Primary HIV-1 and Infectious Molecular Clones Are Differentially Susceptible to Broadly Neutralizing Antibodies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Monoclonal Antibodies
2.3. Media and Cell Culture
2.4. HIV-1 Preparation and Purification
2.5. HIV-1 Capture Assay Using PBMCs
2.6. HIV-1 Replication Experiments
2.7. Isolation of CD4+ T Cells
2.8. RNA Extraction and qRT-PCR
2.9. Statistical Analysis
3. Results
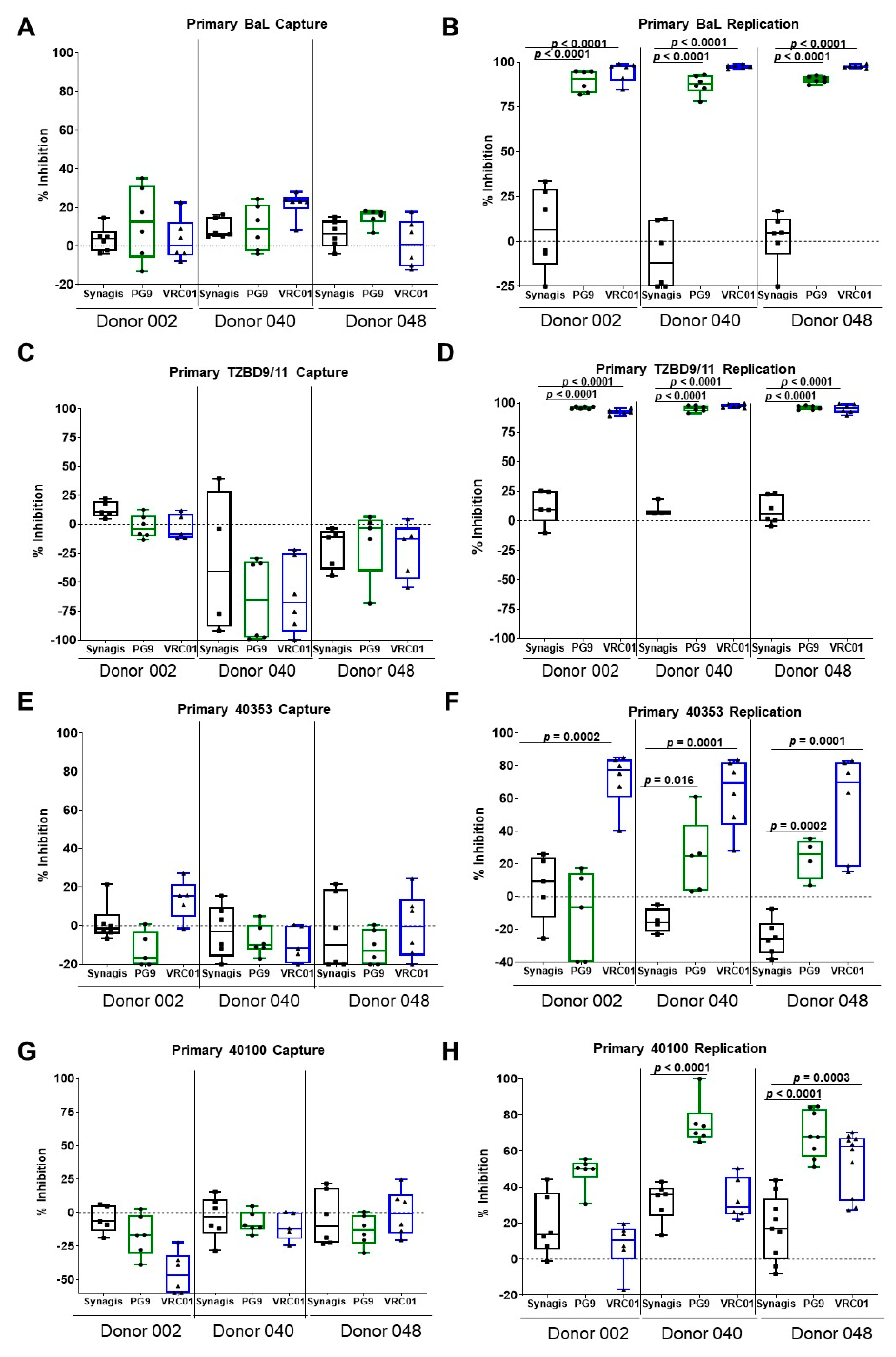
3.1. Capture and Replication of Primary HIV-1 in Human PBMCs
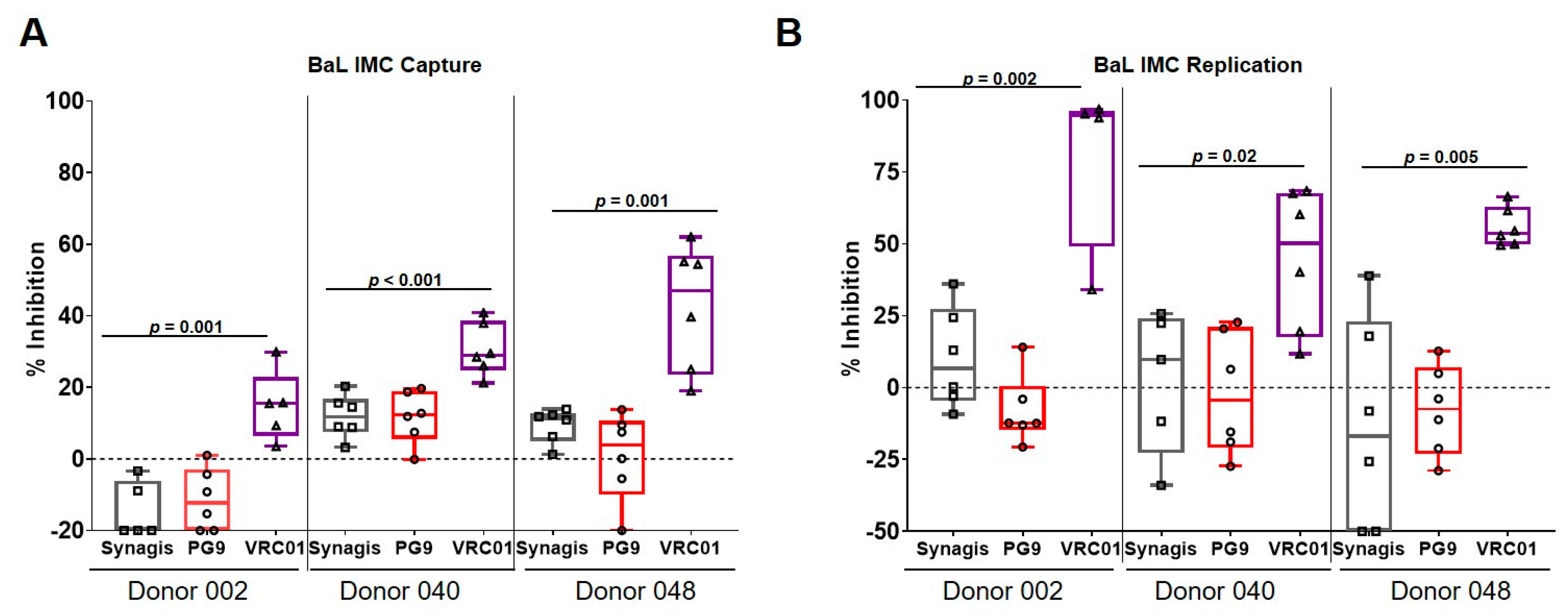
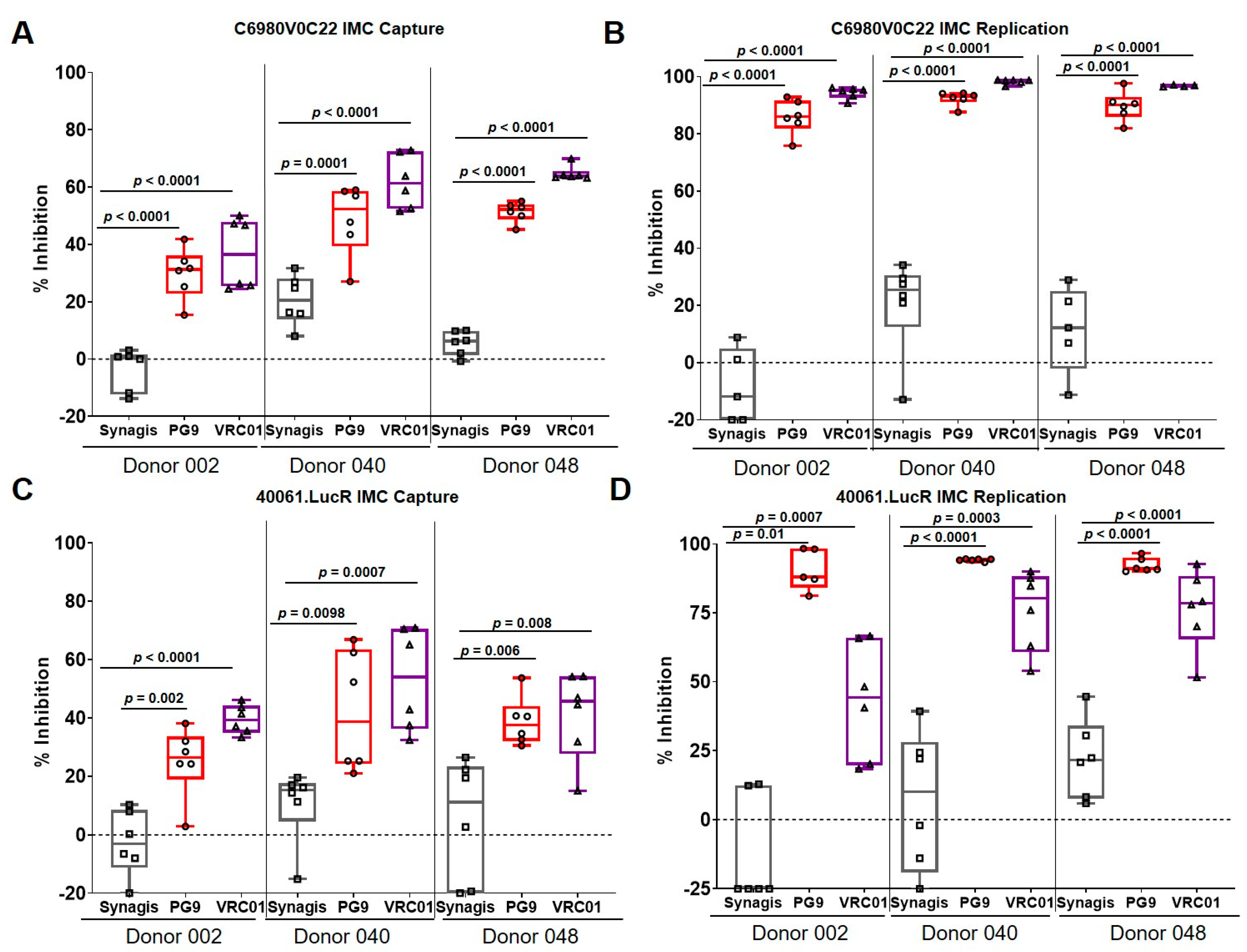
3.2. Capture and Replication of Infectious Molecular Clones in Human PBMCs
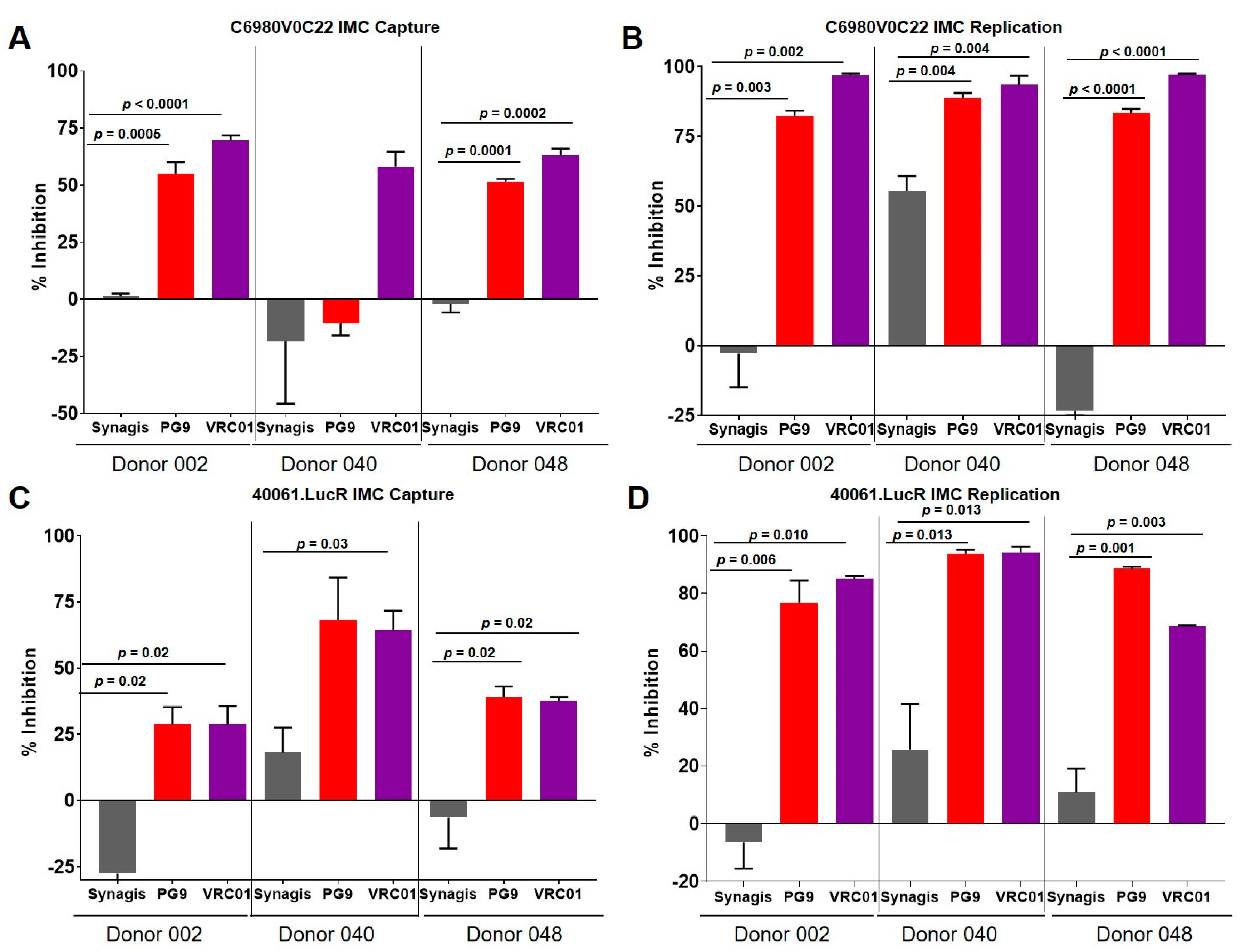
3.3. Virus Capture and Replication of IMCs with Isolated CD4+ T Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Montefiori, D.C. Evaluating Neutralizing Antibodies against Hiv, Siv, and Shiv in Luciferase Reporter Gene Assays. Curr. Protoc. Immunol. 2005. [CrossRef] [PubMed]
- Chuang, G.-Y.; Acharya, P.; Schmidt, S.D.; Yang, Y.; Louder, M.K.; Zhou, T.; Kwon, Y.D.; Pancera, M.; Bailer, R.T.; Doria-Rose, N.A.; et al. Residue-Level Prediction of Hiv-1 Antibody Epitopes Based on Neutralization of Diverse Viral Strains. J. Virol. 2013, 87, 10047–10058. [Google Scholar] [CrossRef]
