
Nucleotide Pool Imbalance and Antibody Gene Diversification


{kind=link}
{kind=link}
Abstract
1. Introduction
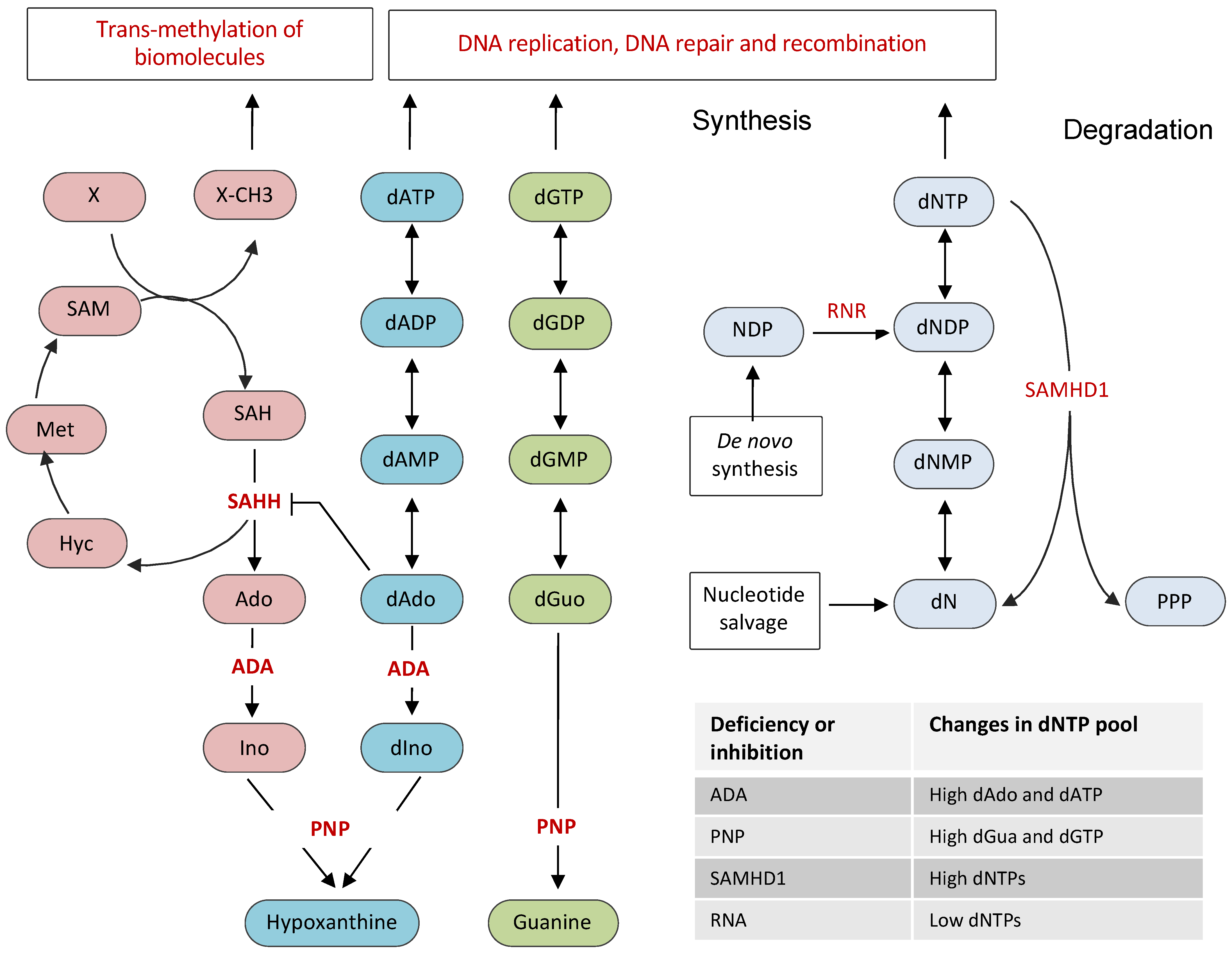
2. Cellular Nucleotide Pool and Its Regulation
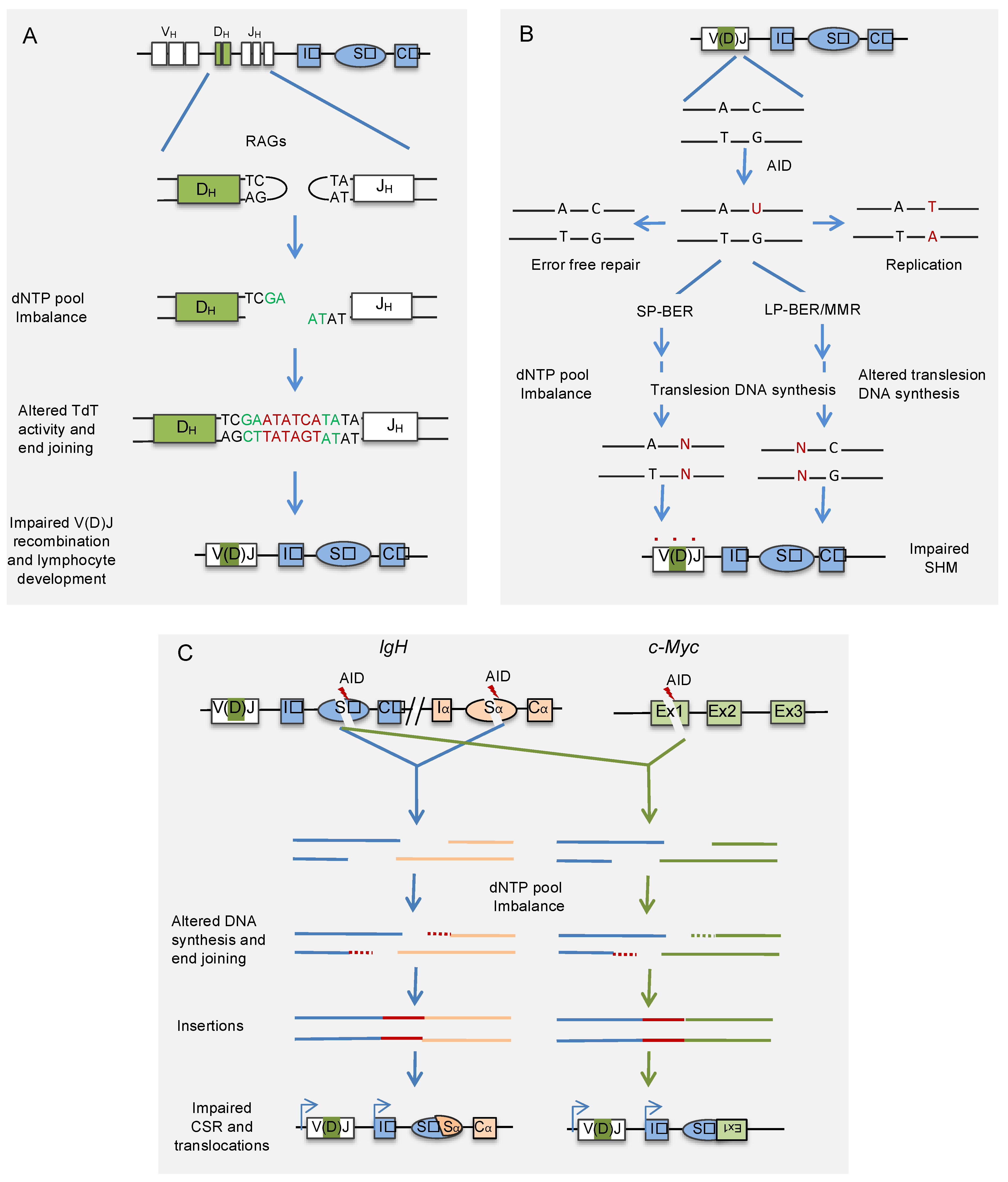
3. Cellular Nucleotide Pool and V(D)J Recombination
4. Cellular Nucleotide Pool and Somatic Hypermutation
5. Cellular Nucleotide Pool and Class Switch Recombination
6. Conclusions
7. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Murphy, K.M.; Weaver, C.; Mowat, A. Janeway’s Immunobiology, 9th ed.; Garland Science: New York, NY, USA, 2017. [Google Scholar]
- Leder, P. The genetics of antibody diversity. Sci. Am. 1982, 246, 102–115. [Google Scholar] [CrossRef]
- Tonegawa, S. Somatic generation of antibody diversity. Nature 1983, 302, 575–581. [Google Scholar] [CrossRef]
- Honjo, T. Immunoglobulin genes. Annu. Rev. Immunol. 1983, 1, 499–528. [Google Scholar] [CrossRef][Green Version]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef]
- Nagaoka, H.; Muramatsu, M.; Yamamura, N.; Kinoshita, K.; Honjo, T. Activation-induced deaminase (AID)-directed hypermutation in the immunoglobulin Sμ region. J. Exp. Med. 2002, 195, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Tarlinton, D.; Good-Jacobson, K. Diversity among memory B cells: Origin, consequences, and utility. Science 2013, 341, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Mathews, C.K. DNA precursor metabolism and genomic stability. FASEB J. 2006, 20, 1300–1314. [Google Scholar] [CrossRef] [PubMed]
- Mathews, C.K. Deoxyribonucleotides as genetic and metabolic regulators. FASEB J. 2014, 28, 3832–3840. [Google Scholar] [CrossRef] [PubMed]
- Mathews, C.K. Deoxyribonucleotide metabolism, mutagenesis and cancer. Nat. Rev. Cancer 2015, 15, 528–539. [Google Scholar] [CrossRef]
- Kunz, B.A.; Kohalmi, S.E.; Kunkel, T.; Mathews, C.K.; McIntosh, E.M.; Reidy, J.A. Deoxyribonucleoside triphosphate levels: A critical factor in the maintenance of genetic stability. Mutat. Res. Genet. Toxicol. 1994, 318, 1–64. [Google Scholar] [CrossRef]
- Meuth, M. The molecular basis of mutations induced by deoxyribonucleoside triphosphate pool imbalances in mammalian cells. Exp. Cell Res. 1989, 181, 305–316. [Google Scholar] [CrossRef]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Nordlund, P.; Reichard, P. Ribonucleotide reductases. Annu. Rev. Biochem. 2006, 75, 681–706. [Google Scholar] [CrossRef] [PubMed]
- Chimploy, K.; Mathews, C.K. Mouse ribonucleotide reductase control. J. Biol. Chem. 2001, 276, 7093–7100. [Google Scholar] [CrossRef] [PubMed]
- Fairman, J.W.; Wijerathna, S.R.; Ahmad, F.; Xu, H.; Nakano, R.; Jha, S.; Prendergast, J.; Welin, R.M.; Flodin, S.; Roos, A.; et al. Structural basis for allosteric regulation of human ribonucleotide reductase by nucleotide-induced oligomerization. Nat. Struct. Mol. Biol. 2011, 18, 316–322. [Google Scholar] [CrossRef]
