
Small Molecule Inhibitors of MERTK and FLT3 Induce Cell Cycle Arrest in Human CD8+ T Cells

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. T Cell Isolation and Stimulation
2.2. Flow Cytometry
2.3. Cytokine Measurements
2.4. Real-Time qPCR
2.5. siRNA Knockdown of FLT3
2.6. Statistical Analysis
3. Results
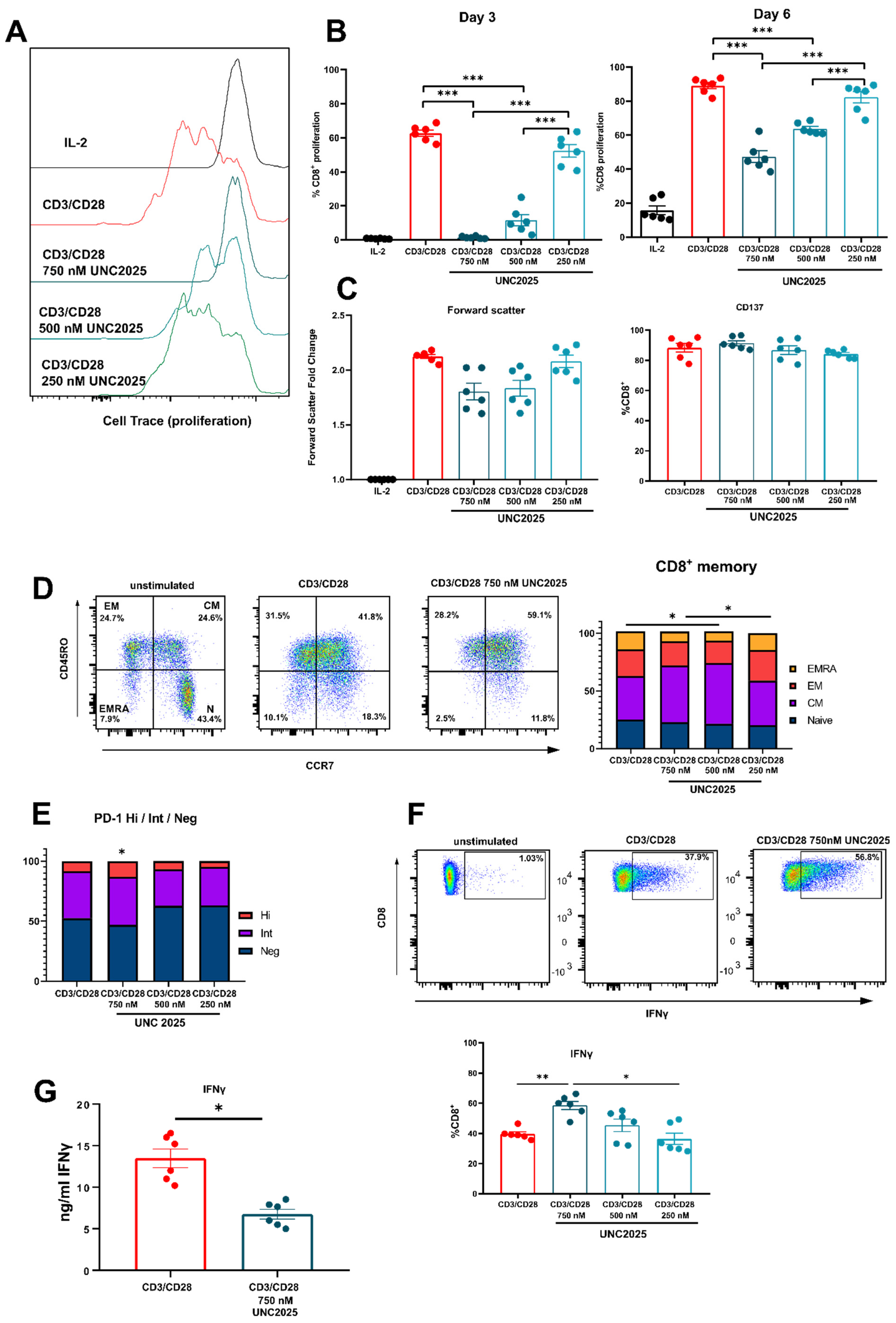
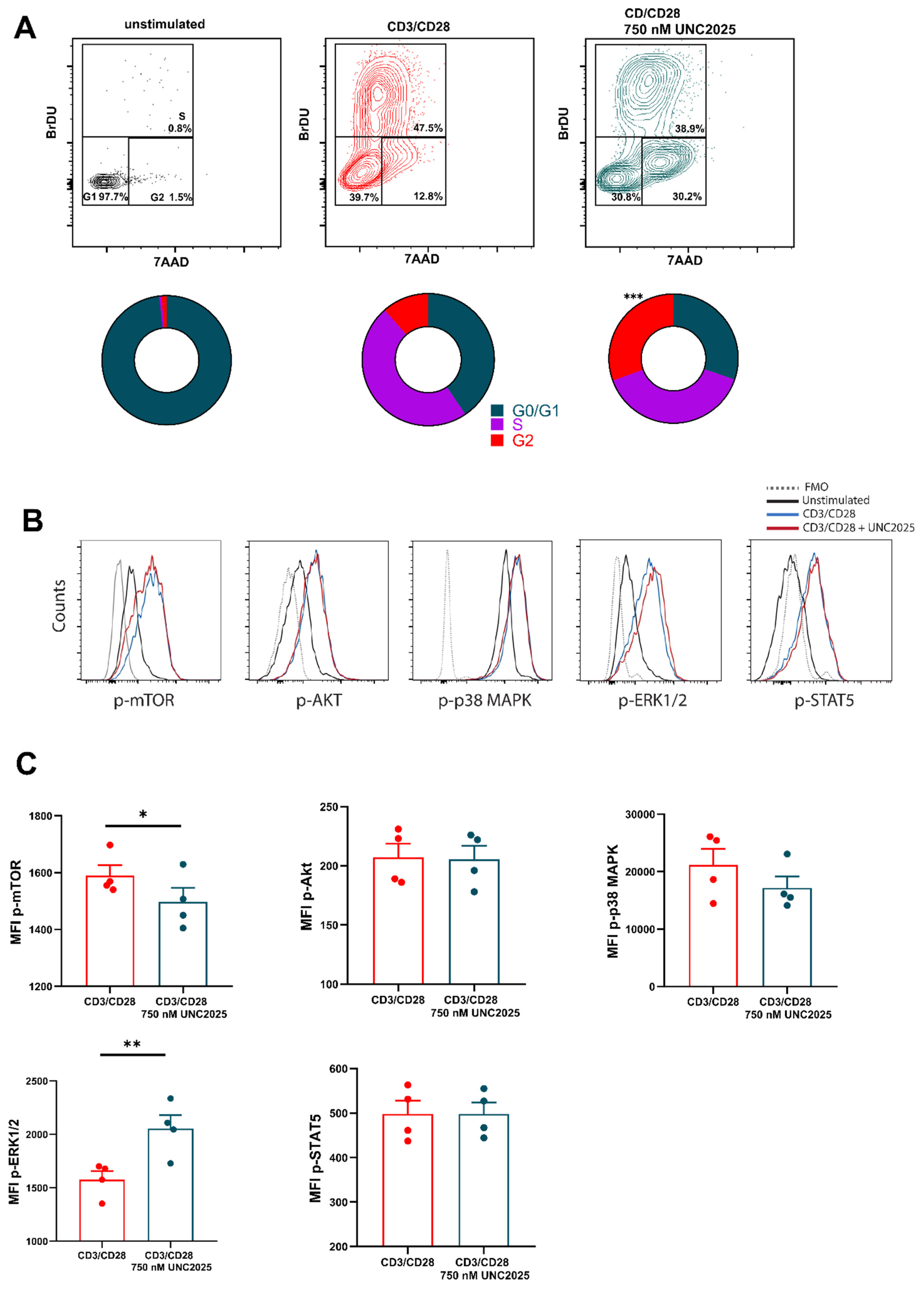
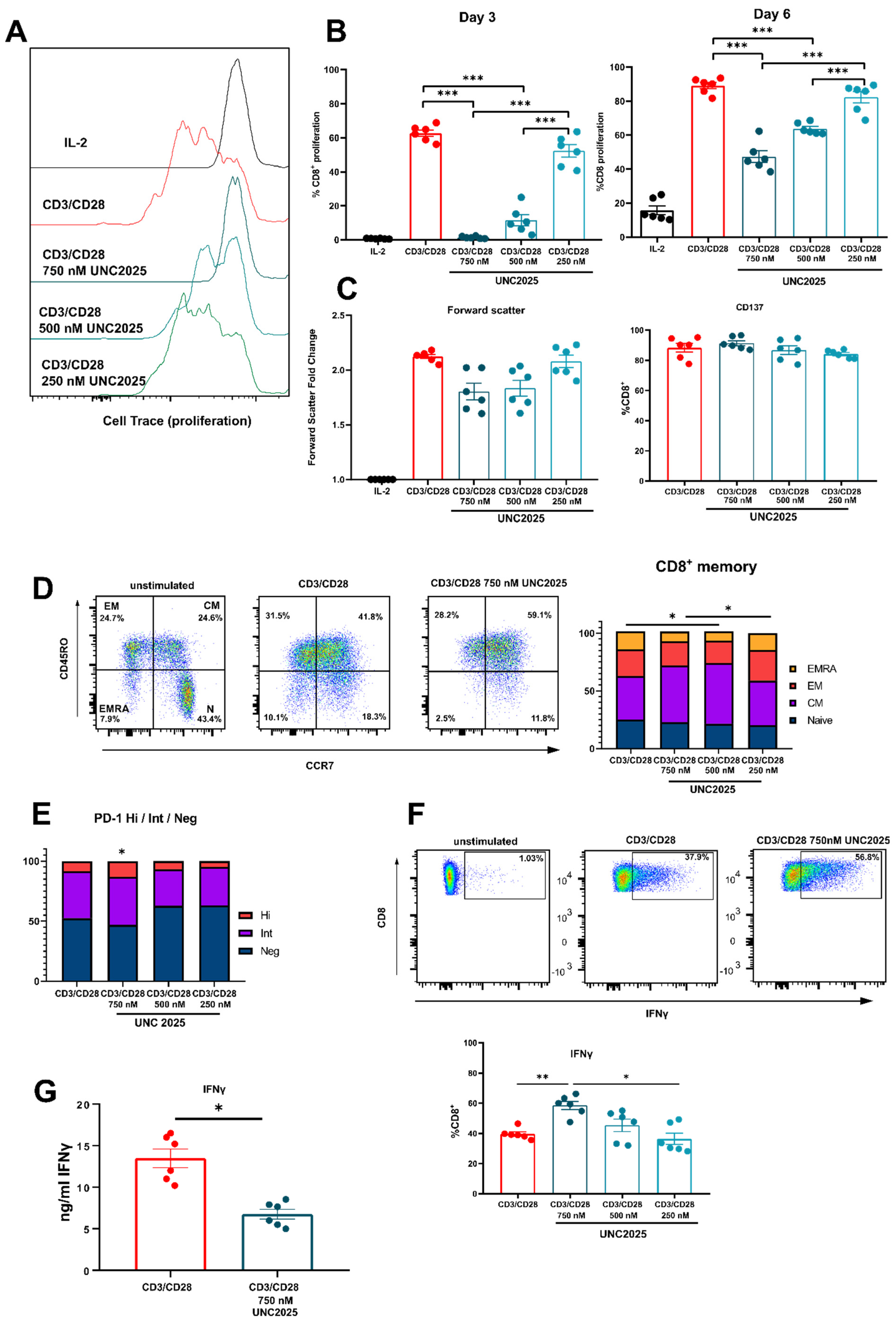
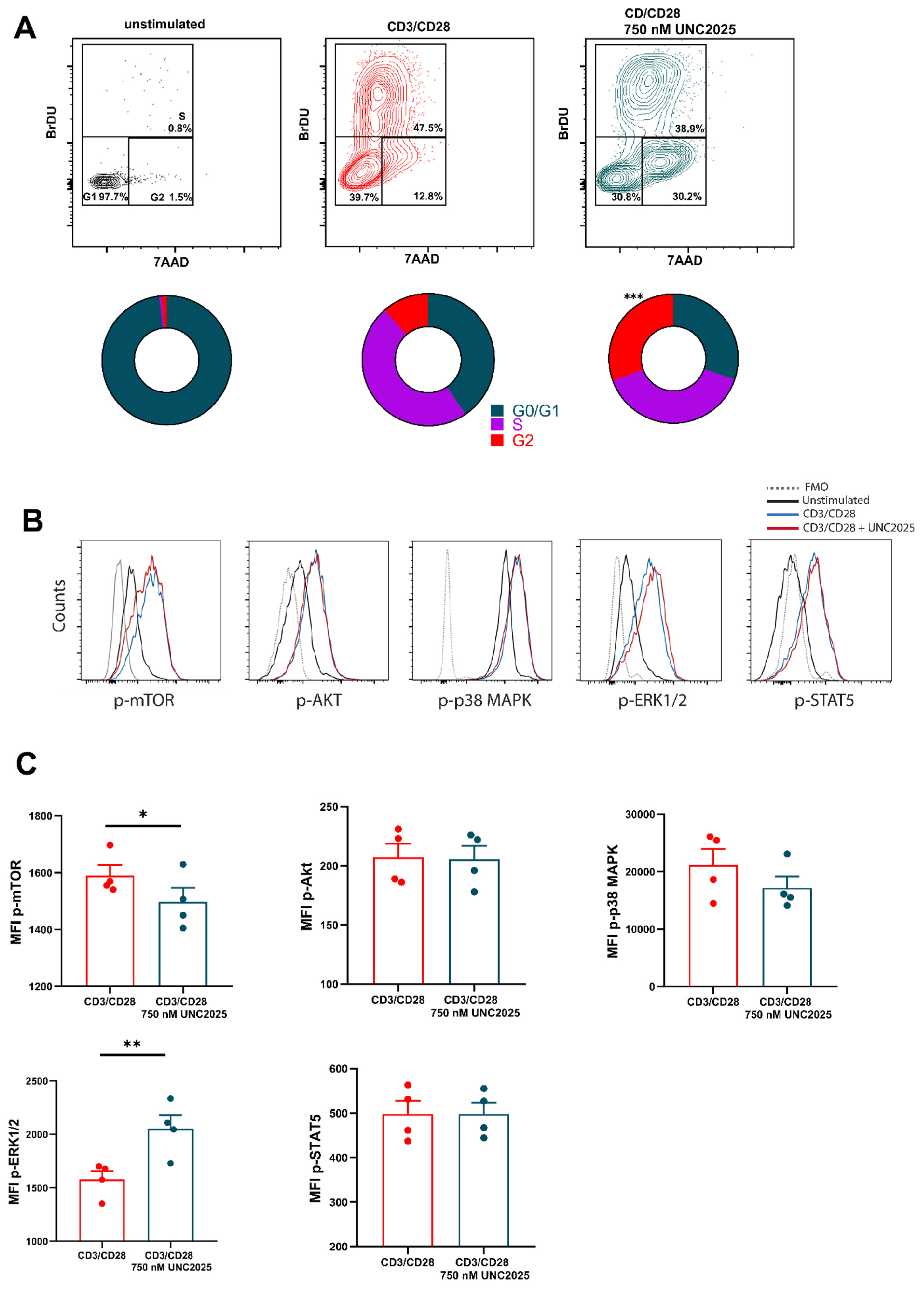
3.1. Dual MERTK/FLT3 Inhibitor UNC2025 Inhibits CD8+ T Cell Proliferation
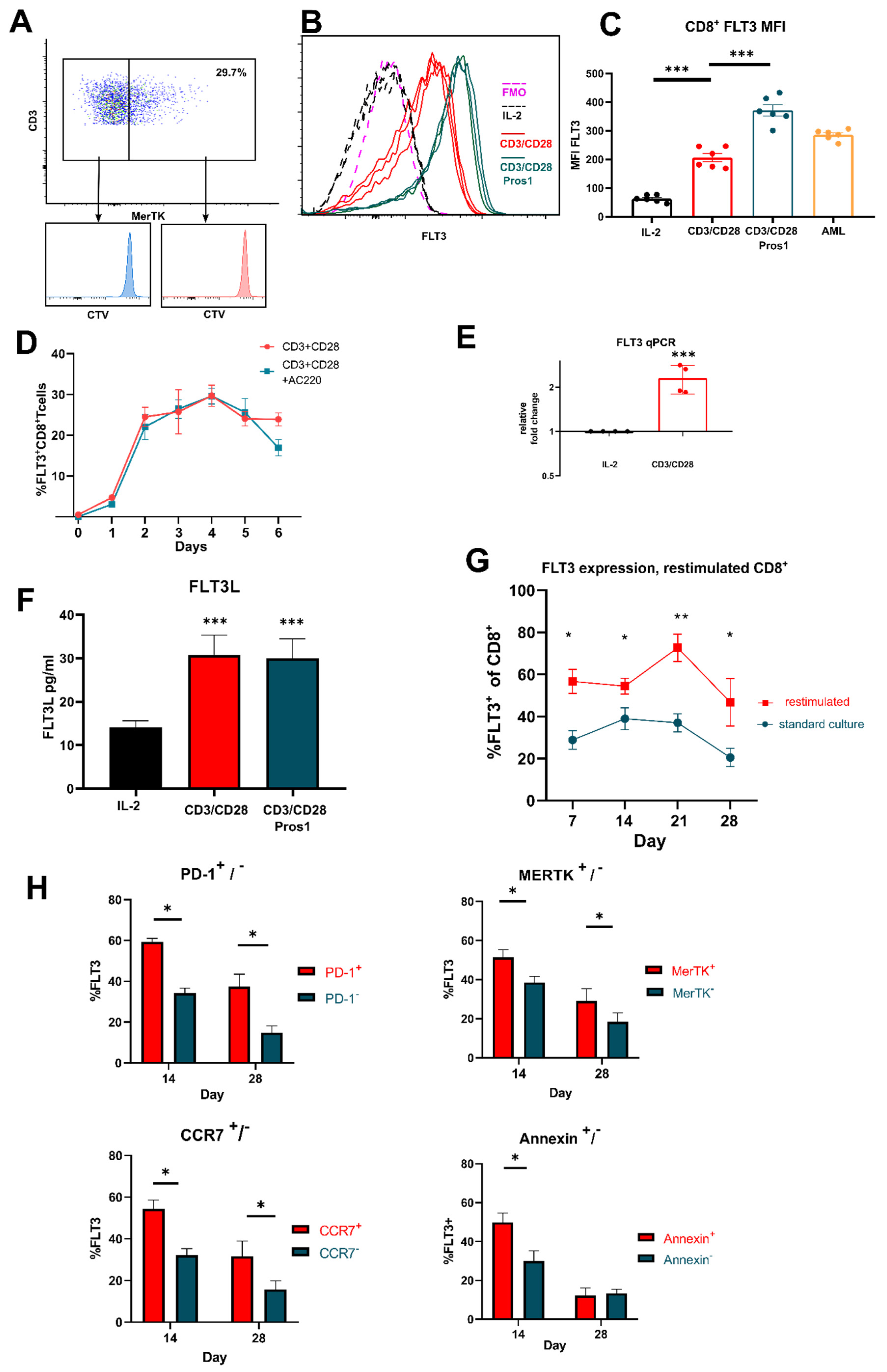
3.2. FLT3 Is Expressed by Activated T Cells
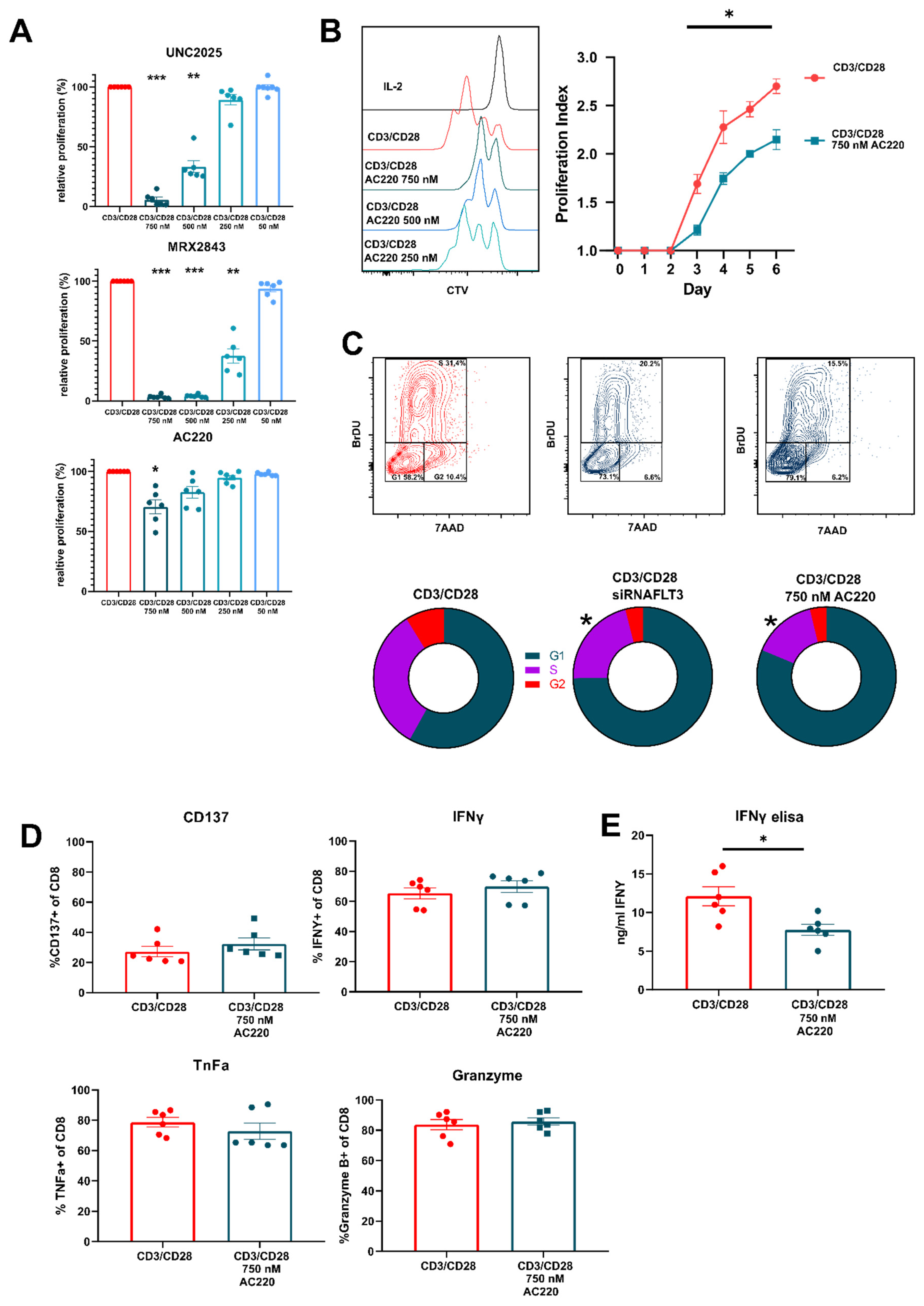
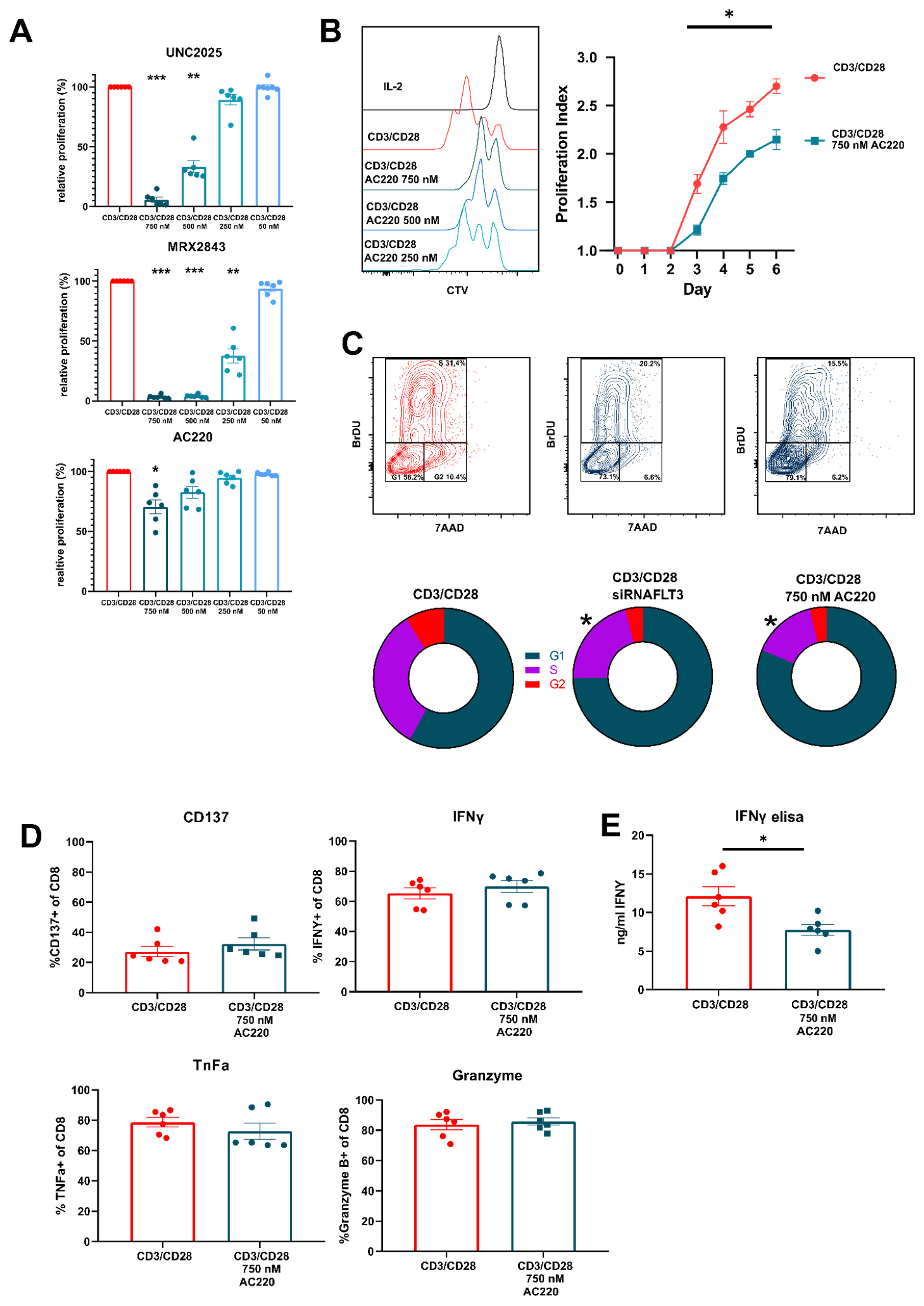
3.3. FLT3 Inhibitor AC220 Impacts CD8+ T Cell Cycle Kinetics
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Fei, F.; Yu, Y.; Schmitt, A.; Rojewski, M.T.; Chen, B.; Greiner, J.; Götz, M.; Guillaume, P.; Döhner, H.; Bunjes, D.; et al. Dasatinib exerts an immunosuppressive effect on CD8+ T cells specific for viral and leukemia antigens. Exp. Hematol. 2008, 36, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Schade, A.E.; Schieven, G.L.; Townsend, R.; Jankowska, A.M.; Susulic, V.; Zhang, R.; Szpurka, H.; Maciejewski, J.P. Dasatinib, a small-molecule protein tyrosine kinase inhibitor, inhibits T-cell activation and proliferation. Blood 2008, 111, 1366. [Google Scholar] [CrossRef] [Green Version]
- Zarrinkar, P.P.; Gunawardane, R.N.; Cramer, M.D.; Gardner, M.F.; Brigham, D.; Belli, B.; Karaman, M.W.; Pratz, K.W.; Pallares, G.; Chao, Q.; et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood 2009, 114, 2984–2992. [Google Scholar] [CrossRef] [PubMed]
