancies: Friends or foes. J. Hematol. Oncol. 2019, 12, 105. [Google Scholar] [CrossRef]
- Johansson, A.; Rudolfsson, S.; Hammarsten, P.; Halin, S.; Pietras, K.; Jones, J.; Stattin, P.; Egevad, L.; Granfors, T.; Wikstrom, P.; et al. Mast cells are novel independent prognostic markers in prostate cancer and represent a target for therapy. Am. J. Pathol. 2010, 177, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Welsh, T.J.; Green, R.H.; Richardson, D.; Waller, D.A.; O’Byrne, K.J.; Bradding, P. Macrophage and mast-cell invasion of tumor cell islets confers a marked survival advantage in non-small-cell lung cancer. J. Clin. Oncol. 2005, 23, 8959–8967. [Google Scholar] [CrossRef]
- Rajput, A.B.; Turbin, D.A.; Cheang, M.C.; Voduc, D.K.; Leung, S.; Gelmon, K.A.; Gilks, C.B.; Huntsman, D.G. Stromal mast cells in invasive breast cancer are a marker of favourable prognosis: A study of 4444 cases. Breast Cancer Res. Treat. 2008, 107, 249–257. [Google Scholar] [CrossRef]
- Varricchi, G.; Galdiero, M.R.; Loffredo, S.; Marone, G.; Iannone, R.; Marone, G.; Granata, F. Are Mast Cells MASTers in Cancer? Front. Immunol. 2017, 8, 424. [Google Scholar] [CrossRef] [PubMed]
- Toth-Jakatics, R.; Jimi, S.; Takebayashi, S.; Kawamoto, N. Cutaneous malignant melanoma: Correlation between neovascularization and peritumor accumulation of mast cells overexpressing vascular endothelial growth factor. Hum. Pathol. 2000, 31, 955–960. [Google Scholar] [CrossRef]
- Malfettone, A.; Silvestris, N.; Saponaro, C.; Ranieri, G.; Russo, A.; Caruso, S.; Popescu, O.; Simone, G.; Paradiso, A.; Mangia, A. High density of tryptase-positive mast cells in human colorectal cancer: A poor prognostic factor related to protease-activated receptor 2 expression. J. Cell. Mol. Med. 2013, 17, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.R.; Ding, Y.; Ricks, T.K.; Gullapalli, A.; Wolfe, B.L.; Trejo, J. Protease-activated receptor-2 is essential for factor VIIa and Xa-induced signaling, migration, and invasion of breast cancer cells. Cancer Res. 2006, 66, 307–314. [Google Scholar] [CrossRef]
- Gooch, J.L.; Lee, A.V.; Yee, D. Interleukin 4 inhibits growth and induces apoptosis in human breast cancer cells. Cancer Res. 1998, 58, 4199–4205. [Google Scholar]
- Henderson, W.R.; Chi, E.Y.; Jong, E.C.; Klebanoff, S.J. Mast cell-mediated tumor-cell cytotoxicity. Role of the peroxidase system. J. Exp. Med. 1981, 153, 520–533. [Google Scholar] [CrossRef]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef]
- Hashimoto, W.; Osaki, T.; Okamura, H.; Robbins, P.D.; Kurimoto, M.; Nagata, S.; Lotze, M.T.; Tahara, H. Differential antitumor effects of administration of recombinant IL-18 or recombinant IL-12 are mediated primarily by Fas-Fas ligand- and perforin-induced tumor apoptosis, respectively. J. Immunol. 1999, 163, 583–589. [Google Scholar] [PubMed]
- Trinchieri, G. Biology of natural killer cells. Adv. Immunol. 1989, 47, 187–376. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Crowe, N.Y.; Godfrey, D.I. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int. Immunol. 2001, 13, 459–463. [Google Scholar] [CrossRef]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e1014. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, T.; Saddawi-Konefka, R.; Vermi, W.; Koebel, C.M.; Arthur, C.; White, J.M.; Uppaluri, R.; Andrews, D.M.; Ngiow, S.F.; Teng, M.W.; et al. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J. Exp. Med. 2012, 209, 1869–1882. [Google Scholar] [CrossRef]
- Cai, G.; Kastelein, R.A.; Hunter, C.A. IL-10 enhances NK cell proliferation, cytotoxicity and production of IFN-γ when combined with IL-18. Eur. J. Immunol. 1999, 29, 2658–2665. [Google Scholar] [CrossRef]
- Jamieson, A.M.; Diefenbach, A.; McMahon, C.W.; Xiong, N.; Carlyle, J.R.; Raulet, D.H. The Role of the NKG2D Immunoreceptor in Immune Cell Activation and Natural Killing. Immunity 2002, 17, 19–29. [Google Scholar] [CrossRef]
- Takanami, I.; Takeuchi, K.; Giga, M. The prognostic value of natural killer cell infiltration in resected pulmonary adenocarcinoma. J. Thorac. Cardiovasc. Surg. 2001, 121, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Villegas, F.R.; Coca, S.; Villarrubia, V.G.; Jiménez, R.; Chillón, M.a.J.; Jareño, J.; Zuil, M.; Callol, L. Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer. Lung Cancer 2002, 35, 23–28. [Google Scholar] [CrossRef]
- Lopez-Soto, A.; Gonzalez, S.; Smyth, M.J.; Galluzzi, L. Control of Metastasis by NK Cells. Cancer Cell 2017, 32, 135–154. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Gangloff, M. Different dimerisation mode for TLR4 upon endosomal acidification? Trends Biochem. Sci. 2012, 37, 92–98. [Google Scholar] [CrossRef]
- Satoh, T.; Akira, S. Toll-Like Receptor Signaling and Its Inducible Proteins. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Adams, S. Toll-like receptor agonists in cancer therapy. Immunotherapy 2009, 1, 949–964. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Rodell, C.B.; Arlauckas, S.P.; Cuccarese, M.F.; Garris, C.S.; Li, R.; Ahmed, M.S.; Kohler, R.H.; Pittet, M.J.; Weissleder, R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018, 2, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Vidyarthi, A.; Khan, N.; Agnihotri, T.; Negi, S.; Das, D.K.; Aqdas, M.; Chatterjee, D.; Colegio, O.R.; Tewari, M.K.; Agrewala, J.N. TLR-3 Stimulation Skews M2 Macrophages to M1 Through IFN-alphabeta Signaling and Restricts Tumor Progression. Front. Immunol. 2018, 9, 1650. [Google Scholar] [CrossRef]