- Hofer, A.; Crona, M.; Logan, D.; Sjöberg, B.-M. DNA building blocks: Keeping control of manufacture. Crit. Rev. Biochem. Mol. Biol. 2011, 47, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Chabes, A.; Thelander, L. Controlled protein degradation regulates ribonucleotide reductase activity in proliferating mammalian cells during the normal cell cycle and in response to DNA damage and replication blocks. J. Biol. Chem. 2000, 275, 17747–17753. [Google Scholar] [CrossRef] [PubMed]
- Engström, Y.; Rozell, B. Immunocytochemical evidence for the cytoplasmic localization and differential expression during the cell cycle of the M1 and M2 subunits of mammalian ribonucleotide reductase. EMBO J. 1988, 7, 1615–1620. [Google Scholar] [CrossRef]
- Fasullo, M.; Endres, L. Nucleotide salvage deficiencies, DNA damage and neurodegeneration. Int. J. Mol. Sci. 2015, 16, 9431–9449. [Google Scholar] [CrossRef]
- Powell, R.D.; Holland, P.J.; Hollis, T.; Perrino, F.W. Aicardi-Goutières syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J. Biol. Chem. 2011, 286, 43596–43600. [Google Scholar] [CrossRef]
- Goldstone, D.; Ennis-Adeniran, V.; Hedden, J.J.; Groom, H.C.T.; Rice, G.; Christodoulou, E.; Walker, P.A.; Kelly, G.; Haire, L.F.; Yap, M.W.; et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 2011, 480, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Franzolin, E.; Pontarin, G.; Rampazzo, C.; Miazzi, C.; Ferraro, P.; Palumbo, E.; Reichard, P.; Bianchi, V. The deoxynucleotide triphosphohydrolase SAMHD1 is a major regulator of DNA precursor pools in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 14272–14277. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, R.; Schumann, T.; Gerbaulet, A.; Nguyen, L.A.; Schubert, N.; Alexopoulou, D.; Berka, U.; Lienenklaus, S.; Peschke, K.; Gibbert, K.; et al. Mouse SAMHD1 has antiretroviral activity and suppresses a spontaneous cell-intrinsic antiviral response. Cell Rep. 2013, 4, 689–696. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Maelfait, J.; Bridgeman, A.; Rigby, R.; Hayward, B.; Liberatore, R.; Bieniasz, P.D.; Towers, G.; Moita, L.; Crow, Y.; et al. SAMHD1-dependent retroviral control and escape in mice. EMBO J. 2013, 32, 2454–2462. [Google Scholar] [CrossRef] [PubMed]
- Baldauf, H.-M.; Pan, X.; Erikson, E.; Schmidt, S.; Daddacha, W.; Burggraf, M.; Schenkova, K.; Ambiel, I.; Wabnitz, G.H.; Gramberg, T.; et al. SAMHD1 restricts HIV-1 infection in resting CD4+ T cells. Nat. Med. 2012, 18, 1682–1688. [Google Scholar] [CrossRef]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Ségéral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Clifford, R.; Louis, T.; Robbe, P.; Ackroyd, S.; Burns, A.; Timbs, A.T.; Colopy, G.W.; Dreau, H.; Sigaux, F.; Judde, J.G.; et al. SAMHD1 is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood 2014, 123, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Rentoft, M.; Lindell, K.; Tran, P.; Chabes, A.L.; Buckland, R.J.; Watt, D.L.; Marjavaara, L.; Nilsson, A.K.; Melin, B.; Trygg, J.; et al. Heterozygous colon cancer-associated mutations of SAMHD1 have functional significance. Proc. Natl. Acad. Sci. USA 2016, 113, 4723–4728. [Google Scholar] [CrossRef]