- DeRyckere, D.; Lee-Sherick, A.B.; Huey, M.G.; Hill, A.A.; Tyner, J.W.; Jacobsen, K.M.; Page, L.S.; Kirkpatrick, G.G.; Eryildiz, F.; Montgomery, S.A.; et al. UNC2025, a MERTK small-molecule inhibitor, is therapeutically effective alone and in combination with methotrexate in leukemia models. Clin. Cancer Res. 2017, 23, 1481–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosurgi, L.; Bernink, J.H.; Cuevas, V.D.; Gagliani, N.; Joannas, L.; Schmid, E.T.; Booth, C.J.; Ghosh, S.; Rothlin, C.V. Paradoxical role of the proto-oncogene Axl and Mer receptor tyrosine kinases in colon cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 13091–13096. [Google Scholar] [CrossRef] [Green Version]
- Tworkoski, K.; Singhal, G.; Szpakowski, S.; Zito, C.I.; Bacchiocchi, A.; Muthusamy, V.; Bosenberg, M.; Krauthammer, M.; Halaban, R.; Stern, D.F. Phosphoproteomic screen identifies potential therapeutic targets in melanoma. Mol. Cancer Res. 2011, 9, 801–812. [Google Scholar] [CrossRef] [Green Version]
- Lee-Sherick, A.B.; Eisenman, K.M.; Sather, S.; McGranahan, A.; Armistead, P.M.; McGary, C.S.; Hunsucker, S.A.; Schlegel, J.; Martinson, H.; Cannon, C.; et al. Aberrant Mer receptor tyrosine kinase expression contributes to leukemogenesis in acute myeloid leukemia. Oncogene 2013, 32, 5359–5368. [Google Scholar] [CrossRef] [Green Version]
- Graham, D.K.; Deryckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef] [PubMed]
- Sinik, L.; Minson, K.A.; Tentler, J.J.; Carrico, J.; Bagby, S.M.; Robinson, W.A.; Kami, R.; Burstyn-Cohen, T.; Gail Eckhardt, S.; Wang, X.; et al. Inhibition of MERTK promotes suppression of tumor growth in BRAF mutant and BRAF wild-type melanoma. Mol. Cancer Ther. 2019, 18, 278–288. [Google Scholar] [CrossRef] [Green Version]
- Minson, K.A.; Smith, C.C.; DeRyckere, D.; Libbrecht, C.; Lee-Sherick, A.B.; Huey, M.G.; Lasater, E.A.; Kirkpatrick, G.D.; Stashko, M.A.; Zhang, W.; et al. The MERTK/FLT3 inhibitor MRX-2843 overcomes resistance-conferring FLT3 mutations in acute myeloid leukemia. JCI Insight 2016, 1, e85630. [Google Scholar] [CrossRef] [PubMed]