- Falkowska, E.; Le, K.M.; Ramos, A.; Doores, K.J.; Lee, J.H.; Blattner, C.; Ramirez, A.; Derking, R.; van Gils, M.J.; Liang, C.H.; et al. Broadly Neutralizing Hiv Antibodies Define a Glycan-Dependent Epitope on the Prefusion Conformation of Gp41 on Cleaved Envelope Trimers. Immunity 2014, 40, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, I.S.; Gordon Joyce, M.; Zhou, T.; Kwong, P.D. Elicitation of Hiv-1-Neutralizing Antibodies against the Cd4-Binding Site. Curr. Opin. HIV AIDS 2013, 8, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Kang, B.H.; Pancera, M.; Lee, J.H.; Tong, T.; Feng, Y.; Imamichi, H.; Georgiev, I.S.; Chuang, G.Y.; Druz, A.; et al. Broad and Potent Hiv-1 Neutralization by a Human Antibody That Binds the Gp41-Gp120 Interface. Nature 2014, 515, 138–142. [Google Scholar] [CrossRef]
- Huang, J.; Ofek, G.; Laub, L.; Louder, M.K.; Doria-Rose, N.A.; Longo, N.S.; Imamichi, H.; Bailer, R.T.; Chakrabarti, B.K.; Sharma, S.K.; et al. Broad and Potent Neutralization of Hiv-1 by a Gp41-Specific Human Antibody. Nature 2012, 491, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Program, N.C.S.; Lynch, R.M.; Zhou, T.; Gao, F.; Alam, S.M.; Boyd, S.D.; Fire, A.Z.; Roskin, K.M.; Schramm, C.A.; et al. Co-Evolution of a Broadly Neutralizing Hiv-1 Antibody and Founder Virus. Nature 2013, 496, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.M.; Huber, M.; Doores, K.J.; Falkowska, E.; Pejchal, R.; Julien, J.