
H2-M and H2-O as Targeting Vehicles for the MHC Class II Processing Compartment Promote Antigen-Specific CD4+ T Cell Activation


Abstract
:1. Introduction
2. Materials and Methods
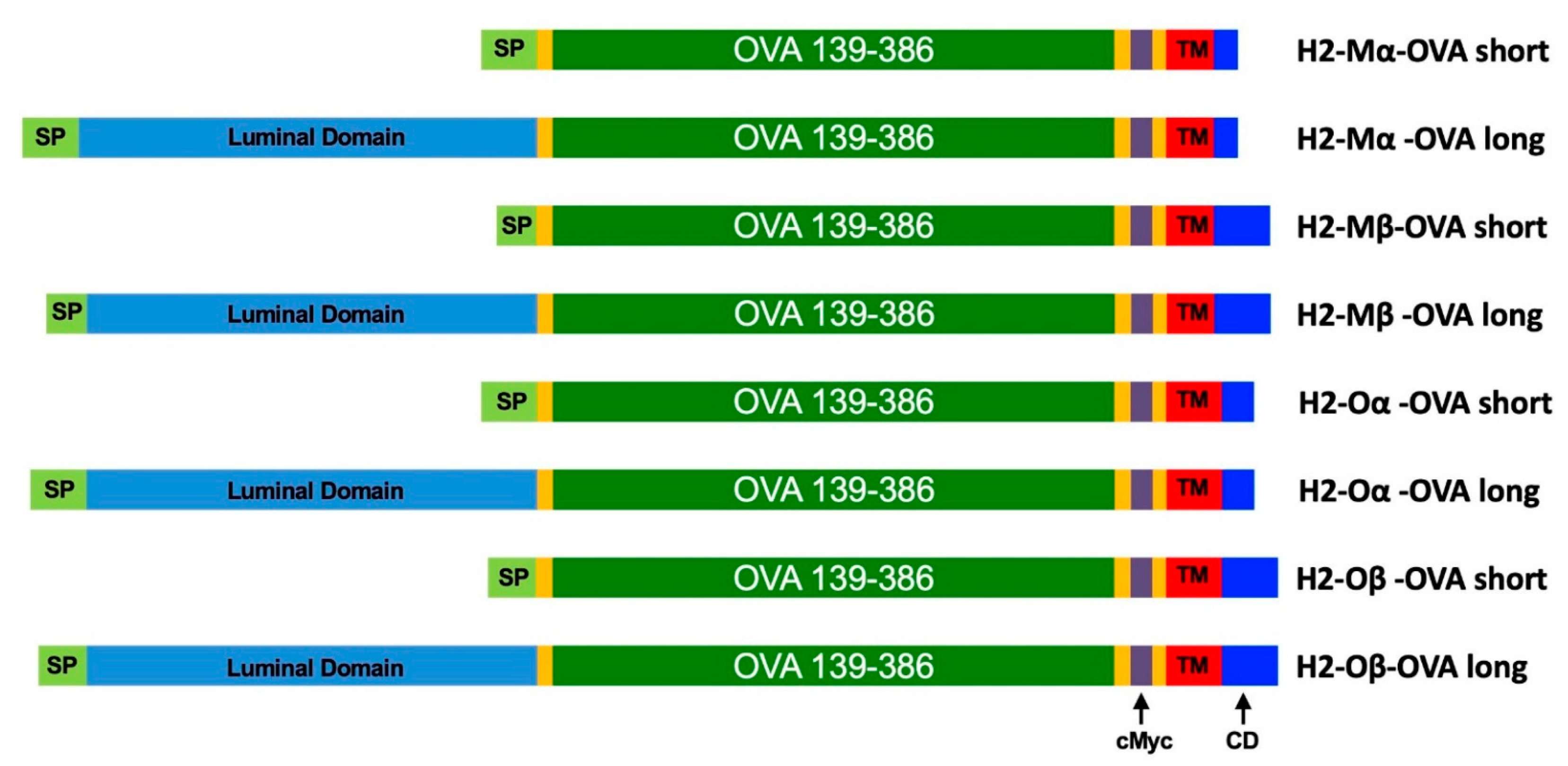
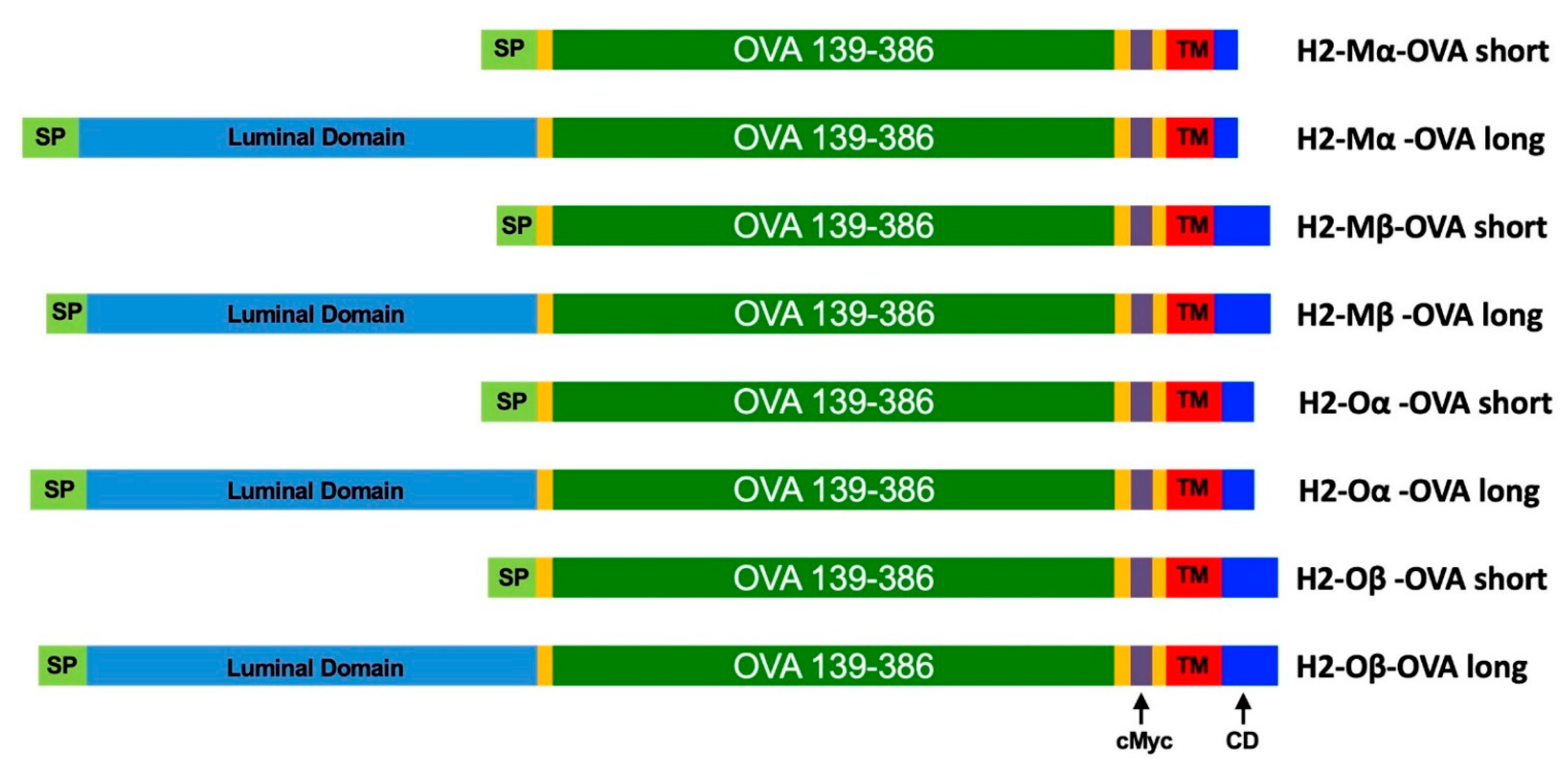
2.1. Design of Fusion Protein Sequences
2.2. mRNA In Vitro Transcription
2.3. Animals
2.4. Cells
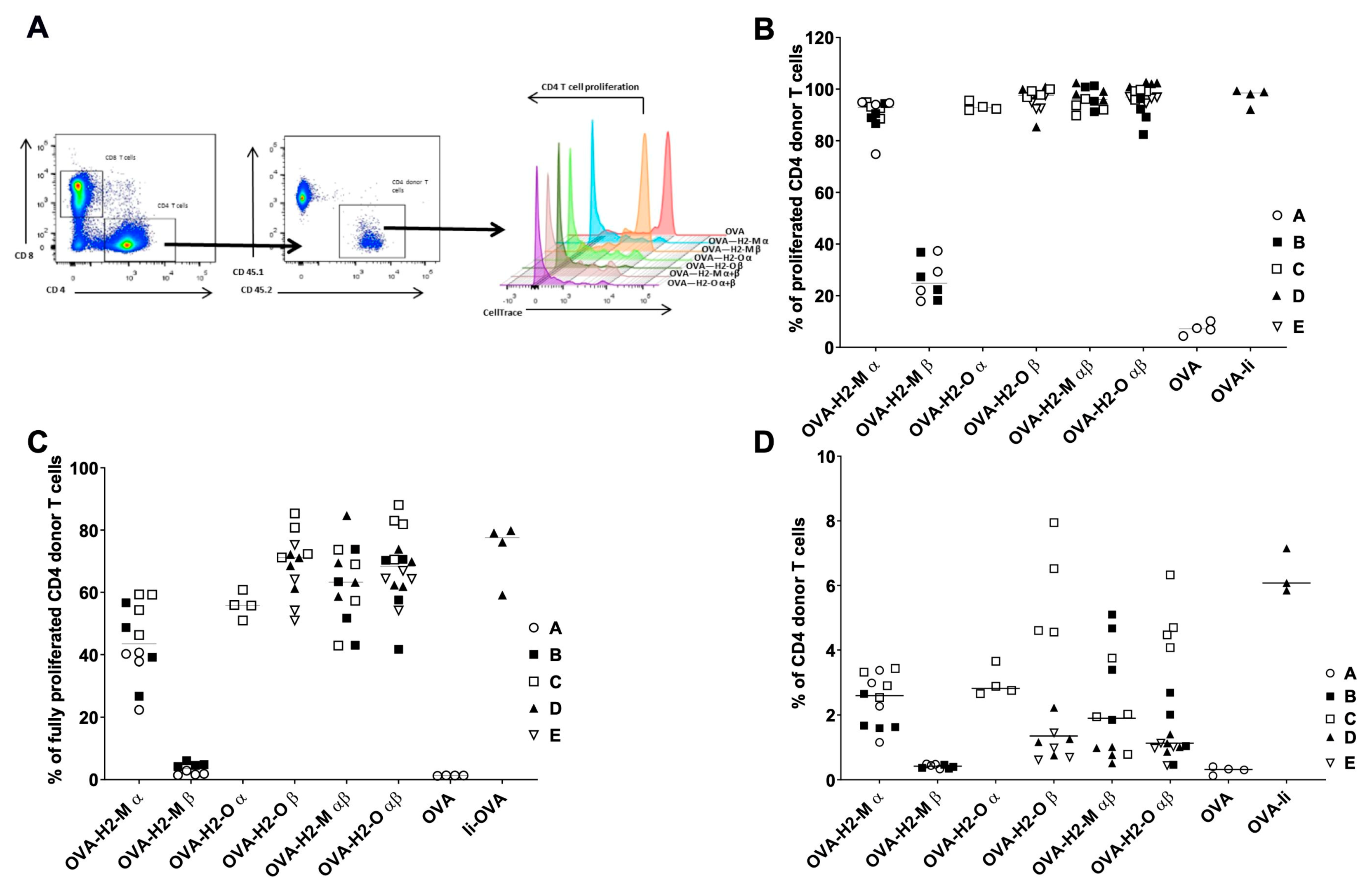
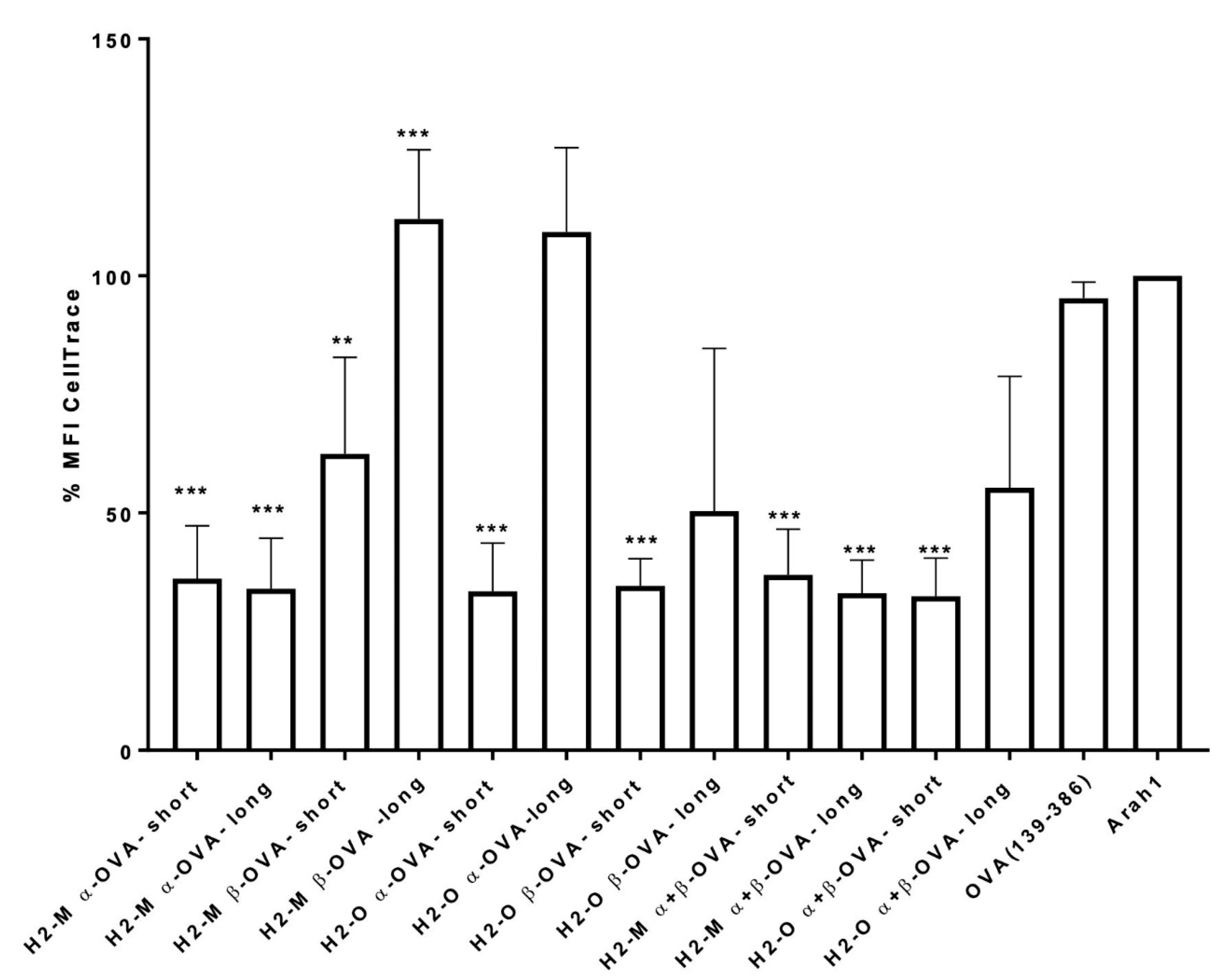
2.5. In Vitro T Cell Proliferation Assay
2.6. In Vivo T Cell Proliferation Model
2.7. Antibodies and Flow Cytometry
2.8. Statistical Analysis
3. Results
3.1. Design and Characterization of mRNAs
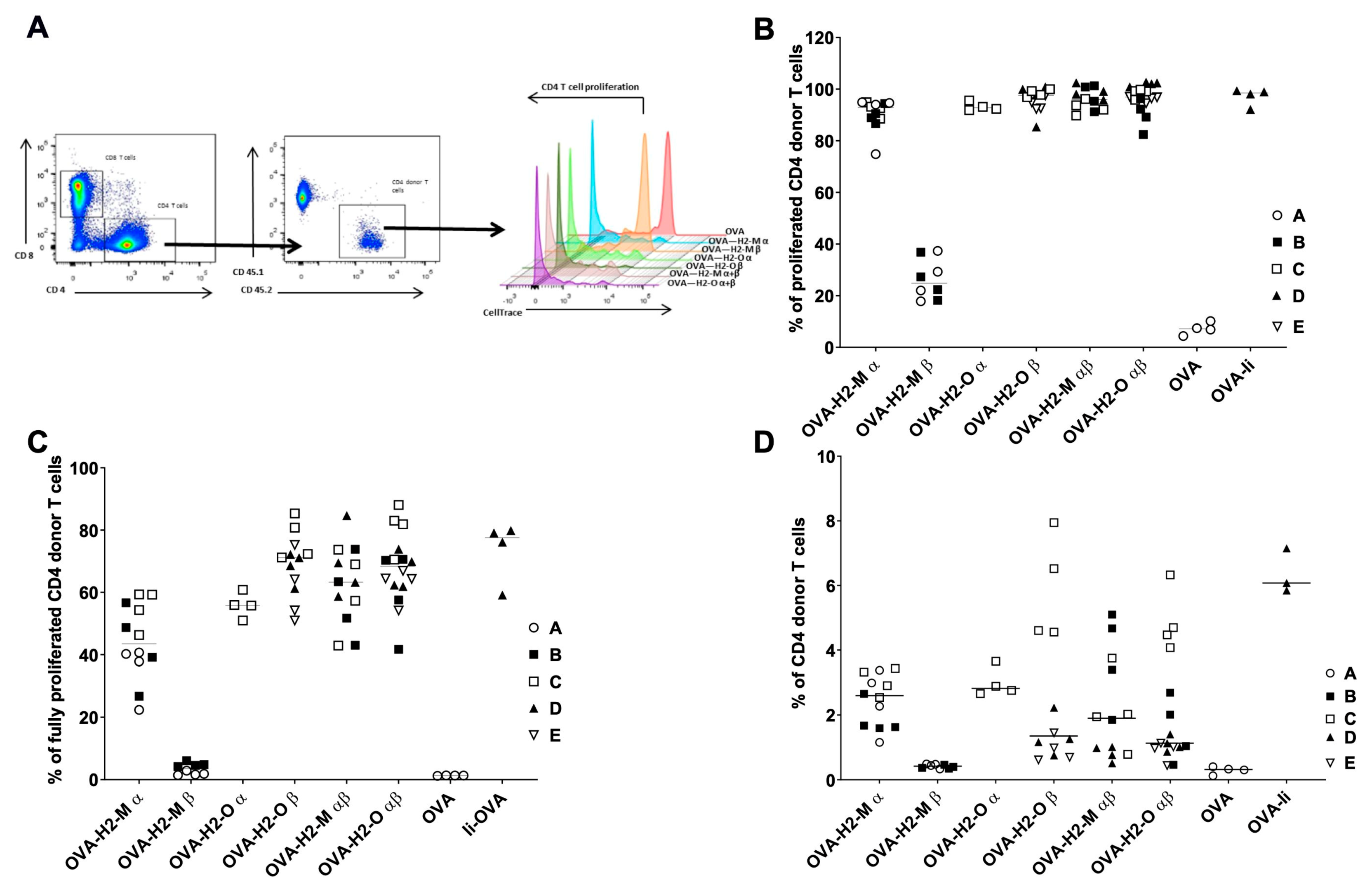
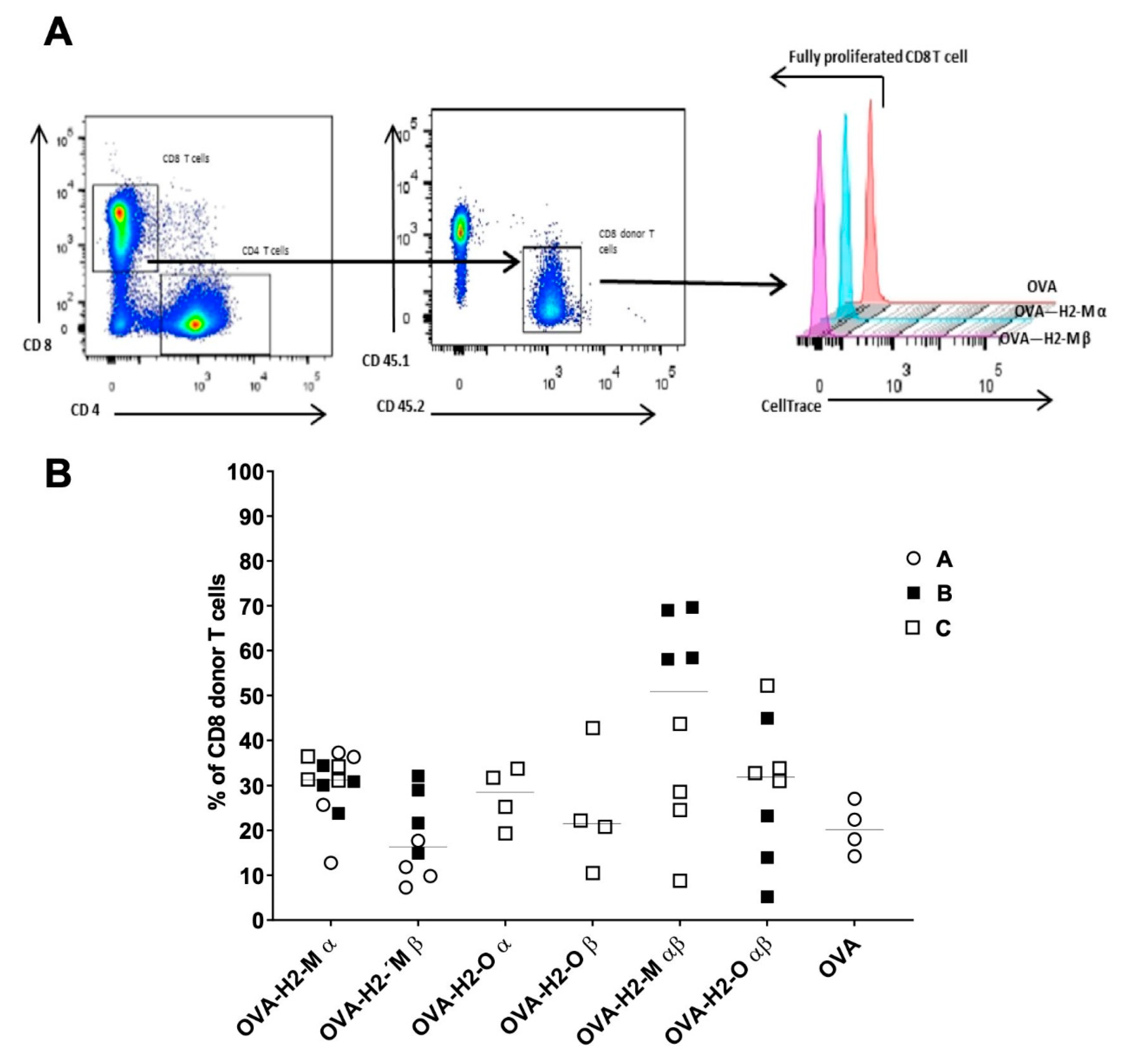
3.2. Enhanced CD4 T-Cell Activation In Vitro Mediated by H2-M and H2-O OVA Fusion mRNAs
3.3. H2-M and H2-O OVA Fusion mRNAs Increase the CD4 T-Cell Activation and Expansion in a Murine In Vivo Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MHC | major histocompatibility complex |
| Ag | antigen |
| APC | Antigen-presenting cell |
| DC | Dendritic cell |
| MFI | Median Fluorescence Intensity |
References
- Bonehill, A.; Heirman, C.; Thielemans, K. Genetic approaches for the induction of a CD4+ T cell response in cancer immunotherapy. J. Gene Med. 2005, 7, 686–695. [Google Scholar] [CrossRef]