- Yang, Y.; Huang, C.T.; Huang, X.; Pardoll, D.M. Persistent Toll-like receptor signals are required for reversal of regulatory T cell-mediated CD8 tolerance. Nat. Immunol. 2004, 5, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, F.; Cai, Y.; Liu, N.; Wang, L.; Xu, D.; Chu, Y. TLR1/TLR2 agonist induces tumor regression by reciprocal modulation of effector and regulatory T cells. J. Immunol. 2011, 186, 1963–1969. [Google Scholar] [CrossRef]
- Jin, B.; Sun, T.; Yu, X.H.; Yang, Y.X.; Yeo, A.E. The effects of TLR activation on T-cell development and differentiation. Clin. Dev. Immunol. 2012, 2012, 836485. [Google Scholar] [CrossRef] [PubMed]
- Salaun, B.; Coste, I.; Rissoan, M.C.; Lebecque, S.J.; Renno, T. TLR3 can directly trigger apoptosis in human cancer cells. J. Immunol. 2006, 176, 4894–4901. [Google Scholar] [CrossRef] [PubMed]
- Dajon, M.; Iribarren, K.; Petitprez, F.; Marmier, S.; Lupo, A.; Gillard, M.; Ouakrim, H.; Victor, N.; Vincenzo, D.B.; Joubert, P.E.; et al. Toll like receptor 7 expressed by malignant cells promotes tumor progression and metastasis through the recruitment of myeloid derived suppressor cells. Oncoimmunology 2019, 8, e1505174. [Google Scholar] [CrossRef]
- Hao, B.; Chen, Z.; Bi, B.; Yu, M.; Yao, S.; Feng, Y.; Yu, Y.; Pan, L.; Di, D.; Luo, G.; et al. Role of TLR4 as a prognostic factor for survival in various cancers: A meta-analysis. Oncotarget 2018, 9, 13088–13099. [Google Scholar] [CrossRef]
- Jouhi, L.; Renkonen, S.; Atula, T.; Makitie, A.; Haglund, C.; Hagstrom, J. Different Toll-Like Receptor Expression Patterns in Progression toward Cancer. Front. Immunol. 2014, 5, 638. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Campos, J.; Gallotta, M.; Gong, M.; Crain, C.; Naik, E.; Coffman, R.L.; Guiducci, C. Intratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8+ T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7240–E7249. [Google Scholar] [CrossRef]
- Sato-Kaneko, F.; Yao, S.; Ahmadi, A.; Zhang, S.S.; Hosoya, T.; Kaneda, M.M.; Varner, J.A.; Pu, M.; Messer, K.S.; Guiducci, C.; et al. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Ribas, A.; Medina, T.; Kummar, S.; Amin, A.; Kalbasi, A.; Drabick, J.J.; Barve, M.; Daniels, G.A.; Wong, D.J.; Schmidt, E.V.; et al. SD-101 in Combination with Pembrolizumab in Advanced Melanoma: Results of a Phase Ib, Multicenter Study. Cancer Discov. 2018, 8, 1250–1257. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Nabell, L.; Wong, D.J.L.; Day, T.A.; Daniels, G.A.; Milhem, M.M.; Deva, S.; Jameson, M.B.; Guntinas-Lichius, O.; Almubarak, M.; et al. Phase 1b/2, open label, multicenter study of intratumoral SD-101 in combination with pembrolizumab in anti-PD-1 treatment naïve patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC). J. Clin. Oncol. 2019, 37, 6039. [Google Scholar] [CrossRef]
- Kivimae, S.; Hennessy, M.; Pena, R.; Kirksey, Y.; Nieves, W.; Quatch, P.; Cetz, J.; Ren, Z.; Cai, H.; Deng, B.L.; et al. Comprehensive antitumor immune activation by a novel TLR7/8 targeting agent NKTR-262 combined with CD122-biased immunostimulatory cytokine NKTR-214. Proc. Am. Assoc. Cancer Res. Annu. Meet. 2018 2018, 78, 3755. [Google Scholar]
- Mullins, S.R.; Vasilakos, J.P.; Deschler, K.; Grigsby, I.; Gillis, P.; John, J.; Elder, M.J.; Swales, J.; Timosenko, E.; Cooper, Z.; et al. Intratumoral immunotherapy with TLR7/8 agonist MEDI9197 modulates the tumor microenvironment leading to enhanced activity when combined with other immunotherapies. J. Immunother. Cancer 2019, 7, 244. [Google Scholar] [CrossRef] [PubMed]
- Morales, A. Long-term results and complications of intracavitary bacillus Calmette-Guerin therapy for bladder cancer. J. Urol. 1984, 132, 457–459. [Google Scholar] [CrossRef]
- Beutner, K.R.; Spruance, S.L.; Hougham, A.J.; Fox, T.L.; Owens, M.L.; Douglas, J.M., Jr. Treatment of genital warts with an immune-response modifier (imiquimod). J. Am. Acad. Dermatol. 1998, 38, 230–239. [Google Scholar] [CrossRef]
- Casella, C.R.; Mitchell, T.C. Putting endotoxin to work for us: Monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell Mol. Life Sci. 2008, 65, 3231–3240. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, V.; Keating, G.M.; Perry, C.M. Imiquimod: In superficial basal cell carcinoma. Am. J. Clin. Dermatol. 2005, 6, 195–200; discussion 192–201. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef]
- Woo, S.R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Zhao, L.; Hu, H.G.; Li, W.H.; Li, Y.M. Agonists and inhibitors of the STING pathway: Potential agents for immunotherapy. Med. Res. Rev. 2020, 40, 1117–1141. [Google Scholar] [CrossRef]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Song, Z.; Shen, A.; Chen, T.; Zhang, A. Small molecules targeting the innate immune cGAS–STING–TBK1 signaling pathway. Acta Pharm. Sin. B 2020, in press, pre-proof. [Google Scholar] [CrossRef]