-P.; Wang, S.-K.; Ramos, A.; Chan-Hui, P.-Y.; Moyle, M.; et al. Broad Neutralization Coverage of Hiv by Multiple Highly Potent Antibodies. Nature 2011, 477, 466–470. [Google Scholar] [CrossRef]
- Walker, L.M.; Phogat, S.K.; Chan-Hui, P.-Y.; Wagner, D.; Phung, P.; Goss, J.L.; Wrin, T.; Simek, M.D.; Fling, S.; Mitcham, J.L.; et al. Broad and Potent Neutralizing Antibodies from an African Donor Reveal a New Hiv-1 Vaccine Target. Science 2009, 326, 285–289. [Google Scholar] [CrossRef]
- Wu, X.; Yang, Z.-Y.; Li, Y.; Hogerkorp, C.-M.; Schief, W.R.; Seaman, M.S.; Zhou, T.; Schmidt, S.D.; Wu, L.; Xu, L.; et al. Rational Design of Envelope Identifies Broadly Neutralizing Human Monoclonal Antibodies to Hiv-1. Science 2010, 329, 856–861. [Google Scholar] [CrossRef]
- Brown, B.K.; Wieczorek, L.; Sanders-Buell, E.; Borges, A.R.; Robb, M.L.; Birx, D.L.; Michael, N.L.; McCutchan, F.E.; Polonis, V.R. Cross-Clade Neutralization Patterns among Hiv-1 Strains from the Six Major Clades of the Pandemic Evaluated and Compared in Two Different Models. Virology 2008, 375, 529–538. [Google Scholar] [CrossRef]