- Marchand, M.; van Baren, N.; Weynants, P.; Brichard, V.; Dreno, B.; Tessier, M.H.; Rankin, E.; Parmiani, G.; Arienti, F.; Humblet, Y.; et al. Tumor regressions observed in patients with metastatic melanoma treated with an antigenic peptide encoded by gene MAGE-3 and presented by HLA-A1. Int. J. Cancer 1999, 80, 219–230. [Google Scholar] [CrossRef]
- Nestle, F.O.; Alijagic, S.; Gilliet, M.; Sun, Y.; Grabbe, S.; Dummer, R.; Burg, G.; Schadendorf, D. Vaccination of melanoma patients with peptide-or tumor lysate-pulsed dendritic cells. Nat. Med. 1998, 4, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Schwartzentruber, D.J.; Hwu, P.; Marincola, F.M.; Topalian, S.L.; Restifo, N.P.; Dudley, M.E.; Schwarz, S.L.; Spiess, P.J.; et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat. Med. 1998, 4, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Roche, P.A.; Furuta, K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat. Rev. Immunol. 2015, 15, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Bonehill, A.; Heirman, C.; Tuyaerts, S.; Michiels, A.; Zhang, Y.; van der Bruggen, P.; Thielemans, K. Efficient presentation of known HLA class II-restricted MAGE-A3 epitopes by dendritic cells electroporated with messenger RNA encoding an invariant chain with genetic exchange of class II-associated invariant chain peptide. Cancer Res. 2003, 63, 5587–5594. [Google Scholar]