- Sufit, A.; Lee-Sherick, A.B.; Deryckere, D.; Rupji, M.; Dwivedi, B.; Varella-Garcia, M.; Pierce, A.M.; Kowalski, J.; Wang, X.; Frye, S.V.; et al. MERTK inhibition induces polyploidy and promotes cell death and cellular senescence in glioblastoma multiforme. PLoS ONE 2016, 11, e0165107. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Lew, E.D.; Seelige, R.; Tindall, E.A.; Walsh, C.; Fagan, P.C.; Lee, J.Y.; Nevarez, R.; Oh, J.; Tucker, K.D.; et al. Immuno-oncological Efficacy of RXDX-106, a Novel TAM (TYRO3, AXL, MER) Family Small-Molecule Kinase Inhibitor. Cancer Res. 2019, 79, 1996–2008. [Google Scholar] [CrossRef] [Green Version]
- Lee-Sherick, A.B.; Jacobsen, K.M.; Henry, C.J.; Huey, M.G.; Parker, R.E.; Page, L.S.; Hill, A.A.; Wang, X.; Frye, S.V.; Earp, H.S.; et al. MERTK inhibition alters the PD-1 axis and promotes anti-leukemia immunity. JCI Insight 2018, 3, e97941. [Google Scholar] [CrossRef]
- Carow, C.E.; Levenstein, M.; Kaufmann, S.H.; Chen, J.; Amin, S.; Rockwell, P.; Witte, L.; Borowitz, M.J.; Civin, C.I.; Small, D. Expression of the Hematopoietic Growth Factor Receptor FLT3 (STK-1/Flk2) in Human Leukemias. Blood 1996, 87, 1089–1096. [Google Scholar] [CrossRef] [Green Version]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Tabe, Y.; Lu, H.; Borthakur, G.; Miida, T.; Kantarjian, H.; Andreeff, M.; Konopleva, M. Mechanisms of apoptosis induction by simultaneous inhibition of PI3K and FLT3-ITD in AML cells in the hypoxic bone marrow microenvironment. Cancer Lett. 2013, 329, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peschel, I.; Podmirseg, S.R.; Taschler, M.; Duyster, J.; Götze, K.S.; Sill, H.; Nachbaur, D.; Jäkel, H.; Hengst, L. FLT3 and FLT3-ITD phosphorylate and inactivate the cyclin-dependent kinase inhibitor p27Kip1 in acute myeloid leukemia. Haematologica 2017, 102, 1378–1389. [Google Scholar] [CrossRef] [Green Version]