- Kim, J.; Jobe, O.; Peachman, K.K.; Michael, N.L.; Robb, M.L.; Rao, M.; Rao, V.B. Quantitative Analyses Reveal Distinct Sensitivities of the Capture of Hiv-1 Primary Viruses and Pseudoviruses to Broadly Neutralizing Antibodies. Virology 2017, 508, 188–198. [Google Scholar] [CrossRef]
- Jobe, O.; Peachman, K.K.; Matyas, G.R.; Asher, L.V.; Alving, C.R.; Rao, M. An Anti-Phosphoinositide-Specific Monoclonal Antibody That Neutralizes Hiv-1 Infection of Human Monocyte-Derived Macrophages. Virology 2012, 430, 110–119. [Google Scholar] [CrossRef]
- Chackerian, B.; Long, E.M.; Luciw, P.A.; Overbaugh, J. Human Immunodeficiency Virus Type 1 Coreceptors Participate in Postentry Stages in the Virus Replication Cycle and Function in Simian Immunodeficiency Virus Infection. J. Virol. 1997, 71, 3932–3939. [Google Scholar] [CrossRef]
- Robb, M.L.; Eller, L.A.; Kibuuka, H.; Rono, K.; Maganga, L.; Nitayaphan, S.; Kroon, E.; Sawe, F.K.; Sinei, S.; Sriplienchan, S.; et al. Prospective Study of Acute Hiv-1 Infection in Adults in East Africa and Thailand. N. Engl. J. Med. 2016, 374, 2120–2130. [Google Scholar] [CrossRef]
- Landais, E.; Murrell, B.; Briney, B.; Murrell, S.; Rantalainen, K.; Berndsen, Z.T.; Ramos, A.; Wickramasinghe, L.; Smith, M.L.; Eren, K.; et al. Hiv Envelope Glycoform Heterogeneity and Localized Diversity Govern the Initiation and Maturation of a V2 Apex Broadly Neutralizing Antibody Lineage. Immunity 2017, 47, 990–1003.e9. [Google Scholar] [CrossRef]