- Diebold, S.S.; Cotten, M.; Koch, N.; Zenke, M. MHC class II presentation of endogenously expressed antigens by transfected dendritic cells. Gene Ther. 2001, 8, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, S.; Frauwirth, K.; Shastri, N. Expression of endogenous peptide-major histocompatibility complex class II complexes derived from invariant chain-antigen fusion proteins. Proc. Natl. Acad. Sci. USA 1995, 92, 7217–7221. [Google Scholar] [CrossRef] [Green Version]
- Thomson, S.A.; Burrows, S.R.; Misko, I.S.; Moss, D.J.; Coupar, B.E.; Khanna, R. Targeting a polyepitope protein incorporating multiple class II-restricted viral epitopes to the secretory/endocytic pathway facilitates immune recognition by CD4+ cytotoxic T lymphocytes: A novel approach to vaccine design. J. Virol. 1998, 72, 2246–2252. [Google Scholar] [CrossRef] [Green Version]
- Van Meirvenne, S.; Straetman, L.; Heirman, C.; Dullaers, M.; De Greef, C.; Van Tendeloo, V.; Thielemans, K. Efficient genetic modification of murine dendritic cells by electroporation with mRNA. Cancer Gene Ther. 2002, 9, 787–797. [Google Scholar] [CrossRef] [Green Version]
- Bonehill, A.; Heirman, C.; Tuyaerts, S.; Michiels, A.; Breckpot, K.; Brasseur, F.; Zhang, Y.; Van Der Bruggen, P.; Thielemans, K. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J. Immunol. 2004, 172, 6649–6657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, D.M.; Vidard, L.; Rock, K.L. Characterization of MHC class II-presented peptides generated from an antigen targeted to different endocytic compartments. Eur. J. Immunol. 2000, 30, 2333–2343. [Google Scholar] [CrossRef]