- Harrington, K.J.; Brody, J.; Ingham, M.; Strauss, J.; Cemerski, S.; Wang, M.; Tse, A.; Khilnani, A.; Marabelle, A.; Golan, T. Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Sandhu, S.K.; Hamid, O.; Spreafico, A.; Kasper, S.; Dummer, R.; Shimizu, T.; Steeghs, N.; Lewis, N.; Talluto, C.C.; et al. Phase Ib study of MIW815 (ADU-S100) in combination with spartalizumab (PDR001) in patients (pts) with advanced/metastatic solid tumors or lymphomas. J. Clin. Oncol. 2019, 37, 2507. [Google Scholar] [CrossRef]
- Sivick, K.E.; Desbien, A.L.; Glickman, L.H.; Reiner, G.L.; Corrales, L.; Surh, N.H.; Hudson, T.E.; Vu, U.T.; Francica, B.J.; Banda, T.; et al. Magnitude of Therapeutic STING Activation Determines CD8(+) T Cell-Mediated Anti-tumor Immunity. Cell Rep. 2019, 29, 785–789. [Google Scholar] [CrossRef]
- Schieven, G.; Brown, J.; Swanson, J.; Stromko, B.S.C.; Ho, C.-P.; Zhang, R.; Li, W.B.; Qiu, H.; Sun, H.; Fink, B.; et al. Preclinical characterization of BMS-986301, a differentiated STING agonist with robust antitumor activity as monotherapy or in combination with anti-PD-1. In Proceedings of the 33rd Annual Meeting & Pre-Conference Programs of the Society for Immunotherapy of Cancer (SITC 2018), Washington, DC, USA, 7–11 November 2018. [Google Scholar]
- Ramanjulu, J.M.; Pesiridis, G.S.; Yang, J.; Concha, N.; Singhaus, R.; Zhang, S.Y.; Tran, J.L.; Moore, P.; Lehmann, S.; Eberl, H.C.; et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 2018, 564, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Flood, B.A.; Higgs, E.F.; Li, S.; Luke, J.J.; Gajewski, T.F. STING pathway agonism as a cancer therapeutic. Immunol. Rev. 2019, 290, 24–38. [Google Scholar] [CrossRef]
- Kinkead, H.L.; Hopkins, A.; Lutz, E.; Wu, A.A.; Yarchoan, M.; Cruz, K.; Woolman, S.; Vithayathil, T.; Glickman, L.H.; Ndubaku, C.O.; et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Fu, J.; Kanne, D.B.; Leong, M.; Glickman, L.H.; McWhirter, S.M.; Lemmens, E.; Mechette, K.; Leong, J.J.; Lauer, P.; Liu, W.; et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med. 2015, 7, 283ra252. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, A.; Peterson, N.; Khalaj, K.; Vitkin, N.; Robinson, A.; Francis, J.A.; Koti, M. STING agonist therapy in combination with PD-1 immune checkpoint blockade enhances response to carboplatin chemotherapy in high-grade serous ovarian cancer. Br. J. Cancer 2018, 119, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Poeck, H.; Bscheider, M.; Gross, O.; Finger, K.; Roth, S.; Rebsamen, M.; Hannesschlager, N.; Schlee, M.; Rothenfusser, S.; Barchet, W.; et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat. Immunol. 2010, 11, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Elion, D.L.; Cook, R.S. Harnessing RIG-I and intrinsic immunity in the tumor microenvironment for therapeutic cancer treatment. Oncotarget 2018, 9, 29007–29017. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Kinch, L.N.; Brautigam, C.A.; Chen, X.; Du, F.; Grishin, N.V.; Chen, Z.J. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 2012, 36, 959–973. [Google Scholar] [CrossRef]
- Heidegger, S.; Wintges, A.; Stritzke, F.; Bek, S.; Steiger, K.; Koenig, P.A.; Gottert, S.; Engleitner, T.; Ollinger, R.; Nedelko, T.; et al. RIG-I activation is critical for responsiveness to checkpoint blockade. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]
- Terheyden, P.; Weishaupt, C.; Heinzerling, L.; Klinkhardt, U.; Krauss, J.; Mohr, P.; Kiecker, F.; Becker, J.C.; Dähling, A.; Döner, F.; et al. Phase I dose-escalation and expansion study of intratumoral CV8102, a RNA-based TLR- and RIG-1 agonist in patients with advanced solid tumors. Ann. Oncol. 2018, 29, viii466. [Google Scholar] [CrossRef]
- Barsoum, J.; Renn, M.; Schuberth, C.; Jakobs, C.; Schwickart, A.; Schlee, M.; van den Boorn, J.; Hartmann, G. Abstract B44: Selective stimulation of RIG-I with a novel synthetic RNA induces strong anti-tumor immunity in mouse tumor models. Cancer Immunol. Res. 2017. [Google Scholar] [CrossRef]
- Middleton, M.R.; Wermke, M.; Calvo, E.; Chartash, E.; Zhou, H.; Zhao, X.; Niewel, M.; Dobrenkov, K.; Moreno, V. Phase I/II, multicenter, open-label study of intratumoral/intralesional administration of the retinoic acid–inducible gene I (RIG-I) activator MK-4621 in patients with advanced or recurrent tumors. Ann. Oncol. 2018, 29, viii712. [Google Scholar] [CrossRef]
- Linehan, M.M.; Dickey, T.H.; Molinari, E.S.; Fitzgerald, M.E.; Potapova, O.; Iwasaki, A.; Pyle, A.M. A minimal RNA ligand for potent RIG-I activation in living mice. Sci. Adv. 2018, 4, e1701854. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Muthusamy, V.; Fedorova, O.; Kong, Y.; Kim, D.J.; Bosenberg, M.; Pyle, A.M.; Iwasaki, A. Intratumoral delivery of RIG-I agonist SLR14 induces robust antitumor responses. J. Exp. Med. 2019, 216, 2854–2868. [Google Scholar] [CrossRef]
- Jones, M.; Cunningham, M.E.; Wing, P.; DeSilva, S.; Challa, R.; Sheri, A.; Padmanabhan, S.; Iyer, R.P.; Korba, B.E.; Afdhal, N.; et al. SB 9200, a novel agonist of innate immunity, shows potent antiviral activity against resistant HCV variants. J. Med. Virol. 2017, 89, 1620–1628. [Google Scholar] [CrossRef]