- Wieczorek, L.; Krebs, S.J.; Kalyanaraman, V.; Whitney, S.; Tovanabutra, S.; Moscoso, C.G.; Sanders-Buell, E.; Williams, C.; Slike, B.; Molnar, S.; et al. Comparable Antigenicity and Immunogenicity of Oligomeric Forms of a Novel, Acute Hiv-1 Subtype C Gp145 Envelope for Use in Preclinical and Clinical Vaccine Research. J. Virol. 2015, 89, 7478–7493. [Google Scholar] [CrossRef][Green Version]
- Lehmann, M.J.; Sherer, N.M.; Marks, C.B.; Pypaert, M.; Mothes, W. Actin- and Myosin-Driven Movement of Viruses Along Filopodia Precedes Their Entry into Cells. J. Cell Biol. 2005, 170, 317–325. [Google Scholar] [CrossRef]
- Sherer, N.M.; Lehmann, M.J.; Jimenez-Soto, L.F.; Horensavitz, C.; Pypaert, M.; Mothes, W. Retroviruses Can Establish Filopodial Bridges for Efficient Cell-to-Cell Transmission. Nat. Cell Biol. 2007, 9, 310–315. [Google Scholar] [CrossRef]
- Endres, T.; Lampe, M.; Briggs, J.A.G.; Kräusslich, H.-G.; Bräuchle, C.; Müller, B.; Lamb, D.C. Hiv-1-Cellular Interactions Analyzed by Single Virus Tracing. Eur. Biophys. J. 2008, 37, 1291–1301. [Google Scholar] [CrossRef]
- Burckhardt, C.J.; Greber, U.F. Virus Movements on the Plasma Membrane Support Infection and Transmission between Cells. PLoS Pathog. 2009, 5, e1000621. [Google Scholar] [CrossRef]