- Kim, T.W.; Hung, C.F.; Boyd, D.; Juang, J.; He, L.; Kim, J.W.; Hardwick, J.M.; Wu, T.C. Enhancing DNA vaccine potency by combining a strategy to prolong dendritic cell life with intracellular targeting strategies. J. Immunol. 2003, 171, 2970–2976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lautwein, A.; Burster, T.; Lennon-Dumenil, A.M.; Overkleeft, H.S.; Weber, E.; Kalbacher, H.; Driessen, C. Inflammatory stimuli recruit cathepsin activity to late endosomal compartments in human dendritic cells. Eur. J. Immunol. 2002, 32, 3348–3357. [Google Scholar] [CrossRef]
- Lin, X.; Dashti, A.; Schinazi, R.F.; Tang, J. Intracellular diversion of glycoprotein GP160 of human immunodeficiency virus to lysosomes as a strategy of AIDS gene therapy. FASEB J. 1993, 7, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Parra-Lopez, C.A.; Lindner, R.; Vidavsky, I.; Gross, M.; Unanue, E.R. Presentation on class II MHC molecules of endogenous lysozyme targeted to the endocytic pathway. J. Immunol. 1997, 158, 2670–2679. [Google Scholar]
- Shi, G.P.; Villadangos, J.A.; Dranoff, G.; Small, C.; Gu, L.; Haley, K.J.; Riese, R.; Ploegh, H.L.; Chapman, H.A. Cathepsin S required for normal MHC class II peptide loading and germinal center development. Immunity 1999, 10, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Kreiter, S.; Selmi, A.; Diken, M.; Sebastian, M.; Osterloh, P.; Schild, H.; Huber, C.; Tureci, O.; Sahin, U. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J. Immunol. 2008, 180, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Denzin, L.K.; Cresswell, P. HLA-DM induces CLIP dissociation from MHC class II alpha beta dimers and facilitates peptide loading. Cell 1995, 82, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Amria, S.; Hajiaghamohseni, L.M.; Harbeson, C.; Zhao, D.; Goldstein, O.; Blum, J.S.; Haque, A. HLA-DM negatively regulates HLA-DR4-restricted collagen pathogenic peptide presentation and T cell recognition. Eur. J. Immunol. 2008, 38, 1961–1970. [Google Scholar] [CrossRef]
- Hartman, I.Z.; Kim, A.; Cotter, R.J.; Walter, K.; Dalai, S.K.; Boronina, T.; Griffith, W.; Lanar, D.E.; Schwenk, R.; Krzych, U.; et al. A reductionist cell-free major histocompatibility complex class II antigen processing system identifies immunodominant epitopes. Nat. Med. 2010, 16, 1333–1340. [Google Scholar] [CrossRef] [Green Version]
- Kremer, A.N.; van der Meijden, E.D.; Honders, M.W.; Goeman, J.J.; Wiertz, E.J.; Falkenburg, J.H.; Griffioen, M. Endogenous HLA class II epitopes that are immunogenic in vivo show distinct behavior toward HLA-DM and its natural inhibitor HLA-DO. Blood 2012, 120, 3246–3255. [Google Scholar] [CrossRef] [PubMed]