- Akimova, E.; Gassner, F.J.; Schubert, M.; Rebhandl, S.; Arzt, C.; Rauscher, S.; Tober, V.; Zaborsky, N.; Greil, R.; Geisberger, R. SAMHD1 restrains aberrant nucleotide insertions at repair junctions generated by DNA end joining. Nucleic Acids Res. 2021, 49, 2598–2608. [Google Scholar] [CrossRef]
- Husain, A.; Xu, J.; Fujii, H.; Nakata, M.; Kobayashi, M.; Wang, J.; Rehwinkel, J.; Honjo, T.; A Begum, N. SAMHD 1-mediated dNTP degradation is required for efficient DNA repair during antibody class switch recombination. EMBO J. 2020, 39, e102931. [Google Scholar] [CrossRef]
- Daddacha, W.; Koyen, A.E.; Bastien, A.J.; Head, P.; Dhere, V.R.; Nabeta, G.N.; Connolly, E.C.; Werner, E.; Madden, M.; Daly, M.B.; et al. SAMHD1 promotes DNA end resection to facilitate DNA repair by homologous recombination. Cell Rep. 2017, 20, 1921–1935. [Google Scholar] [CrossRef] [PubMed]
- Beloglazova, N.; Flick, R.; Tchigvintsev, A.; Brown, G.; Popovic, A.; Nocek, B.; Yakunin, A.F. Nuclease activity of the human SAMHD1 protein implicated in the Aicardi-Goutières syndrome and HIV-1 restriction. J. Biol. Chem. 2013, 288, 8101–8110. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, J.; Choi, J.; Oh, C.; Kim, S.; Seo, M.; Kim, S.-Y.; Seo, D.; Kim, J.; White, T.E.; Brandariz-Nuñez, A.; et al. The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat. Med. 2014, 20, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, J.; Gelais, C.S.; De Silva, S.; Yount, J.; Tang, C.; Ji, X.; Shepard, C.; Xiong, Y.; Kim, B.; Wu, L. SAMHD1-mediated HIV-1 restriction in cells does not involve ribonuclease activity. Nat. Med. 2016, 22, 1072–1074. [Google Scholar] [CrossRef] [PubMed]
- Seamon, K.J.; Bumpus, N.N.; Stivers, J.T. Single-stranded nucleic acids bind to the tetramer interface of samhd1 and prevent formation of the catalytic homotetramer. Biochemistry 2016, 55, 6087–6099. [Google Scholar] [CrossRef]
- Welbourn, S.; Strebel, K. Low dNTP levels are necessary but may not be sufficient for lentiviral restriction by SAMHD1. Virology 2015, 488, 271–277. [Google Scholar] [CrossRef]
- Amie, S.M.; Bambara, R.A.; Kim, B. GTP is the primary activator of the anti-HIV restriction factor SAMHD1. J. Biol. Chem. 2013, 288, 25001–25006. [Google Scholar] [CrossRef]
- Chun, J.J.; Schatz, D.G.; Oettinger, M.A.; Jaenisch, R.; Baltimore, D. The recombination activating gene-1 (RAG-1) transcript is present in the murine central nervous system. Cell 1991, 64, 189–200. [Google Scholar] [CrossRef]
- Oettinger, M.; Schatz, D.; Gorka, C.; Baltimore, D. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science 1990, 248, 1517–1523. [Google Scholar] [CrossRef] [PubMed]
- Schatz, D.G.; Oettinger, M.A.; Baltimore, D. The V(D)J recombination activating gene, RAG-1. Cell 1989, 59, 1035–1048. [Google Scholar] [CrossRef]
- Jung, D.; Alt, F.W. Unraveling V(D)J recombination: Insights into gene regulation. Cell 2004, 116, 299–311. [Google Scholar] [CrossRef]