- Sherer, N.M.; Jin, J.; Mothes, W. Directional Spread of Surface-Associated Retroviruses Regulated by Differential Virus-Cell Interactions. J. Virol. 2010, 84, 3248–3258. [Google Scholar] [CrossRef]
- Kukura, P.; Ewers, H.; Müller, C.; Renn, A.; Helenius, A.; Sandoghdar, V. High-Speed Nanoscopic Tracking of the Position and Orientation of a Single Virus. Nat. Methods 2009, 6, 923–927. [Google Scholar] [CrossRef]
- Chand, S.; Messina, E.L.; AlSalmi, W.; Ananthaswamy, N.; Gao, G.; Uritskiy, G.; Padilla-Sanchez, V.; Mahalingam, M.; Peachman, K.K.; Robb, M.L.; et al. Glycosylation and Oligomeric State of Envelope Protein Might Influence Hiv-1 Virion Capture by Alpha4beta7 Integrin. Virology 2017, 508, 199–212. [Google Scholar] [CrossRef]
- Blumenthal, R.; Durell, S.R.; Viard, M. Hiv Entry and Envelope Glycoprotein-Mediated Fusion. J. Biol. Chem. 2012, 287, 40841–40849. [Google Scholar] [CrossRef]
- Herold, N.; Anders-Osswein, M.; Glass, B.; Eckhardt, M.; Müller, B.; Kräusslich, H.-G.; Anders-Ößwein, M. Hiv-1 Entry in Supt1-R5, Cem-Ss, and Primary Cd4+ T Cells Occurs at the Plasma Membrane and Does Not Require Endocytosis. J. Virol. 2014, 88, 13956–13970. [Google Scholar] [CrossRef]
- Fackler, O.T.; Peterlin, B. Endocytic Entry of Hiv-1. Curr. Biol. 2000, 10, 1005–1008. [Google Scholar] [CrossRef]