- Lazarski, C.A.; Chaves, F.A.; Sant, A.J. The impact of DM on MHC class II-restricted antigen presentation can be altered by manipulation of MHC-peptide kinetic stability. J. Exp. Med. 2006, 203, 1319–1328. [Google Scholar] [CrossRef] [Green Version]
- Pu, Z.; Lovitch, S.B.; Bikoff, E.K.; Unanue, E.R. T cells distinguish MHC-peptide complexes formed in separate vesicles and edited by H2-DM. Immunity 2004, 20, 467–476. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Calvo-Calle, J.M.; Dominguez-Amorocho, O.; Stern, L.J. HLA-DM constrains epitope selection in the human CD4 T cell response to vaccinia virus by favoring the presentation of peptides with longer HLA-DM-mediated half-lives. J. Immunol. 2012, 189, 3983–3994. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Reed-Loisel, L.M.; Karlsson, L.; Jensen, P.E. H2-O expression in primary dendritic cells. J. Immunol. 2006, 176, 3548–3556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welsh, R.; Song, N.; Sadegh-Nasseri, S. What to do with HLA-DO/H-2O two decades later? Immunogenetics 2019, 71, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, S.; Azizian, K.T.; Haque, A.; Henderson, J.M.; Hendel, A.; Shore, S.; Antony, J.S.; Hogrefe, R.I.; Kormann, M.S.D.; Porteus, M.H.; et al. Uridine Depletion and Chemical Modification Increase Cas9 mRNA Activity and Reduce Immunogenicity without HPLC Purification. Mol. Ther. Nucleic Acids 2018, 12, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Broos, K.; Van