- Motea, E.A.; Berdis, A.J. Terminal deoxynucleotidyl transferase: The story of a misguided DNA polymerase. Biochim. Biophys. Acta 2010, 1804, 1151–1166. [Google Scholar] [CrossRef]
- Sadofsky, M.J. The RAG proteins in V(D)J recombination: More than just a nuclease. Nucleic Acids Res. 2001, 29, 1399–1409. [Google Scholar] [CrossRef]
- Schatz, D.G.; Ji, Y. Recombination centres and the orchestration of V(D)J recombination. Nat. Rev. Immunol. 2011, 11, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, D.; Baetz, K.; Wu, G.E. Conservation of sequence in recombination signal sequence spacers. Nucleic Acids Res. 1994, 22, 1785–1796. [Google Scholar] [CrossRef] [PubMed]
- van Gent, D.C.; Ramsden, D.; Gellert, M. The RAG1 and RAG2 proteins establish the 12/23 rule in V(D)J recombination. Cell 1996, 85, 107–113. [Google Scholar] [CrossRef]
- Hiom, K.; Gellert, M. Assembly of a 12/23 paired signal complex: A critical control point in V(D)J recombination. Mol. Cell 1998, 1, 1011–1019. [Google Scholar] [CrossRef]
- Van Gent, D.C.; Hiom, K.; Paull, T.T.; Gellert, M. Stimulation of V(D)J cleavage by high mobility group proteins. EMBO J. 1997, 16, 2665–2670. [Google Scholar] [CrossRef]
- Agrawal, A.; Schatz, D.G. RAG1 and RAG2 form a stable postcleavage synaptic complex with DNA containing signal ends in V(D)J recombination. Cell 1997, 89, 43–53. [Google Scholar] [CrossRef]
- Fugmann, S.D.; Lee, A.I.; Shockett, P.E.; Villey, I.J.; Schatz, D.G. The RAG proteins and V(D)J recombination: Complexes, ends, and transposition. Annu. Rev. Immunol. 2000, 18, 495–527. [Google Scholar] [CrossRef]
- Malu, S.; Malshetty, V.; Francis, D.; Cortes, P. Role of non-homologous end joining in V(D)J recombination. Immunol. Res. 2012, 54, 233–246. [Google Scholar] [CrossRef]
- Lu, H.; Schwarz, K.; Lieber, M.R. Extent to which hairpin opening by the Artemis: DNA-PKcs complex can contribute to junctional diversity in V(D)J recombination. Nucleic Acids Res. 2007, 35, 6917–6923. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.L.; Gilfillan, S.; Thai, T.H.; Kearney, J.F. Terminal deoxynucleotidyl transferase and repertoire development. Immunol. Rev. 2000, 175, 150–157. [Google Scholar] [CrossRef]
- Edwards, N.L. Immunodeficiencies associated with errors in purine metabolism. Med. Clin. North. Am. 1985, 69, 505–518. [Google Scholar] [CrossRef]
- Apasov, S.G.; Blackburn, M.R.; Kellems, R.E.; Smith, P.T.; Sitkovsky, M.V. Adenosine deaminase deficiency increases thymic apoptosis and causes defective T cell receptor signaling. J. Clin. Investig. 2001, 108, 131–141. [Google Scholar] [CrossRef]
- Aldrich, M.B.; Chen, W.; Blackburn, M.R.; Martinez-Valdez, H.; Datta, S.K.; Kellems, R.E. Impaired germinal center maturation in adenosine deaminase deficiency. J. Immunol. 2003, 171, 5562–5570. [Google Scholar] [CrossRef]
- Gangi-Peterson, L.; Sorscher, D.H.; Reynolds, J.W.; Kepler, T.B.; Mitchell, B.S. Nucleotide pool imbalance and adenosine deaminase deficiency induce alterations of N-region insertions during V(D)J recombination. J. Clin. Investig. 1999, 103, 833–841. [Google Scholar] [CrossRef]