- Kyi, C.; Roudko, V.; Sabado, R.; Saenger, Y.; Loging, W.; Mandeli, J.; Thin, T.H.; Lehrer, D.; Donovan, M.; Posner, M.; et al. Therapeutic Immune Modulation against Solid Cancers with Intratumoral Poly-ICLC: A Pilot Trial. Clin. Cancer Res. 2018, 24, 4937–4948. [Google Scholar] [CrossRef]
- Dauletbaev, N.; Cammisano, M.; Herscovitch, K.; Lands, L.C. Stimulation of the RIG-I/MAVS Pathway by Polyinosinic:Polycytidylic Acid Upregulates IFN-beta in Airway Epithelial Cells with Minimal Costimulation of IL-8. J. Immunol. 2015, 195, 2829–2841. [Google Scholar] [CrossRef]
- Salazar, A.M.; Erlich, R.B.; Mark, A.; Bhardwaj, N.; Herberman, R.B. Therapeutic in situ autovaccination against solid cancers with intratumoral poly-ICLC: Case report, hypothesis, and clinical trial. Cancer Immunol. Res. 2014, 2, 720–724. [Google Scholar] [CrossRef]
- van Kooten, C.; Banchereau, J. CD40-CD40 ligand. J. Leukoc. Biol. 2000, 67, 2–17. [Google Scholar] [CrossRef]
- Todryk, S.M.; Tutt, A.L.; Green, M.H.; Smallwood, J.A.; Halanek, N.; Dalgleish, A.G.; Glennie, M.J. CD40 ligation for immunotherapy of solid tumours. J. Immunol. Methods 2001, 248, 139–147. [Google Scholar] [CrossRef]
- van Mierlo, G.J.; den Boer, A.T.; Medema, J.P.; van der Voort, E.I.; Fransen, M.F.; Offringa, R.; Melief, C.J.; Toes, R.E. CD40 stimulation leads to effective therapy of CD40(-) tumors through induction of strong systemic cytotoxic T lymphocyte immunity. Proc. Natl. Acad. Sci. USA 2002, 99, 5561–5566. [Google Scholar] [CrossRef]
- Buhtoiarov, I.N.; Lum, H.; Berke, G.; Paulnock, D.M.; Sondel, P.M.; Rakhmilevich, A.L. CD40 ligation activates murine macrophages via an IFN-gamma-dependent mechanism resulting in tumor cell destruction in vitro. J. Immunol. 2005, 174, 6013–6022. [Google Scholar] [CrossRef] [PubMed]
- Elgueta, R.; Benson, M.J.; de Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.R.; Carbone, F.R.; Karamalis, F.; Flavell, R.A.; Miller, J.F.; Heath, W.R. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 1998, 393, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Byrne, K.T.; Vonderheide, R.H. CD40 Stimulation Obviates Innate Sensors and Drives T Cell Immunity in Cancer. Cell Rep. 2016, 15, 2719–2732. [Google Scholar] [CrossRef]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed]
- Long, K.B.; Gladney, W.L.; Tooker, G.M.; Graham, K.; Fraietta, J.A.; Beatty, G.L. IFNgamma and CCL2 Cooperate to Redirect Tumor-Infiltrating Monocytes to Degrade Fibrosis and Enhance Chemotherapy Efficacy in Pancreatic Carcinoma. Cancer Discov. 2016, 6, 400–413. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Burg, J.M.; Mick, R.; Trosko, J.A.; Li, D.; Shaik, M.N.; Tolcher, A.W.; Hamid, O. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology 2013, 2, e23033. [Google Scholar] [CrossRef]
- Nowak, A.K.; Cook, A.M.; McDonnell, A.M.; Millward, M.J.; Creaney, J.; Francis, R.J.; Hasani, A.; Segal, A.; Musk, A.W.; Turlach, B.A.; et al. A phase 1b clinical trial of the CD40-activating antibody CP-870,893 in combination with cisplatin and pemetrexed in malignant pleural mesothelioma. Ann. Oncol. 2015, 26, 2483–2490. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.S.; Poudel, B.; Torres, E.R.; Sidhom, J.W.; Robinson, T.M.; Christmas, B.; Scott, B.; Cruz, K.; Woolman, S.; Wall, V.Z.; et al. A CD40 Agonist and PD-1 Antagonist Antibody Reprogram the Microenvironment of Nonimmunogenic Tumors to Allow T-cell-Mediated Anticancer Activity. Cancer Immunol. Res. 2019, 7, 428–442. [Google Scholar] [CrossRef]
- Byrne, K.T.; Leisenring, N.H.; Bajor, D.L.; Vonderheide, R.H. CSF-1R-Dependent Lethal Hepatotoxicity When Agonistic CD40 Antibody Is Given before but Not after Chemotherapy. J. Immunol. 2016, 197, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Winograd, R.; Byrne, K.T.; Evans, R.A.; Odorizzi, P.M.; Meyer, A.R.; Bajor, D.L.; Clendenin, C.; Stanger, B.Z.; Furth, E.E.; Wherry, E.J.; et al. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol. Res. 2015, 3, 399–411. [Google Scholar] [CrossRef]
- O’Hara, M.H.; O’Reilly, E.M.; Rosemarie, M.; Varadhachary, G.; Wainberg, Z.A.; Ko, A.; Fisher, G.A.; Rahma, O.; Lyman, J.P.; Cabanski, C.R.; et al. A phase 1b study of CD40 agonistic monoclonal antibody APX005M together with gemcitabine and nab-paclitaxel with or without nivolumab in untreated metastatic pancreatic ductal adenocarcinoma (PDAC) patients. In Proceedings of the American Association for Cancer Research Annual Meeting, Atlanta, GA, USA, 29 March–3 April 2019. [Google Scholar]
- Ngiow, S.F.; Young, A.; Blake, S.J.; Hill, G.R.; Yagita, H.; Teng, M.W.; Korman, A.J.; Smyth, M.J. Agonistic CD40 mAb-Driven IL12 Reverses Resistance to Anti-PD1 in a T-cell-Rich Tumor. Cancer Res. 2016, 76, 6266–6277. [Google Scholar] [CrossRef]
- Bajor, D.L.; Mick, R.; Riese, M.J.; Huang, A.C.; Sullivan, B.; Richman, L.P.; Torigian, D.A.; George, S.M.; Stelekati, E.; Chen, F.; et al. Long-term outcomes of a phase I study of agonist CD40 antibody and CTLA-4 blockade in patients with metastatic melanoma. Oncoimmunology 2018, 7, e1468956. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chan, H.T.C.; Orr, C.M.; Dadas, O.; Booth, S.G.; Dahal, L.N.; Penfold, C.A.; O’Brien, L.; Mockridge, C.I.; French, R.R.; et al. Complex Interplay between Epitope Specificity and Isotype Dictates the Biological Activity of Anti-human CD40 Antibodies. Cancer Cell 2018, 33, 664–675.e664. [Google Scholar] [CrossRef]
