
Annotation of Potential Vaccine Targets and Design of a Multi-Epitope Subunit Vaccine against Yersinia pestis through Reverse Vaccinology and Validation through an Agent-Based Modeling Approach

 ,
,  ,
,  , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Retrieval of Proteomes
2.2. Removal of Homologous Proteins
2.3. Removal of Paralogous Proteins
2.4. Non-Essential Protein Removal
2.5. Subcellular Localization of Proteins
2.6. Prioritizing Potential Vaccine Candidates
2.7. Collection of Virulent Proteins
2.8. Antigenicity of the Proteins
2.9. Prioritization of Proteins
2.10. Epitope Prediction
2.11. Vaccine Construct Design
2.12. Allergenicity Prediction
2.13. Antigenicity Evaluation
2.14. Evaluation of Physiochemical Parameters
2.15. Homology Modeling of the Vaccine Construct
2.16. Tertiary Structure Refinement
2.17. Structure Validation
2.18. B-Cell Epitope Prediction
2.19. Molecular Docking of the Vaccine Constructs and TLR-4
2.20. Molecular Dynamics Simulation
2.21. Codon Optimization and Cloning of the Vaccine Construct
2.22. Immune Simulation
3. Results
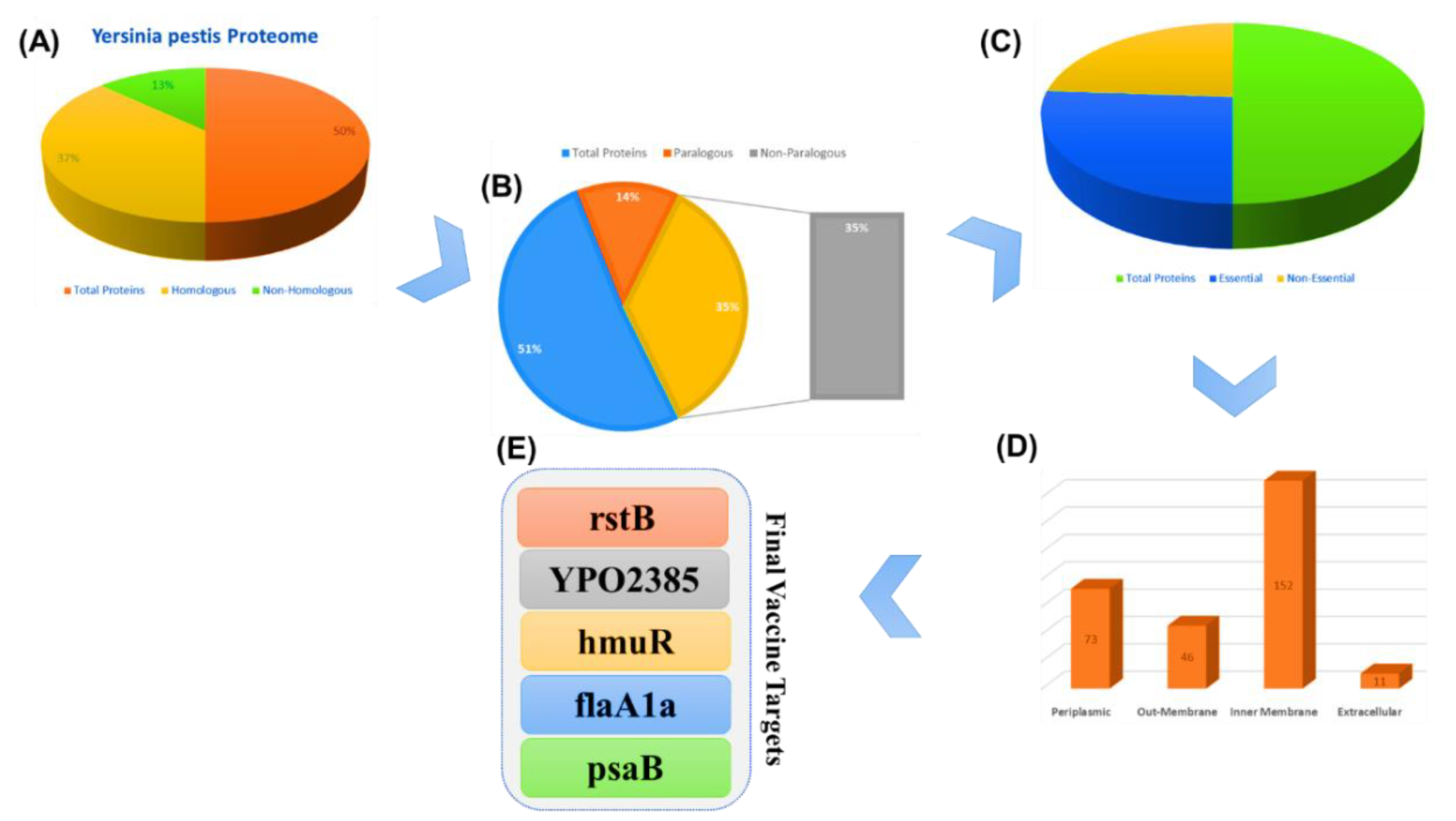
3.1. Retrieval of Proteomes
3.2. Removal of Homologous Proteins
3.3. Removal of Paralogous Proteins
3.4. Removal of Non-Essential Proteins
3.5. Subcellular Localization
3.6. Collection of Virulent Proteins
3.7. Antigenicity and Physiochemical Properties of the Selected Vaccine Targets
3.8. CTL and HTL Epitope Prediction
3.9. B-Cell Epitope Prediction
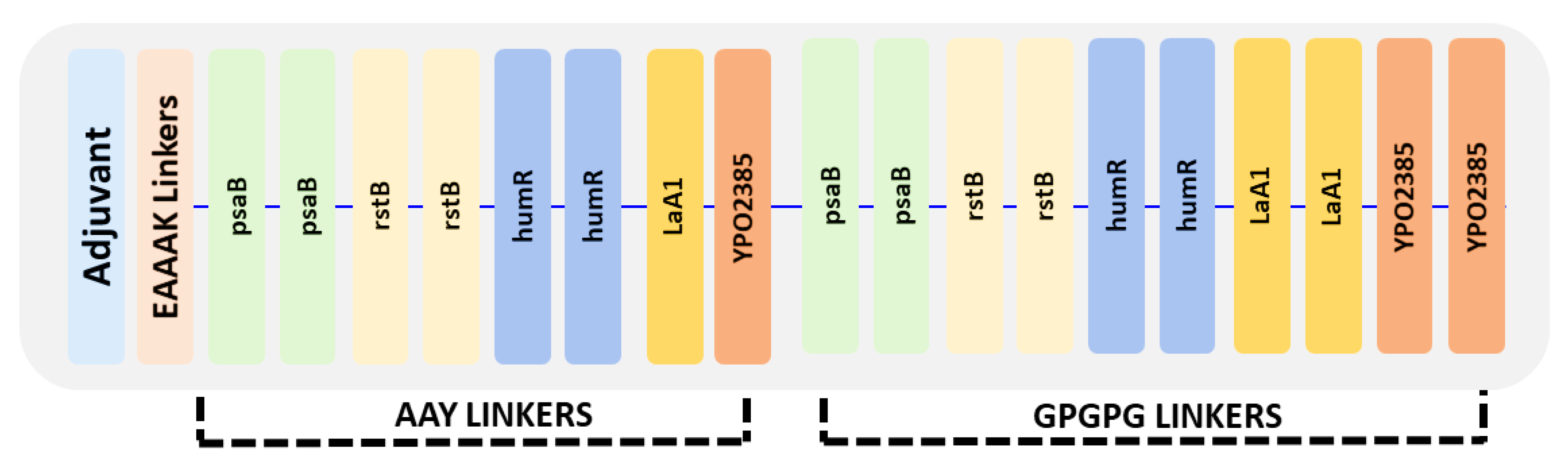
3.10. Vaccine Construct Design
3.11. D Structure Prediction Using Robetta
3.12. Refinement of the Vaccine Tertiary Structure
3.13. Vaccine 3D Structure Validation
3.14. Physiochemical Properties of the Final Vaccine
3.15. Secondary Structure Prediction
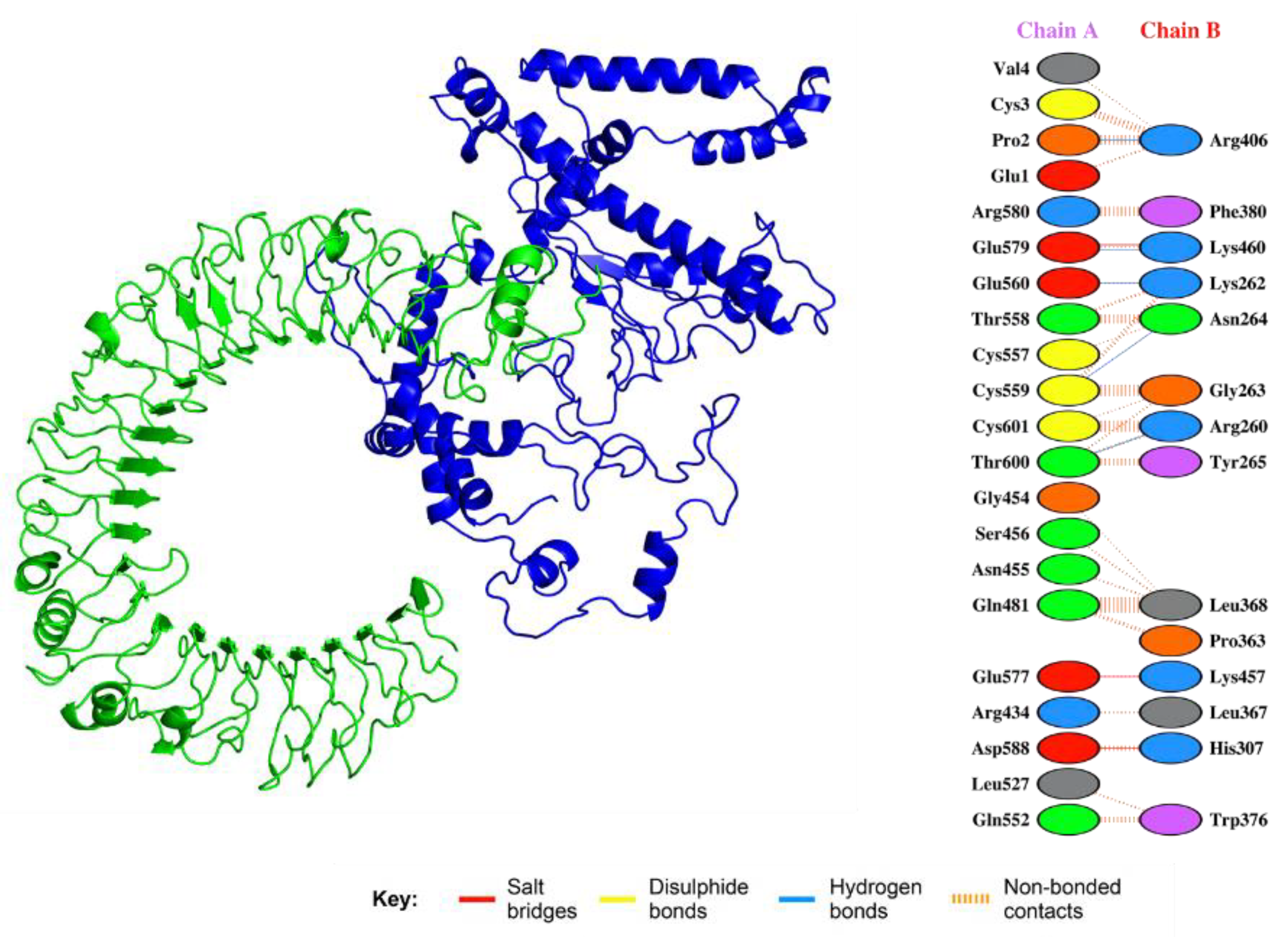
3.16. Analysis of Vaccine Construct Interaction with TLRs
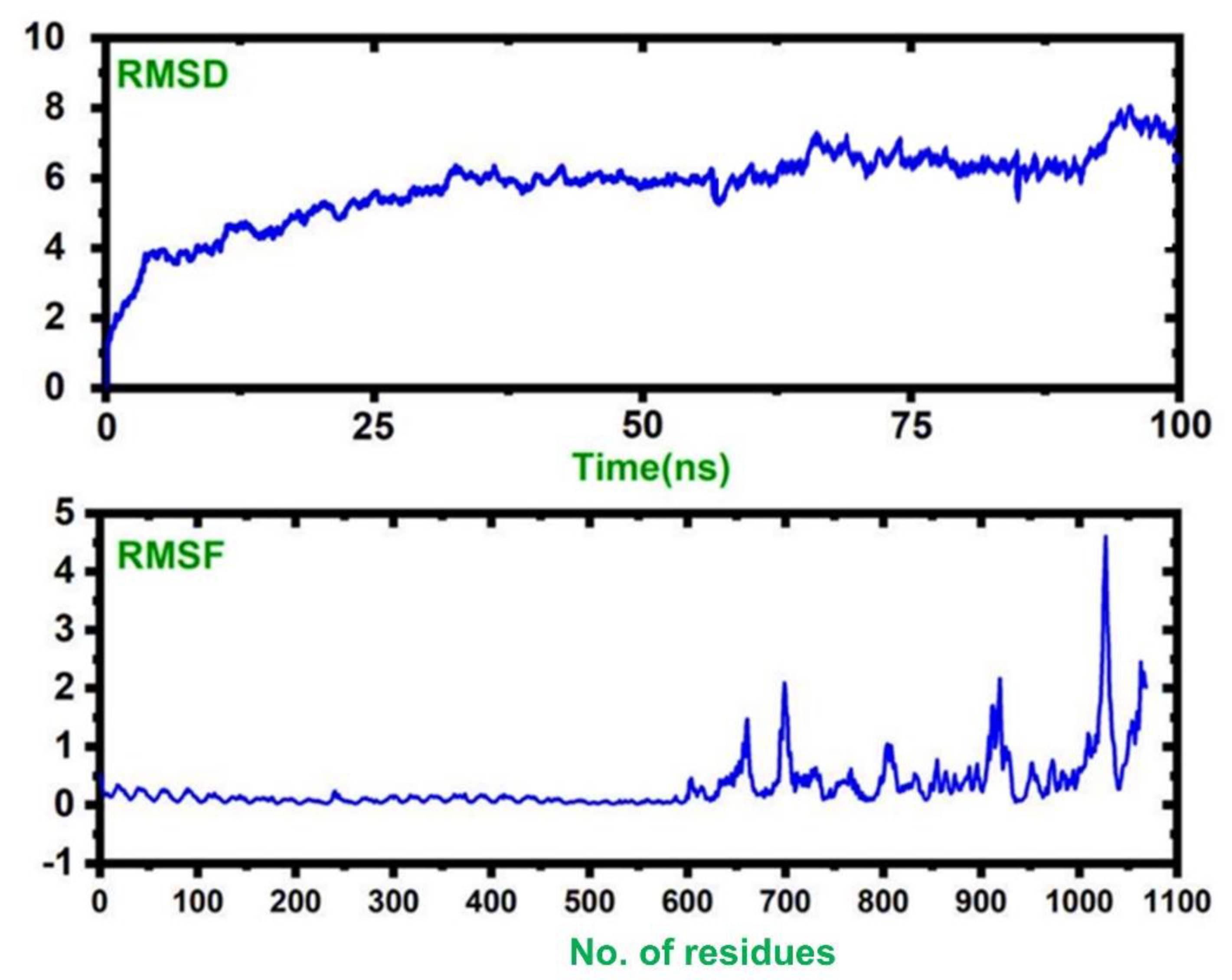
3.17. Molecular Simulations of TLR–Vaccine Complexes
3.18. Codon Optimization and Cloning of the Vaccine Construct
3.19. Immune Simulation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pechous, R.D.; Sivaraman, V.; Stasulli, N.M.; Goldman, W.E. Pneumonic plague: The darker side of Yersinia pestis. Trends Microbiol. 2016, 24, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, R.; Drancourt, M.; Raoult, D. Plague, camels, and lice. Proc. Natl. Acad. Sci. USA 2019, 116, 7620–7621. [Google Scholar] [CrossRef] [Green Version]