- Benveniste, P.; Zhu, W.; Cohen, A. Interference with thymocyte differentiation by an inhibitor of S-adenosylhomocysteine hydrolase. J. Immunol. 1995, 155, 536–544. [Google Scholar] [PubMed]
- Hershfield, M.S.; Kredich, N.M.; Ownby, D.R.; Ownby, H.; Buckley, R. In vivo inactivation of erythrocyte S-adenosylhomocysteine hydrolase by 2’-deoxyadenosine in adenosine deaminase-deficient patients. J. Clin. Investig. 1979, 63, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.; Gudas, L.J.; Ammann, A.J.; Staal, G.E.J.; Martin, D.W. Deoxyguanosine triphosphate as a possible toxic metabolite in the immunodeficiency associated with purine nucleoside phosphorylase deficiency. J. Clin. Investig. 1978, 61, 1405–1409. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.-S. Deoxyguanosine toxicity on lymphoid cells as a cause for immunosuppression in purine nucleoside phosphorylase deficiency. Cell 1978, 14, 523–530. [Google Scholar] [CrossRef]
- Somech, R.; Lev, A.; Simon, A.J.; Hanna, S.; Etzioni, A. T and B-cell defects in a novel purine nucleoside phosphorylase mutation. J. Allergy Clin. Immunol. 2012, 130, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Hanft, V.N.; Fruchtman, S.R.; Pickens, C.V.; Rosse, W.F.; Howard, T.A.; Ware, R.E. Acquired DNA mutations associated with in vivo hydroxyurea exposure. Blood 2000, 95, 3589–3593. [Google Scholar] [CrossRef]
- Feng, Y.; Seija, N.; Di Noia, J.M.; Martin, A. AID in antibody diversification: There and back again. Trends Immunol. 2020, 41, 586–600. [Google Scholar] [CrossRef]
- Revy, P.; Muto, T.; Levy, Y.; Geissmann, F.; Plebani, A.; Sanal, O.; Catalan, N.; Forveille, M.; Dufourcq-Lagelouse, R.; Gennery, A.; et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the hyper-IgM syndrome (HIGM2). Cell 2000, 102, 565–575. [Google Scholar] [CrossRef]
- Methot, S.; Di Noia, J. Molecular mechanisms of somatic hypermutation and class switch recombination. Adv. Immunol. 2017, 133, 37–87. [Google Scholar] [CrossRef]
- Di Noia, J.; Neuberger, M.S. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature 2002, 419, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Rada, C.; Ehrenstein, M.; Neuberger, M.S.; Milstein, C. Hot spot focusing of somatic hypermutation in MSH2-deficient mice suggests two stages of mutational targeting. Immunity 1998, 9, 135–141. [Google Scholar] [CrossRef]
- Dingler, F.A.; Kemmerich, K.; Neuberger, M.S.; Rada, C. Uracil excision by endogenous SMUG 1 glycosylase promotes efficient I g class switching and impacts on A: T substitutions during somatic mutation. Eur. J. Immunol. 2014, 44, 1925–1935. [Google Scholar] [CrossRef] [PubMed]
- Rada, C.; Williams, G.T.; Nilsen, H.; Barnes, D.; Lindahl, T.; Neuberger, M.S. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr. Biol. 2002, 12, 1748–1755. [Google Scholar] [CrossRef]
- Weill, J.-C.; Reynaud, C.-A. DNA polymerases in adaptive immunity. Nat. Rev. Immunol. 2008, 8, 302–312. [Google Scholar] [CrossRef]