- Beatty, G.L.; Li, Y.; Long, K.B. Cancer immunotherapy: Activating innate and adaptive immunity through CD40 agonists. Expert Rev. Anticancer. Ther. 2017, 17, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Luqman, M.; Klabunde, S.; Lin, K.; Georgakis, G.V.; Cherukuri, A.; Holash, J.; Goldbeck, C.; Xu, X.; Kadel, E.E., 3rd; Lee, S.H.; et al. The antileukemia activity of a human anti-CD40 antagonist antibody, HCD122, on human chronic lymphocytic leukemia cells. Blood 2008, 112, 711–720. [Google Scholar] [CrossRef]
- Khubchandani, S.; Czuczman, M.S.; Hernandez-Ilizaliturri, F.J. Dacetuzumab, a humanized mAb against CD40 for the treatment of hematological malignancies. Curr. Opin. Investig. Drugs 2009, 10, 579–587. [Google Scholar]
- Franchi, L.; Warner, N.; Viani, K.; Nuñez, G. Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 2009, 227, 106–128. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Gong, T.; Yang, Y.; Jin, T.; Jiang, W.; Zhou, R. Orchestration of NLRP3 Inflammasome Activation by Ion Fluxes. Trends Immunol. 2018, 39, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.M.; O’Neill, L.A.J. Metabolic regulation of NLRP3. Immunol. Rev. 2018, 281, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Garcia, J.J.; Martinez-Banaclocha, H.; Angosto-Bazarra, D.; de Torre-Minguela, C.; Baroja-Mazo, A.; Alarcon-Vila, C.; Martinez-Alarcon, L.; Amores-Iniesta, J.; Martin-Sanchez, F.; Ercole, G.A.; et al. P2X7 receptor induces mitochondrial failure in monocytes and compromises NLRP3 inflammasome activation during sepsis. Nat. Commun. 2019, 10, 2711. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Man, S.M.; Kanneganti, T.D. Inflammasomes and Cancer. Cancer Immunol. Res. 2017, 5, 94–99. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Salcedo, R.; Worschech, A.; Cardone, M.; Jones, Y.; Gyulai, Z.; Dai, R.M.; Wang, E.; Ma, W.; Haines, D.; O’HUigin, C.; et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: Role of interleukin 18. J. Exp. Med. 2010, 207, 1625–1636. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Allen, I.C.; TeKippe, E.M.; Woodford, R.-M.T.; Uronis, J.M.; Holl, E.K.; Rogers, A.B.; Herfarth, H.H.; Jobin, C.; Ting, J.P.Y. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 2010, 207, 1045–1056. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Zhu, R.; Zhu, J.; Zhao, R.; Li, M. E2-Induced Activation of the NLRP3 Inflammasome Triggers Pyroptosis and Inhibits Autophagy in HCC Cells. Oncol. Res. 2019, 27, 827–834. [Google Scholar] [CrossRef]
- Dupaul-Chicoine, J.; Arabzadeh, A.; Dagenais, M.; Douglas, T.; Champagne, C.; Morizot, A.; Rodrigue-Gervais, I.G.; Breton, V.; Colpitts, S.L.; Beauchemin, N.; et al. The Nlrp3 Inflammasome Suppresses Colorectal Cancer Metastatic Growth in the Liver by Promoting Natural Killer Cell Tumoricidal Activity. Immunity 2015, 43, 751–763. [Google Scholar] [CrossRef]
- Wei, Q.; Mu, K.; Li, T.; Zhang, Y.; Yang, Z.; Jia, X.; Zhao, W.; Huai, W.; Guo, P.; Han, L. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab. Investig. 2014, 94, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.T.; Sceneay, J.; Paget, C.; Wong, C.S.; Duret, H.; Tschopp, J.; Moller, A.; Smyth, M.J. NLRP3 suppresses NK cell-mediated responses to carcinogen-induced tumors and metastases. Cancer Res. 2012, 72, 5721–5732. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Luo, Q.; Feng, X.; Zhang, R.; Li, J.; Chen, F. NLRP3 promotes tumor growth and metastasis in human oral squamous cell carcinoma. BMC Cancer 2018, 18, 500. [Google Scholar] [CrossRef] [PubMed]
- Daley, D.; Mani, V.R.; Mohan, N.; Akkad, N.; Pandian, G.S.D.B.; Savadkar, S.; Lee, K.B.; Torres-Hernandez, A.; Aykut, B.; Diskin, B.; et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J. Exp. Med. 2017, 214, 1711–1724. [Google Scholar] [CrossRef]
- Guo, B.; Fu, S.; Zhang, J.; Liu, B.; Li, Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci. Rep. 2016, 6, 36107. [Google Scholar] [CrossRef] [PubMed]
- Theivanthiran, B.; Evans, K.S.; DeVito, N.C.; Plebanek, M.; Sturdivant, M.; Wachsmuth, L.P.; Salama, A.K.; Kang, Y.; Hsu, D.; Balko, J.M.; et al. A tumor-intrinsic PD-L1/NLRP3 inflammasome signaling pathway drives resistance to anti-PD-1 immunotherapy. J. Clin. Investig. 2020, 130, 2570–2586. [Google Scholar] [CrossRef] [PubMed]
- Marty-Roix, R.; Vladimer, G.I.; Pouliot, K.; Weng, D.; Buglione-Corbett, R.; West, K.; MacMicking, J.D.; Chee, J.D.; Wang, S.; Lu, S.; et al. Identification of QS-21 as an Inflammasome-activating Molecular Component of Saponin Adjuvants. J. Biol. Chem. 2016, 291, 1123–1136. [Google Scholar] [CrossRef]