- Kampa-Schittenhelm, K.M.; Heinrich, M.C.; Akmut, F.; Döhner, H.; Döhner, K.; Schittenhelm, M.M. Quizartinib (AC220) is a potent second generation class III tyrosine kinase inhibitor that displays a distinct inhibition profile against mutant-FLT3, -PDGFRA and -KIT isoforms. Mol. Cancer 2013, 12, 19. [Google Scholar] [CrossRef]
- Majothi, S.; Adams, D.; Loke, J.; Stevens, S.P.; Wheatley, K.; Wilson, J.S. FLT3 inhibitors in acute myeloid leukaemia: Assessment of clinical effectiveness, adverse events and future research-a systematic review and meta-analysis. Syst. Rev. 2020, 9, 285. [Google Scholar] [CrossRef]
- Scott, R.S.; McMahon, E.J.; Pop, S.M.; Reap, E.A.; Caricchio, R.; Cohen, P.L.; Earp, H.S.; Matsushima, G.K. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 2001, 411, 207–211. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.A.; Lemke, G. TAM Receptors Are Pleiotropic Inhibitors of the Innate Immune Response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabezón, R.; Carrera-Silva, E.A.; Flórez-Grau, G.; Errasti, A.E.; Calderón-Gómez, E.; Lozano, J.J.; España, C.; Ricart, E.; Panés, J.; Rothlin, C.V.; et al. MERTK as negative regulator of human T cell activation. J. Leukoc. Biol. 2015, 97, 751–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, M.J.W.; Dulkeviciute, D.; Draghi, A.; Ritter, C.; Rahbech, A.; Skadborg, S.K.; Seremet, T.; Carnaz Simões, A.M.; Martinenaite, E.; Halldórsdóttir, H.R.; et al. MERTK Acts as a Costimulatory Receptor on Human CD8+ T Cells. Cancer Immunol. Res. 2019, 7, 1472–1484. [Google Scholar] [CrossRef] [Green Version]
- Matthews, W.; Jordan, C.; Wiegand, G.W.; Pardoll, D.; Lemischka, I.R. A receptor tyrosine kinase specific to hematopoietic stem and progenitor cell-enriched populations. Cell 1991, 65, 1143–1152. [Google Scholar] [CrossRef]
- Ray, R.J.; Paige, C.J.; Furlonger, C.; Lyman, S.D.; Rottapel, R. Flt3 ligand supports the differentiation of early B cell progenitors in the presence of interleukin-11 and interleukin-7. Eur. J. Immunol. 1996, 26, 1504–1510. [Google Scholar] [CrossRef]