- Yang, R. Plague: Recognition, treatment, and prevention. J. Clin. Microbiol. 2018, 56, e01519-17. [Google Scholar] [CrossRef] [Green Version]
- Eichelberger, K.R.; Goldman, W.E. Human Neutrophil Isolation and Degranulation Responses to Yersinia pestis Infection. In Pathogenic Yersinia; Springer: Berlin/Heidelberg, Germany, 2019; pp. 197–209. [Google Scholar]
- Smiley, S.T.; Szaba, F.M.; Kummer, L.W.; Duso, D.K.; Lin, J.-S. Yersinia pestis Pla protein thwarts T cell defense against plague. Infect. Immun. 2019, 87, e00126-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.; Burland, V.; Plunkett III, G.; Boutin, A.; Mayhew, G.F.; Liss, P.; Perna, N.T.; Rose, D.J.; Mau, B.; Zhou, S. Genome sequence of Yersinia pestis KIM. J. Bacteriol. 2002, 184, 4601–4611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zauberman, A.; Cohen, S.; Mamroud, E.; Flashner, Y.; Tidhar, A.; Ber, R.; Elhanany, E.; Shafferman, A.; Velan, B. Interaction of Yersinia pestis with macrophages: Limitations in YopJ-dependent apoptosis. Infect. Immun. 2006, 74, 3239–3250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Ke, Y.; Wang, J.; Tan, Y.; Myeni, S.K.; Li, D.; Shi, Q.; Yan, Y.; Chen, H.; Guo, Z. Insight into bacterial virulence mechanisms against host immune response via the Yersinia pestis-human protein-protein interaction network. Infect. Immun. 2011, 79, 4413–4424. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Yang, R. Interaction between Yersinia pestis and the host immune system. Infect. Immun. 2008, 76, 1804–1811. [Google Scholar] [CrossRef] [Green Version]
- Kelly, D.F.; Rappuoli, R. Reverse vaccinology and vaccines for serogroup B Neisseria meningitidis. In Hot Topics in Infection and Immunity in Children II; Springer: Erlin/Heidelberg, Germany, 2005; pp. 217–223. [Google Scholar]
- Ahmad, I.; Ali, S.S.; Shah, I.; Khan, S.; Khan, M.; Ullah, S.; Ali, S.; Khan, J.; Ali, M.; Khan, A. Computational vaccinology based development of multi-epitope subunit vaccine for protection against the Norovirus infections. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Khan, A.; Kaushik, A.C.; Wang, Y.; Ali, S.S.; Junaid, M.; Saleem, S.; Cho, W.C.; Mao, X.; Wei, D.-Q. Immunoinformatic and systems biology approaches to predict and validate peptide vaccines against Epstein–Barr virus (EBV). Sci. Rep. 2019, 9, 1–12. [Google Scholar]