- Vermaelen, K. Vaccine Strategies to Improve Anti-cancer Cellular Immune Responses. Front. Immunol. 2019, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Kopalli, S.R.; Kang, T.-B.; Lee, K.-H.; Koppula, S. NLRP3 Inflammasome Activation Inhibitors in Inflammation-Associated Cancer Immunotherapy: An Update on the Recent Patents. Recent. Pat Anticancer Drug Discov. 2018, 13, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, C.-F.; Li, Y.-C.; Deng, W.-W.; Mao, L.; Wu, L.; Zhang, W.-F.; Zhang, L.; Sun, Z.-J. Blockage of the NLRP3 inflammasome by MCC950 improves anti-tumor immune responses in head and neck squamous cell carcinoma. Cell. Mol. Life Sci. 2018, 75, 2045–2058. [Google Scholar] [CrossRef]
- Xia, X.; Wang, X.; Cheng, Z.; Qin, W.; Lei, L.; Jiang, J.; Hu, J. The role of pyroptosis in cancer: Pro-cancer or pro-”host”? Cell Death Dis. 2019, 10, 650. [Google Scholar] [CrossRef]
- Gao, J.; Qiu, X.; Xi, G.; Liu, H.; Zhang, F.; Lv, T.; Song, Y. Downregulation of GSDMD attenuates tumor proliferation via the intrinsic mitochondrial apoptotic pathway and inhibition of EGFR/Akt signaling and predicts a good prognosis in nonsmall cell lung cancer. Oncol. Rep. 2018, 40, 1971–1984. [Google Scholar] [CrossRef]
- Nagarajan, K.; Soundarapandian, K.; Thorne, R.F.; Li, D.; Li, D. Activation of Pyroptotic Cell Death Pathways in Cancer: An Alternative Therapeutic Approach. Transl. Oncol. 2019, 12, 925–931. [Google Scholar] [CrossRef]
- Saito, T.; Takayama, T.; Osaki, T.; Nagai, S.; Suzuki, T.; Sato, M.; Kuwano, H.; Tahara, H. Combined mobilization and stimulation of tumor-infiltrating dendritic cells and natural killer cells with Flt3 ligand and IL-18 in vivo induces systemic antitumor immunity. Cancer Sci. 2008, 99, 2028–2036. [Google Scholar] [CrossRef]
- Nefedova, Y.; Cheng, P.; Gilkes, D.; Blaskovich, M.; Beg, A.A.; Sebti, S.M.; Gabrilovich, D.I. Activation of dendritic cells via inhibition of Jak2/STAT3 signaling. J. Immunol. 2005, 175, 4338–4346. [Google Scholar] [CrossRef]
- Moon, Y.W.; Hajjar, J.; Hwu, P.; Naing, A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J. Immunother. Cancer 2015, 3, 51. [Google Scholar] [CrossRef]
- Cheever, M.A.; Higano, C.S. PROVENGE (Sipuleucel-T) in prostate cancer: The first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 2011, 17, 3520–3526. [Google Scholar] [CrossRef]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef]
- Autio, K.A.; Klebanoff, C.A.; Schaer, D.; Kauh, J.S.; Slovin, S.F.; Blinder, V.S.; Comen, E.A.; Danila, D.C.; Hoffman, D.M.J.; Kang, S.; et al. Phase 1 study of LY3022855, a colony-stimulating factor-1 receptor (CSF-1R) inhibitor, in patients with metastatic breast cancer (MBC) or metastatic castration-resistant prostate cancer (MCRPC). J. Clin. Oncol. 2019, 37, 2548. [Google Scholar] [CrossRef]
- Jeannin, P.; Paolini, L.; Adam, C.; Delneste, Y. The roles of CSFs on the functional polarization of tumor-associated macrophages. FEBS J. 2018, 285, 680–699. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Nguyen, A.V.; Russell, R.G.; Pollard, J.W. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 2001, 193, 727–740. [Google Scholar] [CrossRef]
- Escamilla, J.; Schokrpur, S.; Liu, C.; Priceman, S.J.; Moughon, D.; Jiang, Z.; Pouliot, F.; Magyar, C.; Sung, J.L.; Xu, J.; et al. CSF1 receptor targeting in prostate cancer reverses macrophage-mediated resistance to androgen blockade therapy. Cancer Res. 2015, 75, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, K.P.; Gluck, L.; Martin, L.P.; Olszanski, A.J.; Tolcher, A.W.; Ngarmchamnanrith, G.; Rasmussen, E.; Amore, B.M.; Nagorsen, D.; Hill, J.S.; et al. First-in-Human Study of AMG 820, a Monoclonal Anti-Colony-Stimulating Factor 1 Receptor Antibody, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 5703–5710. [Google Scholar] [CrossRef] [PubMed]
- Wang-Gillam, A.; O’Reilly, E.M.; Bendell, J.C.; Wainberg, Z.A.; Borazanci, E.H.; Bahary, N.; O’Hara, M.H.; Beatty, G.L.; Pant, S.; Cohen, D.J.; et al. A randomized phase II study of cabiralizumab (cabira) + nivolumab (nivo) ± chemotherapy (chemo) in advanced pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 2019, 37, TPS465. [Google Scholar] [CrossRef]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.M.; Cappello, P.; Nguyen, A.V.; Ralainirina, N.; Hardamon, C.R.; Foubert, P.; Schmid, M.C.; Sun, P.; Mose, E.; Bouvet, M.; et al. Macrophage PI3Kgamma Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discov. 2016, 6, 870–885. [Google Scholar] [CrossRef] [PubMed]
- Varner, J.A. Taming the beast: Strategies to target the immune-suppressive macrophage to enhance cancer immune therapy. Proc. Am. Assoc. Cancer Res. Annu. Meet. 2019, 2019, 79. [Google Scholar]
- Liu, J.; Wang, L.; Zhao, F.; Tseng, S.; Narayanan, C.; Shura, L.; Willingham, S.; Howard, M.; Prohaska, S.; Volkmer, J.; et al. Pre-Clinical Development of a Humanized Anti-CD47 Antibody with Anti-Cancer Therapeutic Potential. PLoS ONE 2015, 10, e0137345. [Google Scholar] [CrossRef]
- Tseng, D.; Volkmer, J.P.; Willingham, S.B.; Contreras-Trujillo, H.; Fathman, J.W.; Fernhoff, N.B.; Seita, J.; Inlay, M.A.; Weiskopf, K.; Miyanishi, M.; et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. USA 2013, 110, 11103–11108. [Google Scholar] [CrossRef]