- Waskow, C.; Liu, K.; Darrasse-Jeze, G.; Guermonprez, P.; Ginhoux, F.; Merad, M.; Shengelia, T.; Yao, K.; Nussenzweig, M. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat. Immunol. 2008, 9, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Liu, K.; Helft, J.; Bogunovic, M.; Greter, M.; Hashimoto, D.; Price, J.; Yin, N.; Bromberg, J.; Lira, S.A.; et al. The origin and development of nonlymphoid tissue CD103+ DCs. J. Exp. Med. 2009, 206, 3115–3130. [Google Scholar] [CrossRef] [Green Version]
- Astier, A.L.; Beriou, G.; Eisenhaure, T.M.; Anderton, S.M.; Hafler, D.A.; Hacohen, N. RNA Interference Screen in Primary Human T Cells Reveals FLT3 as a Modulator of IL-10 Levels. J. Immunol. 2010, 184, 685–693. [Google Scholar] [CrossRef] [Green Version]
- Chklovskaia, E.; Nissen, C.; Landmann, L.; Rahner, C.; Pfister, O.; Wodnar-Filipowicz, A. Cell-surface trafficking and release of flt3 ligand from T lymphocytes is induced by common cytokine receptor γ-chain signaling and inhibited by cyclosporin A. Blood 2001, 97, 1027–1034. [Google Scholar] [CrossRef]
- Kasikara, C.; Davra, V.; Calianese, D.; Geng, K.; Spires, T.E.; Quigley, M.; Wichroski, M.; Sriram, G.; Suarez-Lopez, L.; Yaffe, M.B.; et al. Pan-TAM tyrosine kinase inhibitor BMS-777607 Enhances Anti-PD-1 mAb efficacy in a murine model of triple-negative breast cancer. Cancer Res. 2019, 79, 2669–2683. [Google Scholar] [CrossRef] [Green Version]
- Caetano, M.S.; Younes, A.I.; Barsoumian, H.B.; Quigley, M.; Menon, H.; Gao, C.; Spires, T.; Reilly, T.P.; Cadena, A.P.; Cushman, T.R.; et al. Triple therapy with MeRTK and PD1 inhibition plus radiotherapy promotes abscopal antitumor immune responses. Clin. Cancer Res. 2019, 25, 7576–7584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Fei, M.; Zhang, G.; Liang, W.C.; Lin, W.Y.; Wu, Y.; Piskol, R.; Ridgway, J.; McNamara, E.; Huang, H.; et al. Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived cGAMP. Immunity 2020, 52, 357–373.e9. [Google Scholar] [CrossRef]