- Khan, A.; Ali, S.S.; Khan, M.T.; Saleem, S.; Ali, A.; Suleman, M.; Babar, Z.; Shafiq, A.; Khan, M.; Wei, D.-Q. Combined drug repurposing and virtual screening strategies with molecular dynamics simulation identified potent inhibitors for SARS-CoV-2 main protease (3CLpro). J. Biomol. Struct. Dyn. 2021, 39, 4659–4670. [Google Scholar] [CrossRef]
- Khan, A.; Junaid, M.; Kaushik, A.C.; Ali, A.; Ali, S.S.; Mehmood, A.; Wei, D.-Q. Computational identification, characterization and validation of potential antigenic peptide vaccines from hrHPVs E6 proteins using immunoinformatics and computational systems biology approaches. PLoS ONE 2018, 13, e0196484. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Khan, M.; Saleem, S.; Babar, Z.; Ali, A.; Khan, A.A.; Sardar, Z.; Hamayun, F.; Ali, S.S.; Wei, D.-Q. Phylogenetic analysis and structural perspectives of RNA-dependent RNA-polymerase inhibition from SARs-CoV-2 with natural products. Interdiscip. Sci. Comput. Life Sci. 2020, 12, 335–348. [Google Scholar] [CrossRef]
- Khan, A.; Khan, M.T.; Saleem, S.; Junaid, M.; Ali, A.; Ali, S.S.; Khan, M.; Wei, D.-Q. Structural Insights into the mechanism of RNA recognition by the N-terminal RNA-binding domain of the SARS-CoV-2 nucleocapsid phosphoprotein. Comput. Struct. Biotechnol. J. 2020, 18, 2174–2184. [Google Scholar] [CrossRef]
- Khan, M.; Khan, S.; Ali, A.; Akbar, H.; Sayaf, A.M.; Khan, A.; Wei, D.-Q. Immunoinformatics approaches to explore Helicobacter Pylori proteome (Virulence Factors) to design B and T cell multi-epitope subunit vaccine. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Khan, S.; Khan, A.; Rehman, A.U.; Ahmad, I.; Ullah, S.; Khan, A.A.; Ali, S.S.; Afridi, S.G.; Wei, D.-Q. Immunoinformatics and structural vaccinology driven prediction of multi-epitope vaccine against Mayaro virus and validation through in-silico expression. Infect. Genet. Evol. 2019, 73, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Bueno, L.L.; Lobo, F.P.; Morais, C.G.; Mourão, L.C.; de Ávila, R.A.M.; Soares, I.S.; Fontes, C.J.; Lacerda, M.V.; Olortegui, C.C.; Bartholomeu, D.C. Identification of a highly antigenic linear B cell epitope within Plasmodium vivax apical membrane antigen 1 (AMA-1). PLoS ONE 2011, 6, e21289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, S.; Guan, L.; Dong, Y.; Wang, C.; Feng, L.; Xie, Y. Design and evaluation of a multi-epitope assembly peptide vaccine against Acinetobacter baumannii infection in mice. Swiss Med. Wkly. 2019, 149. [Google Scholar] [CrossRef] [PubMed]