- Petrova, P.S.; Viller, N.N.; Wong, M.; Pang, X.; Lin, G.H.; Dodge, K.; Chai, V.; Chen, H.; Lee, V.; House, V.; et al. TTI-621 (SIRPalphaFc): A CD47-Blocking Innate Immune Checkpoint Inhibitor with Broad Antitumor Activity and Minimal Erythrocyte Binding. Clin. Cancer Res. 2017, 23, 1068–1079. [Google Scholar] [CrossRef]
- Sikic, B.I.; Lakhani, N.; Patnaik, A.; Shah, S.A.; Chandana, S.R.; Rasco, D.; Colevas, A.D.; O’Rourke, T.; Narayanan, S.; Papadopoulos, K.; et al. First-in-Human, First-in-Class Phase I Trial of the Anti-CD47 Antibody Hu5F9-G4 in Patients With Advanced Cancers. J. Clin. Oncol. 2019, 37, 946–953. [Google Scholar] [CrossRef]
- Hu, B.; Wang, Z.; Zeng, H.; Qi, Y.; Chen, Y.; Wang, T.; Wang, J.; Chang, Y.; Bai, Q.; Xia, Y.; et al. Blockade of DC-SIGN+ tumor-associated macrophages reactivates anti-tumor immunity and improves immunotherapy in muscle-invasive bladder cancer. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Halama, N.; Zoernig, I.; Berthel, A.; Kahlert, C.; Klupp, F.; Suarez-Carmona, M.; Suetterlin, T.; Brand, K.; Krauss, J.; Lasitschka, F.; et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell 2016, 29, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef]
- Raghavan, S.; Mehta, P.; Xie, Y.; Lei, Y.L.; Mehta, G. Ovarian cancer stem cells and macrophages reciprocally interact through the WNT pathway to promote pro-tumoral and malignant phenotypes in 3D engineered microenvironments. J. Immunother. Cancer 2019, 7, 190. [Google Scholar] [CrossRef]
- Fang, W.B.; Yao, M.; Brummer, G.; Acevedo, D.; Alhakamy, N.; Berkland, C.; Cheng, N. Targeted gene silencing of CCL2 inhibits triple negative breast cancer progression by blocking cancer stem cell renewal and M2 macrophage recruitment. Oncotarget 2016, 7, 49349–49367. [Google Scholar] [CrossRef]
- Pienta, K.J.; Machiels, J.P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Investig. New Drugs 2013, 31, 760–768. [Google Scholar] [CrossRef]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef]
- Diefenbach, A.; Jensen, E.R.; Jamieson, A.M.; Raulet, D.H. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 2001, 413, 165–171. [Google Scholar] [CrossRef]
- Chitadze, G.; Lettau, M.; Bhat, J.; Wesch, D.; Steinle, A.; Furst, D.; Mytilineos, J.; Kalthoff, H.; Janssen, O.; Oberg, H.H.; et al. Shedding of endogenous MHC class I-related chain molecules A and B from different human tumor entities: Heterogeneous involvement of the “a disintegrin and metalloproteases” 10 and 17. Int. J. Cancer 2013, 133, 1557–1566. [Google Scholar] [CrossRef]
- Cathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting Matrix Metalloproteinases in Cancer: Bringing New Life to Old Ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Ferrari de Andrade, L.; Tay, R.E.; Pan, D.; Luoma, A.M.; Ito, Y.; Badrinath, S.; Tsoucas, D.; Franz, B.; May, K.F., Jr.; Harvey, C.J.; et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science 2018, 359, 1537–1542. [Google Scholar] [CrossRef]
- Vales-Gomez, M.; Chisholm, S.E.; Cassady-Cain, R.L.; Roda-Navarro, P.; Reyburn, H.T. Selective induction of expression of a ligand for the NKG2D receptor by proteasome inhibitors. Cancer Res. 2008, 68, 1546–1554. [Google Scholar] [CrossRef]
- Lopez-Soto, A.; Folgueras, A.R.; Seto, E.; Gonzalez, S. HDAC3 represses the expression of NKG2D ligands ULBPs in epithelial tumour cells: Potential implications for the immunosurveillance of cancer. Oncogene 2009, 28, 2370–2382. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, T.; Dunn, G.P.; Lacoursiere, D.Y.; Schreiber, R.D.; Bui, J.D. Cancer immunoediting of the NK group 2D ligand H60a. J. Immunol. 2011, 187, 3538–3545. [Google Scholar] [CrossRef] [PubMed]
- Armeanu, S.; Bitzer, M.; Lauer, U.M.; Venturelli, S.; Pathil, A.; Krusch, M.; Kaiser, S.; Jobst, J.; Smirnow, I.; Wagner, A.; et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 2005, 65, 6321–6329. [Google Scholar] [CrossRef]
- Hervieu, A.; Rebe, C.; Vegran, F.; Chalmin, F.; Bruchard, M.; Vabres, P.; Apetoh, L.; Ghiringhelli, F.; Mignot, G. Dacarbazine-mediated upregulation of NKG2D ligands on tumor cells activates NK and CD8 T cells and restrains melanoma growth. J. Investig. Dermatol. 2013, 133, 499–508. [Google Scholar] [CrossRef]
- Anagnostou, E.; Kosmopoulou, M.N.; Chrysina, E.D.; Leonidas, D.D.; Hadjiloi, T.; Tiraidis, C.; Zographos, S.E.; Gyorgydeak, Z.; Somsak, L.; Docsa, T.; et al. Crystallographic studies on two bioisosteric analogues, N-acetyl-beta-D-glucopyranosylamine and N-trifluoroacetyl-beta-D-glucopyranosylamine, potent inhibitors of muscle glycogen phosphorylase. Bioorg. Med. Chem. 2006, 14, 181–189. [Google Scholar] [CrossRef]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Blery, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743.e1713. [Google Scholar] [CrossRef]
- van Montfoort, N.; Borst, L.; Korrer, M.J.; Sluijter, M.; Marijt, K.A.; Santegoets, S.J.; van Ham, V.J.; Ehsan, I.; Charoentong, P.; Andre, P.; et al. NKG2A Blockade Potentiates CD8 T Cell Immunity Induced by Cancer Vaccines. Cell 2018, 175, 1744–1755. [Google Scholar] [CrossRef]
- Creelan, B.C.; Antonia, S.J. The NKG2A immune checkpoint - a new direction in cancer immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 277–278. [Google Scholar] [CrossRef]