- Giroud, P.; Renaudineau, S.; Gudefin, L.; Calcei, A.; Menguy, T.; Rozan, C.; Mizrahi, J.; Caux, C.; Duong, V.; Valladeau-Guilemond, J. Expression of TAM-R in Human Immune Cells and Unique Regulatory Function of MerTK in IL-10 Production by Tolerogenic DC. Front. Immunol. 2020, 11, 564133. [Google Scholar] [CrossRef] [PubMed]
- Guy, C.S.; Vignali, K.M.; Temirov, J.; Bettini, M.; Overacre, A.E.; Smeltzer, M.; Zhang, H.; Huppa, J.B.; Tsai, Y.-H.; Lobry, C.; et al. Distinct T cell receptor signaling pathways drive proliferation and cytokine production in T cells. Nat. Immunol. 2013, 14, 262. [Google Scholar] [CrossRef] [Green Version]
- Wolleschak, D.; Mack, T.S.; Perner, F.; Frey, S.; Schnöder, T.M.; Wagner, M.C.; Höding, C.; Pils, M.C.; Parkner, A.; Kliche, S.; et al. Clinically relevant doses of FLT3-kinase inhibitors quizartinib and midostaurin do not impair T-cell reactivity and function. Haematologica 2014, 99, e90–e93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes, J.; Foran, J.; Ghirdaladze, D.; DeVetten, M.P.; Zodelava, M.; Holman, P.; Levis, M.J.; Kantarjian, H.M.; Borthakur, G.; James, J.; et al. AC220, a Potent, Selective, Second Generation FLT3 Receptor Tyrosine Kinase (RTK) Inhibitor, in a First-in-Human (FIH) Phase 1 AML Study. Blood 2009, 114, 636. [Google Scholar] [CrossRef]
- Zhang, W.; Deryckere, D.; Hunter, D.; Liu, J.; Stashko, M.A.; Minson, K.A.; Cummings, C.T.; Lee, M.; Glaros, T.G.; Newton, D.L.; et al. UNC2025, a Potent and Orally Bioavailable MER/FLT3 Dual Inhibitor. J. Med. Chem. 2014, 57, 7031–7041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, C.T.; Zhang, W.; Davies, K.D.; Kirkpatrick, G.D.; Zhang, D.; DeRyckere, D.; Wang, X.; Frye, S.V.; Earp, H.S.; Graham, D.K. Small molecule inhibition of MERTK is efficacious in non-small cell lung cancer models independent