- Delbos, F.; Aoufouchi, S.; Faili, A.; Weill, J.-C.; Reynaud, C.-A. DNA polymerase η is the sole contributor of A/T modifications during immunoglobulin gene hypermutation in the mouse. J. Exp. Med. 2006, 204, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Winter, D.B.; Kasmer, C.; Kraemer, K.; Lehmann, A.R.; Gearhart, P.J. DNA polymerase η is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nat. Immunol. 2001, 2, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Kano, C.; Hanaoka, F.; Wang, J.-Y. Analysis of mice deficient in both REV1 catalytic activity and POLH reveals an unexpected role for POLH in the generation of C to G and G to C transversions during Ig gene hypermutation. Int. Immunol. 2012, 24, 169–174. [Google Scholar] [CrossRef]
- Krijger, P.H.; Tsaalbi-Shtylik, A.; Wit, N.; Berk, P.C.M.V.D.; de Wind, N.; Jacobs, H. Rev1 is essential in generating G to C transversions downstream of the Ung2 pathway but not the Msh2+Ung2 hybrid pathway. Eur. J. Immunol. 2013, 43, 2765–2770. [Google Scholar] [CrossRef]
- Stavnezer, J.; Linehan, E.K.; Thompson, M.R.; Habboub, G.; Ucher, A.J.; Kadungure, T.; Tsuchimoto, D.; Nakabeppu, Y.; Schrader, C.E. Differential expression of APE1 and APE2 in germinal centers promotes error-prone repair and A:T mutations during somatic hypermutation. Proc. Natl. Acad. Sci. USA 2014, 111, 9217–9222. [Google Scholar] [CrossRef]
- Bardwell, P.; Woo, C.J.; Wei, K.; Li, Z.; Martin, A.; Sack, S.Z.; Parris, T.; Edelmann, W.; Scharff, M.D. Altered somatic hypermutation and reduced class-switch recombination in exonuclease 1–mutant mice. Nat. Immunol. 2004, 5, 224–229. [Google Scholar] [CrossRef]
- Zubani, G.G.; Zivojnovic, M.; De Smet, A.; Albagli-Curiel, O.; Huetz, F.; Weill, J.-C.; Reynaud, C.-A.; Storck, S. Pms2 and uracil-DNA glycosylases act jointly in the mismatch repair pathway to generate Ig gene mutations at A-T base pairs. J. Exp. Med. 2017, 214, 1169–1180. [Google Scholar] [CrossRef]
- Wang, Q.; Kieffer-Kwon, K.-R.; Oliveira, T.Y.; Mayer, C.T.; Yao, K.; Pai, J.; Cao, Z.; Dose, M.; Casellas, R.; Jankovic, M.; et al. The cell cycle restricts activation-induced cytidine deaminase activity to early G1. J. Exp. Med. 2016, 214, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Sharbeen, G.; Yee, C.W.; Smith, A.L.; Jolly, C.J. Ectopic restriction of DNA repair reveals that UNG2 excises AID-induced uracils predominantly or exclusively during G1 phase. J. Exp. Med. 2012, 209, 965–974. [Google Scholar] [CrossRef]
- Thientosapol, E.; Bosnjak, D.; Durack, T.; Stevanovski, I.; van Geldermalsen, M.; Holst, J.; Jahan, Z.; Shepard, C.; Weninger, W.; Kim, B.; et al. SAMHD1 enhances immunoglobulin hypermutation by promoting transversion mutation. Proc. Natl. Acad. Sci. USA 2018, 115, 4921–4926. [Google Scholar] [CrossRef] [PubMed]
- Thomas-Claudepierre, A.-S.; Schiavo, E.; Heyer, V.; Fournier, M.; Page, A.; Robert, I.; Reina-San-Martin, B. The cohesin complex regulates immunoglobulin class switch recombination. J. Exp. Med. 2013, 210, 2495–2502. [Google Scholar] [CrossRef] [PubMed]