- Guedes, R.L.M.; Rodrigues, C.M.F.; Coatnoan, N.; Cosson, A.; Cadioli, F.A.; Garcia, H.A.; Gerber, A.L.; Machado, R.Z.; Minoprio, P.M.C.; Teixeira, M.M.G. A comparative in silico linear B-cell epitope prediction and characterization for South American and African Trypanosoma vivax strains. Genomics 2019, 111, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Bazhan, S.I.; Antonets, D.V.; Karpenko, L.I.; Oreshkova, S.F.; Kaplina, O.N.; Starostina, E.V.; Dudko, S.G.; Fedotova, S.A.; Ilyichev, A.A. In silico designed ebola virus T-cell multi-epitope DNA vaccine constructions are immunogenic in mice. Vaccines 2019, 7, 34. [Google Scholar] [CrossRef] [Green Version]
- Hasan, M.; Azim, K.F.; Begum, A.; Khan, N.A.; Shammi, T.S.; Imran, A.S.; Chowdhury, I.M.; Urme, S.R.A. Vaccinomics strategy for developing a unique multi-epitope monovalent vaccine against Marburg marburgvirus. Infect. Genet. Evol. 2019, 70, 140–157. [Google Scholar] [CrossRef]
- Nosrati, M.; Behbahani, M.; Mohabatkar, H. Towards the first multi-epitope recombinant vaccine against Crimean-Congo hemorrhagic fever virus: A computer-aided vaccine design approach. J. Biomed. Inform. 2019, 93, 103160. [Google Scholar] [CrossRef]
- Mohammed, A.; Hashim, O.; Elrahman, K.; Hamdi, A.; Hassan, M. Epitope-based peptide vaccine design against Mokola rabies virus glycoprotein G utilizing in silico approaches. Immunome Res 2017, 13, 2. [Google Scholar]
- Chen, Z.; Ruan, P.; Wang, L.; Nie, X.; Ma, X.; Tan, Y. T and B cell Epitope analysis of SARS-CoV-2 S protein based on immunoinformatics and experimental research. J. Cell. Mol. Med. 2021, 25, 1274–1289. [Google Scholar] [CrossRef]
- Magrane, M. UniProt Knowledgebase: A hub of integrated protein data. Database 2011, 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahram, A.; Herbordt, M.C. Fast and accurate NCBI BLASTP: Acceleration with multiphase FPGA-based prefiltering. In Proceedings of the 24th ACM International Conference on Supercomputing, Tsukuba, Japan, 2–4 June 2010; pp. 73–82. [Google Scholar]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ou, H.Y.; Zhang, C.T. DEG: A database of essential genes. Nucleic Acids Res. 2004, 32, D271–D272. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-S.; Cheng, C.-W.; Su, W.-C.; Chang, K.-C.; Huang, S.-W.; Hwang, J.-K.; Lu, C.-H. CELLO2GO: A web server for protein subCELlular LOcalization prediction with functional gene ontology annotation. PLoS ONE 2014, 9, e99368. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Khan, S.; Saleem, S.; Nizam-Uddin, N.; Mohammad, A.; Khan, T.; Ahmad, S.; Arshad, M.; Ali, S.S.; Suleman, M. Immunogenomics Guided Design of Immunomodulatory Multi-Epitope Subunit Vaccine against the SARS-CoV-2 new Variants, and its Validation through in Silico Cloning and Immune Simulation. Comput. Biol. Med. 2021, 133, 104420. [Google Scholar] [CrossRef]
- Demeure, C.E.; Dussurget, O.; Fiol, G.M.; Le Guern, A.-S.; Savin, C.; Pizarro-Cerdá, J. Yersinia pestis and plague: An updated view on evolution, virulence determinants, immune subversion, vaccination, and diagnostics. Genes Immun. 2019, 20, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Zhang, M. Genetic diversity and antigenicity analysis of Streptococcus pneumoniae pneumolysin isolated from children with pneumococcal infection. Int. J. Infect. Dis. 2019, 86, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Zaharieva, N.; Dimitrov, I.; Flower, D.; Doytchinova, I. VaxiJen Dataset of Bacterial Immunogens: An Update. Curr. Comput. Aided Drug Des. 2019, 15, 398–400. [Google Scholar] [CrossRef]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007, 8, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Raghava, G. AlgPred: Prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006, 34, W202–W209. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Maheshwari, N.; Chauhan, R.; Sen, N.K.; Sharma, A. Structure prediction and functional characterization of secondary metabolite proteins of Ocimum. Bioinformation 2011, 6, 315. [Google Scholar] [CrossRef] [Green Version]
- Chivian, D.; Kim, D.E.; Malmström, L.; Schonbrun, J.; Rohl, C.A.; Baker, D. Prediction of CASP6 structures using automated Robetta protocols. Proteins Struct. Funct. Bioinform. 2005, 61, 157–166. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef]
- Ko, J.; Park, H.; Heo, L.; Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef]
- Wang, W.; Xia, M.; Chen, J.; Deng, F.; Yuan, R.; Zhang, X.; Shen, F. Data set for phylogenetic tree and RAMPAGE Ramachandran plot analysis of SODs in Gossypium raimondii and G. arboreum. Data Brief 2016, 9, 345–348. [Google Scholar] [CrossRef] [Green Version]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [Green Version]
- Lengths, M.; Angles, M. Limitations of structure evaluation tools errat. Quick Guidel. Comput. Drug Des. 2018, 16, 75. [Google Scholar]