of driver oncogene status. Mol. Cancer Ther. 2015, 14, 2014–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.J.; Dagger, S.A.; Thien, C.B.F.; Wikstrom, M.E.; Langdon, W.Y. Flt3 inhibitor AC220 is a potent therapy in a mouse model of myeloproliferative disease driven by enhanced wild-type Flt3 signaling. Blood 2012, 120, 4049–4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunawardane, R.N.; Nepomuceno, R.R.; Rooks, A.M.; Hunt, J.P.; Ricono, J.M.; Belli, B.; Armstrong, R.C. Transient exposure to quizartinib mediates sustained inhibition of FLT3 signaling while specifically inducing apoptosis in FLT3-activated leukemia cells. Mol. Cancer Ther. 2013, 12, 438–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Kumar, P.; Anreddy, N.; Zhang, Y.K.; Wang, Y.J.; Chen, Y.; Talele, T.T.; Gupta, K.; Trombetta, L.D.; Chen, Z.S. Quizartinib (AC220) reverses ABCG2-mediated multidrug resistance: In vitro and in vivo studies. Oncotarget 2017, 8, 93785–93799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Yang, X.; Knapper, S.; White, P.; Smith, B.D.; Galkin, S.; Small, D.; Burnett, A.; Levis, M. FLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and in vivo. Blood 2011, 117, 3286–3293. [Google Scholar] [CrossRef] [Green Version]
- Sandmaier, B.M.; Khaled, S.; Oran, B.; Gammon, G.; Trone, D.; Frankfurt, O. Results of a phase 1 study of quizartinib as maintenance therapy in subjects with acute myeloid leukemia in remission following allogeneic hematopoietic stem cell transplant. Am. J. Hematol. 2018, 93, 222–231. [Google Scholar] [CrossRef]
- Wu, J.; Frady, L.N.; Bash, R.E.; Cohen, S.M.; Schorzman, A.N.; Su, Y.-T.; Irvin, D.M.; Zamboni, W.C.; Wang, X.; Frye, S.V.; et al. MerTK as a therapeutic target in glioblastoma. Neuro Oncol. 2018, 20, 92. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J.E.; Kantarjian, H.; Foran, J.M.; Ghirdaladze, D.; Zodelava, M.; Borthakur, G.; Gammon, G.; Trone, D.; Armstrong, R.C.; James, J.; et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J. Clin. Oncol. 2013, 31, 3681–3867. [Google Scholar] [CrossRef]