- Boboila, C.; Alt, F.W.; Schwer, B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv. Immunol. 2012, 116, 1–49. [Google Scholar] [CrossRef]
- Küppers, R.; Dalla-Favera, R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene 2001, 20, 5580–5594. [Google Scholar] [CrossRef]
- Leder, P.; Battey, J.; Lenoir, G.; Moulding, C.; Murphy, W.; Potter, H.; Stewart, T.; Taub, R. Translocations among antibody genes in human cancer. Science 1983, 222, 765–771. [Google Scholar] [CrossRef]
- Robbiani, D.F.; Nussenzweig, M.C. Chromosome translocation, B cell lymphoma, and activation-induced cytidine deaminase. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 79–103. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Neumeister, P.; Goossens, T.; Nanjangud, G.; Chaganti, R.S.K.; Küppers, R.; Dalla-Favera, R. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nat. Cell Biol. 2001, 412, 341–346. [Google Scholar] [CrossRef]
- Fujita, T.; Fujii, H. Direct identification of insulator components by insertional chromatin immunoprecipitation. PLoS ONE 2011, 6, e26109. [Google Scholar] [CrossRef]
- Rush, J.S.; Fugmann, S.D.; Schatz, D.G. Staggered AID-dependent DNA double strand breaks are the predominant DNA lesions targeted to S in Ig class switch recombination. Int. Immunol. 2004, 16, 549–557. [Google Scholar] [CrossRef]
- Chen, J.-M.; Cooper, D.N.; Chuzhanova, N.; Férec, C.; Patrinos, G.P. Gene conversion: Mechanisms, evolution and human disease. Nat. Rev. Genet. 2007, 8, 762–775. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.C.; Desiderio, S. Cell cycle regulation of V(D)J recombination-activating protein RAG-2. Proc. Natl. Acad. Sci. USA 1994, 91, 2733–2737. [Google Scholar] [CrossRef] [PubMed]
- Pontarin, G.; Fijolek, A.; Pizzo, P.; Ferraro, P.; Rampazzo, C.; Pozzan, T.; Thelander, L.; Reichard, P.A.; Bianchi, V. Ribonucleotide reduction is a cytosolic process in mammalian cells independently of DNA damage. Proc. Natl. Acad. Sci. USA 2008, 105, 17801–17806. [Google Scholar] [CrossRef]
- D’Angiolella, V.; Donato, V.; Forrester, F.M.; Jeong, Y.-T.; Pellacani, C.; Kudo, Y.; Saraf, A.; Florens, L.; Washburn, M.; Pagano, M. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 2012, 149, 1023–1034. [Google Scholar] [CrossRef]
- Hu, C.-M.; Yeh, M.-T.; Tsao, N.; Chen, C.-W.; Gao, Q.-Z.; Chang, C.-Y.; Lee, M.-H.; Fang, J.-M.; Sheu, S.-Y.; Lin, C.-J.; et al. Tumor cells require thymidylate kinase to prevent dUTP incorporation during DNA repair. Cancer Cell 2012, 22, 36–50. [Google Scholar] [CrossRef]
- Niida, H.; Katsuno, Y.; Sengoku, M.; Shimada, M.; Yukawa, M.; Ikura, M.; Ikura, T.; Kohno, K.; Shima, H.; Suzuki, H.; et al. Essential role of Tip60-dependent recruitment of ribonucleotide reductase at DNA damage sites in DNA repair during G1 phase. Genes Dev. 2010, 24, 333–338. [Google Scholar] [CrossRef]
- Tanaka, H.; Arakawa, H.; Yamaguchi, T.; Shiraishi, K.; Fukuda, S.; Matsui, K.; Takei, Y.; Nakamura, Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 2000, 404, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Watt, D.L.; Buckland, R.J.; Lujan, S.A.; Kunkel, T.A.; Chabes, A. Genome-wide analysis of the specificity and mechanisms of replication infidelity driven by imbalanced dNTP pools. Nucleic Acids Res. 2015, 44, 1669–1680. [Google Scholar] [CrossRef]