- Leung, S.-O.; Hansen, H.; Qu, Z. Glycosylated Humanized B-Cell Specific Antibodies. U.S. Patent 6,254,868, 3 July 2001. [Google Scholar]
- EL-Manzalawy, Y.; Dobbs, D.; Honavar, V. Predicting linear B-cell epitopes using string kernels. J. Mol. Recognit. Interdiscip. J. 2008, 21, 243–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponomarenko, J.; Bui, H.-H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008, 9, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Khan, M.T.; Khan, A.; Rehman, A.U.; Wang, Y.; Akhtar, K.; Malik, S.I.; Wei, D.-Q. Structural and free energy landscape of novel mutations in ribosomal protein S1 (rpsA) associated with pyrazinamide resistance. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Albert, N.L.; Unterrainer, M.; Fleischmann, D.; Lindner, S.; Vettermann, F.; Brunegraf, A.; Vomacka, L.; Brendel, M.; Wenter, V.; Wetzel, C. TSPO PET for glioma imaging using the novel ligand 18 F-GE-180: First results in patients with glioblastoma. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 2230–2238. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Castiglione, F. Immune system simulation online. Bioinformatics 2011, 27, 2013–2014. [Google Scholar] [CrossRef] [Green Version]
- Castiglione, F.; Ribba, B.; Brass, O. Comparing In Silico Results to In Vivo and Ex Vivo of Influenza-Specific Immune Responses after Vaccination or Infection in Humans. In Innovation in Vaccinology; Springer: Berlin/Heidelberg, Germany, 2012; pp. 17–42. [Google Scholar]
- Mayer, H.; Zaenker, K.S.; An Der Heiden, U. A basic mathematical model of the immune response. Chaos (Woodbury, N.Y.) 1995, 5, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Teijaro, J.R. Cytokine storms in infectious diseases. Semin. Immunopathol. 2017, 39, 501–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein | Virulence (Bit Score) | Virulence Sequence Identity | Antigenicity | Mass | Length |
|---|---|---|---|---|---|---|
| rstB | Sensor kinase protein | 182 bits | 33% | 0.6925 | 48,250 Da | 425 |
| YPO2385 | Putative exported protein | 61.2 bits | 34% | 0.7901 | 32,315 Da | 288 |
| hmuR | Hemin receptor | 886 bits | 68% | 0.6496 | 74,230 Da | 676 |
| flaA1 | Flagellin | 97.4 bits | 40% | 0.7208 | 42,701 Da | 404 |
| psaB | Chaperone protein PsaB | 531 bits | 100% | 0.6251 | 30,648 Da | 273 |
| Protein Name | Epitopes | Sequence | Combined Score |
|---|---|---|---|
| Sensor kinase protein | CTL | RTPLVRLRY | 3.1800 |