- Frumento, G.; Zuo, J.; Verma, K.; Croft, W.; Ramagiri, P.; Chen, F.E.; Moss, P. CD117 (c-Kit) is expressed during CD8+ T cell priming and stratifies sensitivity to apoptosis according to strength of TCR engagement. Front. Immunol. 2019, 10, 468. [Google Scholar] [CrossRef] [Green Version]
- Mondino, A.; Mueller, D.L. Review: mTOR at the crossroads of T cell proliferation and tolerance. Semin. Immunol. 2007, 19, 162. [Google Scholar] [CrossRef] [Green Version]
- Callahan, M.K.; Masters, G.; Pratilas, C.A.; Ariyan, C.; Katz, J.; Kitano, S.; Russell, V.; Gordon, R.A.; Vyas, S.; Yuan, J.; et al. Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Immunol. Res. 2014, 2, 70–79. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Powell, R.M.; Peeters, M.J.W.; Rahbech, A.; Aehnlich, P.; Seremet, T.; thor Straten, P. Small Molecule Inhibitors of MERTK and FLT3 Induce Cell Cycle Arrest in Human CD8+ T Cells. Vaccines 2021, 9, 1294. https://doi.org/10.3390/vaccines9111294
Powell RM, Peeters MJW, Rahbech A, Aehnlich P, Seremet T, thor Straten P. Small Molecule Inhibitors of MERTK and FLT3 Induce Cell Cycle Arrest in Human CD8+ T Cells. Vaccines. 2021; 9(11):1294. https://doi.org/10.3390/vaccines9111294
Chicago/Turabian StylePowell, Richard M., Marlies J. W. Peeters, Anne Rahbech, Pia Aehnlich, Tina Seremet, and Per thor Straten. 2021. "Small Molecule Inhibitors of MERTK and FLT3 Induce Cell Cycle Arrest in Human CD8+ T Cells" Vaccines 9, no. 11: 1294. https://doi.org/10.3390/vaccines9111294
APA StylePowell, R. M., Peeters, M. J. W., Rahbech, A., Aehnlich, P., Seremet, T., & thor Straten, P. (2021). Small Molecule Inhibitors of MERTK and FLT3 Induce Cell Cycle Arrest in Human CD8+ T Cells. Vaccines, 9(11), 1294. https://doi.org/10.3390/vaccines9111294