der Jeught, K.; Puttemans, J.; Goyvaerts, C.; Heirman, C.; Dewitte, H.; Verbeke, R.; Lentacker, I.; Thielemans, K.; Breckpot, K. Particle-mediated Intravenous Delivery of Antigen mRNA Results in Strong Antigen-specific T-cell Responses Despite the Induction of Type I Interferon. Mol. Ther. Nucleic Acids 2016, 5, e326. [Google Scholar] [CrossRef] [Green Version]
- Toribio-Fernandez, R.; Zorita, V.; Herrero-Fernandez, B.; Gonzalez-Granado, J.M. An In Vivo Mouse Model to Measure Naïve CD4 T Cell Activation, Proliferation and Th1 Differentiation Induced by Bone Marrow-derived Dendritic Cells. J. Vis.Exp. JoVE 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnden, M.J.; Allison, J.; Heath, W.R.; Carbone, F.R. Defective TCR expression in transgenic mice constructed using cDNA-based alpha-and beta-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 1998, 76, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines-a new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fusion Protein | ORF Length | AA Length | mRNA Size | Reference Sequences | Amino Acids |
|---|---|---|---|---|---|
| H2-M alpha chain-OVA (short) | 1002 | 333 | 1.278 | NP_ 0345516 | 1–28; 230–261 |
| H2-M alpha chain-OVA (long) | 1611 | 536 | 1887 | 1–231; 230–261 | |
| H2-M beta chain-OVA (short) | 1017 | 338 | 1.293 | NP_034517 | 1–18; 217–261 |
| H2-M beta chain-OVA (long) | 1611 | 536 | 1.887 | 1–218; 217–261 | |