| CTL | MAMLVGLVY | 3.1360 | |
| Putative exported protein | CTL | CSGLIYYAY | 2.6220 |
| Hemin receptor | CTL | QSLSANLRY | 3.0050 |
| CTL | SSSTPQAGY | 3.0610 | |
| Flagellin | CTL | ETPKAVNEY | 2.6930 |
| Chaperone protein PsaB | CTL | SADSLTWRY | 2.7150 |
| CTL | PTPFYMNFY | 2.5490 |
| Protein | Allele | Start–End | Peptide Sequence | Method | Percentile Rank | Antigenicity Score |
|---|---|---|---|---|---|---|
| Sensor kinase protein | HLA-DRB5 * 01:01 | 142–156 | LPVFLWMRPHWKDLL | Consensus (smm/nn/sturniolo) | 0.6 | 0.432 |
| HLA-DRB3 * 02:02 | 404–418 | GGASFRFSWPIKTHL | NetMHCIIpan | 1.4 | 1.043 | |
| HLA-DRB1 * 03:01 | 362–376 | EPFVRLDPSRDRATG | Consensus (smm/nn/sturniolo) | 1.6 | 0.564 | |
| Putative exported protein | HLA-DRB3 * 02:02 | 207–221 | NEMYHLRDAAPVKRT | NetMHCIIpan | 5.1 | 1.042 |
| HLA-DRB5 * 01:01 | 161–175 | AMSKLMKQVGKPYRW | Consensus (smm/nn/sturniolo) | 9.7 | 0.613 | |
| Hemin receptor | HLA-DRB1 * 03:01 | 283–297 | RSTIQRDAQLRYNIK | Consensus (smm/nn/sturniolo) | 0.14 | 0.443 |
| HLA-DRB1 * 07:01 | 340–354 | NRTRLFIESPASHLL | Consensus (comb.lib./smm/nn) | 0.54 | 0.480 | |
| HLA-DRB1 * 15:01 | 434–448 | TDWLMLFGSYAQAFR | Consensus (smm/nn/sturniolo) | 0.86 | 0.679 | |
| Flagellin | HLA-DRB1 * 07:01 | 25–39 | NAKSSQRLSTGFRIN | Consensus (comb.lib./smm/nn) | 3.1 | 0.461 |
| HLA-DRB1 * 15:01 | 382–392 | QSSVMMLKKANAATQ | Consensus (smm/nn/sturniolo) | 7.9 | −0.444 | |
| Chaperone protein PsaB | HLA-DRB5 * 01:01 | 89–103 | APFIVTPPLFRLDAG | Consensus (smm/nn/sturniolo) | 0.77 | 0.722 |
| HLA-DRB1 * 15:01 | 181–195 | ADSLTWRYKGNYLEV | Consensus (smm/nn/sturniolo) | 4.2 | 0.504 |
| Protein | Sequence | Starting Position | Score |
|---|---|---|---|
| Chaperone protein PsaB | APFIVTPPLFRLDAGL | 89 | 0.95 |
| CLTGIPPKNGDAWGNT | 128 | 0.94 | |
| YPSSSTKGVSVSVANP | 54 | 0.91 | |
| SLTWRYKGNYLEVNNP | 183 | 0.90 | |
| Histidine kinase | SWPIKTHLPLSADQNV | 411 | 0.94 |
| SGHLDERTHFDPTSSL | 167 | 0.91 | |
| YERPE Hemin receptor | PVSILAGTRYDNYSGS | 396 | 0.94 |
| YETVDAADMLQPGQNS | 164 | 0.93 | |
| KDYISTRVDMQAMTTT | 520 | 0.92 | |
| SRVSSSTPQAGYGVND | 612 | 0.91 | |
| NWDLAYNRTRGKNQNT | 559 | 0.91 | |
| TRDIGNIRQSNGFNAP | 215 | 0.91 | |
| GLTLTNYWVPNPNLKP | 469 | 0.90 | |
| PTMGEMYNDSKHFAIP | 450 | 0.90 | |
| SGSSDGYADVDADKWS | 409 | 0.90 | |
| GWLQDEITLRDLPVSI | 384 | 0.90 | |
| ARPQGSAEEGREQTTE | 319 | 0.90 | |
| YERPE Flagellin | SDVIDAYGAFRATLGA | 321 | 0.95 |