- van Hall, T.; Andre, P.; Horowitz, A.; Ruan, D.F.; Borst, L.; Zerbib, R.; Narni-Mancinelli, E.; van der Burg, S.H.; Vivier, E. Monalizumab: Inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 2019, 7, 263. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S.; Lanier, L.L. Natural Killer Cells in Cancer Immunotherapy. Annu. Rev. Cancer Biol. 2019, 3, 77–103. [Google Scholar] [CrossRef]
- Bald, T.; Krummel, M.F.; Smyth, M.J.; Barry, K.C. The NK cell-cancer cycle: Advances and new challenges in NK cell-based immunotherapies. Nat. Immunol. 2020, 21, 835–847. [Google Scholar] [CrossRef]
- Torikai, H.; Reik, A.; Soldner, F.; Warren, E.H.; Yuen, C.; Zhou, Y.; Crossland, D.L.; Huls, H.; Littman, N.; Zhang, Z.; et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood 2013, 122, 1341–1349. [Google Scholar] [CrossRef]
- Baginska, J.; Viry, E.; Paggetti, J.; Medves, S.; Berchem, G.; Moussay, E.; Janji, B. The critical role of the tumor microenvironment in shaping natural killer cell-mediated anti-tumor immunity. Front. Immunol. 2013, 4, 490. [Google Scholar] [CrossRef]
- Trzonkowski, P.; Szmit, E.; Mysliwska, J.; Dobyszuk, A.; Mysliwski, A. CD4+CD25+ T regulatory cells inhibit cytotoxic activity of T CD8+ and NK lymphocytes in the direct cell-to-cell interaction. Clin. Immunol. 2004, 112, 258–267. [Google Scholar] [CrossRef]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Labadie, B.W.; Bao, R.; Luke, J.J. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan-Kynurenine-Aryl Hydrocarbon Axis. Clin. Cancer Res. 2019, 25, 1462–1471. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| DC | TAM | Mo-MDSC | PMN-MDSC | Neutrophils | NK Cells | Basophils | Eosinophils | Mast Cells | |
|---|---|---|---|---|---|---|---|---|---|
| TLR7/8 | |||||||||
| TLR9 | |||||||||
| RIG-I | |||||||||
| cGAS-STING | |||||||||
| NLRP3 | |||||||||
| CSF-1R | |||||||||
| CD40 | |||||||||
| NKG2D |
| PRR | Agent | Molecule Type | Route of Administration | Cancer Type(s) | Clinical Phase of Development | |
|---|---|---|---|---|---|---|
| TLR8 | VTX-2337 (motilomod) | Small molecule | Intratumoral | ovarian, HNSCC | I, II | |
| dTLR7/8 | NKTR-262 | Small molecule | Intratumoral | Solid tumors | I, II | |
| TLR9 | SD-101 | CpG-C class ODN | Intratumoral | Solid tumors | I, II | |
| EMD 1201081 | Synthetic ODN | Subcutaneous Injection | HNSCC | I, II | ||
| CPG 7909 | CpG ODN | Subcutaneous Injection | Lymphomas | I, II | ||
| IMO-2125 (Tilsotolimod) | Synthetic ODN | Intratumoral | melanoma, Solid tumors | I, II | ||
| CMP-001 | CpG ODN | Intratumoral | Solid tumors | I, II | ||
| RIG-I | SLR-14 | Synthetic stem loop RNA | Intratumoral | Solid tumors | Pre-clinical | |
| RGT-100 (MK-4621) | Synthetic oligonucleotide | Intratumoral | Solid tumors | |||
| MDA-5 | BO-112 (poly(I:C)) | Synthetic dsRNA | Intratumoral | Solid tumors | I | |
| STING | E7766 | Novel macrocycle-bridged | Intravenous | Solid tumors | I | |
| GSK3745417 | Small molecule | Intravenous | Solid tumors | I | ||
| MIW815 (ADU-S100) | Synthetic CDN | Intratumoral | Solid tumors | I, II | ||
| MK1454 | Small molecule | Intratumoral | Solid tumors | I, II | ||
| BMS-986301 | Small molecule | Intratumoral * | Solid tumors | I | ||
| NLRP3 | BMS-986299 | First in class agonist * | Intratumoral * | I | ||
| Other innate immune targets | ||||||
| CSF-1R | Cabiralizumab | Monoclonal antibody | Intravenous | Solid tumors | I, II | |
| JNJ-40346527 | Monoclonal antibody | Intravenous | Advanced prostate cancer | I, II | ||
| PLX3397 | Small molecule | Oral | Solid tumors | I, II | ||
| MCS110 | Solid tumors | I, II | ||||
| IMC-CS4 | Monoclonal antibody | Intravenous | Solid tumors | I | ||
| CD40 | APX005M | Monoclonal antibody | Intravenous | Solid tumors | I, II | |
| CP-870,893 | Monoclonal antibody | Intravenous | Solid tumors | I | ||
| Selicrelumab | Monoclonal antibody | Intravenous | Solid tumors | I, II | ||
| PI3K (delta) Inhibitors | IPI-549 | Small molecule | Oral | Solid tumors | I, II | |
| Class IIa histone deacetylase inhibitor | TMP-195 | Small molecule | Oral | Solid Tumors | Preclinical | |
| IDO inhibitors | Indoximod | Small molecule | Oral | Solid tumors | I, II | |
| STAT3 inhibitors | Siltuximab | Monoclonal antibody | Intravenous | Solid tumors | I, II | |
| WP1066 | Small molecule | Oral | Solid tumors | I | ||
| TT-101 | Small molecule | Oral | Solid tumors | I | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rameshbabu, S.; Labadie, B.W.; Argulian, A.; Patnaik, A. Targeting Innate Immunity in Cancer Therapy. Vaccines 2021, 9, 138. https://doi.org/10.3390/vaccines9020138
Rameshbabu S, Labadie BW, Argulian A, Patnaik A. Targeting Innate Immunity in Cancer Therapy. Vaccines. 2021; 9(2):138. https://doi.org/10.3390/vaccines9020138
Chicago/Turabian StyleRameshbabu, Srikrishnan, Brian W. Labadie, Anna Argulian, and Akash Patnaik. 2021. "Targeting Innate Immunity in Cancer Therapy" Vaccines 9, no. 2: 138. https://doi.org/10.3390/vaccines9020138
APA StyleRameshbabu, S., Labadie, B. W., Argulian, A., & Patnaik, A. (2021). Targeting Innate Immunity in Cancer Therapy. Vaccines, 9(2), 138. https://doi.org/10.3390/vaccines9020138