| H2-O alpha chain-OVA (short) | 1008 | 335 | 1284 | NP_034518 | 1–27; 216–250 |
| H2-O alpha chain-OVA (long) | 1578 | 525 | 1.854 | 1–217; 216–250 | |
| H2-O beta chain-OVA (short) | 1065 | 354 | 1.284 | NP_034519 | 1–28; 219–271 |
| H2-O beta chain-OVA (long) | 1644 | 547 | 1.920 | 1–221; 219–271 | |
| Ii-OVA | 1434 | 477 | 1.710 | NP_034675 | 1–214 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lapazio, L.; Braun, M.; Grandien, K. H2-M and H2-O as Targeting Vehicles for the MHC Class II Processing Compartment Promote Antigen-Specific CD4+ T Cell Activation. Vaccines 2021, 9, 1053. https://doi.org/10.3390/vaccines9101053
Lapazio L, Braun M, Grandien K. H2-M and H2-O as Targeting Vehicles for the MHC Class II Processing Compartment Promote Antigen-Specific CD4+ T Cell Activation. Vaccines. 2021; 9(10):1053. https://doi.org/10.3390/vaccines9101053
Chicago/Turabian StyleLapazio, Lucia, Monika Braun, and Kaj Grandien. 2021. "H2-M and H2-O as Targeting Vehicles for the MHC Class II Processing Compartment Promote Antigen-Specific CD4+ T Cell Activation" Vaccines 9, no. 10: 1053. https://doi.org/10.3390/vaccines9101053
APA StyleLapazio, L., Braun, M., & Grandien, K. (2021). H2-M and H2-O as Targeting Vehicles for the MHC Class II Processing Compartment Promote Antigen-Specific CD4+ T Cell Activation. Vaccines, 9(10), 1053. https://doi.org/10.3390/vaccines9101053