| GFRINSPADNAAGLQI | 35 | 0.93 | |
| LGSIKDTDFADEMKNH | 359 | 0.92 | |
| KQEIETPKAVNEYVVK | 280 | 0.92 | |
| AESVKTLNAMKKLATQ | 359 | 0.91 | |
| Putative exported protein | HVSQASPDDRKKRKAD | 41 | 0.96 |
| GPVSKKTTEPRKTGNN | 69 | 0.93 | |
| SGLIYYAYKDVVKIKM | 187 | 0.92 | |
| GKFIQSPRTGEEIRIS | 249 | 0.91 | |
| TSSIRTAKTPYGRQRN | 113 | 0.9 |
| Model | GDT-HA | RMSD | MolProbity | Clash Score | Poor Rotamers | Rama Favored |
|---|---|---|---|---|---|---|
| MODEL 1 | 0.949 | 0.42 | 1.42 | 7.81 | 0.62 | 98.31 |
| MODEL 2 | 0.958 | 0.40 | 1.40 | 7.31 | 0.61 | 98.54 |
| MODEL 3 | 0.948 | 0.42 | 1.46 | 8.72 | 0.92 | 98.32 |
| MODEL 4 | 0.961 | 0.39 | 1.46 | 8.63 | 0.64 | 98.52 |
| MODEL 5 | 0.943 | 0.42 | 1.46 | 8.61 | 0.66 | 98.33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haq, A.U.; Khan, A.; Khan, J.; Irum, S.; Waheed, Y.; Ahmad, S.; Nizam-Uddin, N.; Albutti, A.; Zaman, N.; Hussain, Z.; et al. Annotation of Potential Vaccine Targets and Design of a Multi-Epitope Subunit Vaccine against Yersinia pestis through Reverse Vaccinology and Validation through an Agent-Based Modeling Approach. Vaccines 2021, 9, 1327. https://doi.org/10.3390/vaccines9111327
Haq AU, Khan A, Khan J, Irum S, Waheed Y, Ahmad S, Nizam-Uddin N, Albutti A, Zaman N, Hussain Z, et al. Annotation of Potential Vaccine Targets and Design of a Multi-Epitope Subunit Vaccine against Yersinia pestis through Reverse Vaccinology and Validation through an Agent-Based Modeling Approach. Vaccines. 2021; 9(11):1327. https://doi.org/10.3390/vaccines9111327
Chicago/Turabian StyleHaq, Azaz Ul, Abbas Khan, Jafar Khan, Shamaila Irum, Yasir Waheed, Sajjad Ahmad, N. Nizam-Uddin, Aqel Albutti, Nasib Zaman, Zahid Hussain, and et al. 2021. "Annotation of Potential Vaccine Targets and Design of a Multi-Epitope Subunit Vaccine against Yersinia pestis through Reverse Vaccinology and Validation through an Agent-Based Modeling Approach" Vaccines 9, no. 11: 1327. https://doi.org/10.3390/vaccines9111327
APA StyleHaq, A. U., Khan, A., Khan, J., Irum, S., Waheed, Y., Ahmad, S., Nizam-Uddin, N., Albutti, A., Zaman, N., Hussain, Z., Ali, S. S., Waseem, M., Kanwal, F., Wei, D.-Q., & Wang, Q. (2021). Annotation of Potential Vaccine Targets and Design of a Multi-Epitope Subunit Vaccine against Yersinia pestis through Reverse Vaccinology and Validation through an Agent-Based Modeling Approach. Vaccines, 9(11), 1327. https://doi.org/10.3390/vaccines9111327