- Bertocci, B.; De Smet, A.; Weill, J.-C.; Reynaud, C.-A. nonoverlapping functions of DNA polymerases Mu, Lambda, and Terminal Deoxynucleotidyltransferase during immunoglobulin V(D)J recombination in vivo. Immunity 2006, 25, 31–41. [Google Scholar] [CrossRef]
- Zelensky, A.; Schimmel, J.; Kool, H.; Kanaar, R.; Tijsterman, M. Inactivation of Pol θ and C-NHEJ eliminates off-target integration of exogenous DNA. Nat. Commun. 2017, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Wyatt, D.; Takata, K.-I.; Mu, Y.; Hensley, S.C.; Tomida, J.; Bylund, G.O.; Doublie, S.; Johansson, E.; Ramsden, D.; et al. Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLoS Genet. 2014, 10, e1004654. [Google Scholar] [CrossRef]
- Wyatt, D.; Feng, W.; Conlin, M.P.; Yousefzadeh, M.J.; Roberts, S.A.; Mieczkowski, P.; Wood, R.; Gupta, G.P.; Ramsden, D.A. Essential roles for polymerase θ-mediated end joining in the repair of chromosome breaks. Mol. Cell 2016, 63, 662–673. [Google Scholar] [CrossRef]
- Babbe, H.; McMenamin, J.; Hobeika, E.; Wang, J.; Rodig, S.J.; Reth, M.; Leder, P. Genomic instability resulting from Blm deficiency compromises development, maintenance, and function of the B cell lineage. J. Immunol. 2009, 182, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Giri, P.K.; Kazadi, D.; Laffleur, B.; Zhang, W.; Grinstein, V.; Pefanis, E.; Brown, L.M.; Ladewig, E.; Martin, O.A.; et al. Nuclear proximity of Mtr4 to RNA exosome restricts DNA mutational asymmetry. Cell 2017, 169, 523–537.e5. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, C.R.; Dhir, S.; Dhir, A.; Moghaddam, A.E.; Sattentau, Q.; Meinhart, A.; Proudfoot, N.J. RNA helicase DDX1 converts RNA G-quadruplex structures into R-loops to promote IgH class switch recombination. Mol. Cell 2018, 70, 650–662.e8. [Google Scholar] [CrossRef] [PubMed]
- Pieper, K.D.; Tan, J.; Piccoli, L.; Foglierini, M.; Barbieri, S.; Chen, Y.; Silacci-Fregni, C.; Wolf, T.; Jarrossay, D.; Anderle, M.; et al. Public antibodies to malaria antigens generated by two LAIR1 insertion modalities. Nature 2017, 548, 597–601. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azhar, A.; Begum, N.A.; Husain, A. Nucleotide Pool Imbalance and Antibody Gene Diversification. Vaccines 2021, 9, 1050. https://doi.org/10.3390/vaccines9101050
Azhar A, Begum NA, Husain A. Nucleotide Pool Imbalance and Antibody Gene Diversification. Vaccines. 2021; 9(10):1050. https://doi.org/10.3390/vaccines9101050
Chicago/Turabian StyleAzhar, Asim, Nasim A. Begum, and Afzal Husain. 2021. "Nucleotide Pool Imbalance and Antibody Gene Diversification" Vaccines 9, no. 10: 1050. https://doi.org/10.3390/vaccines9101050
APA StyleAzhar, A., Begum, N. A., & Husain, A. (2021). Nucleotide Pool Imbalance and Antibody Gene Diversification. Vaccines, 9(10), 1050. https://doi.org/10.3390/vaccines9101050