- Schaeffer, E.; Soros, V.B.; Greene, W.C. Compensatory Link between Fusion and Endocytosis of Human Immunodeficiency Virus Type 1 in Human Cd4 T Lymphocytes. J. Virol. 2004, 78, 1375–1383. [Google Scholar] [CrossRef]
- Daecke, J.; Fackler, O.T.; Dittmar, M.T.; Kräusslich, H.-G. Involvement of Clathrin-Mediated Endocytosis in Human Immunodeficiency Virus Type 1 Entry. J. Virol. 2005, 79, 1581–1594. [Google Scholar] [CrossRef]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. Hiv Enters Cells Via Endocytosis and Dynamin-Dependent Fusion with Endosomes. Cell 2009, 137, 433–444. [Google Scholar] [CrossRef]
- Pritschet, K.; Donhauser, N.; Schuster, P.; Ries, M.; Haupt, S.; Kittan, N.A.; Korn, K.; Pöhlmann, S.; Holland, G.; Bannert, N.; et al. Cd4− and Dynamin-Dependent Endocytosis of Hiv-1 into Plasmacytoid Dendritic Cells. Virology 2012, 423, 152–164. [Google Scholar] [CrossRef]
- Van Wilgenburg, B.; Moore, M.D.; James, W.S.; Cowley, S.A. The Productive Entry Pathway of Hiv-1 in Macrophages Is Dependent on Endocytosis through Lipid Rafts Containing Cd4. PLoS ONE 2014, 9, e86071. [Google Scholar] [CrossRef]
- Maréchal, V.; Prevost, M.-C.; Petit, C.; Perret, E.; Heard, J.-M.; Schwartz, O. Human Immunodeficiency Virus Type 1 Entry into Macrophages Mediated by Macropinocytosis. J. Virol. 2001, 75, 11166–11177. [Google Scholar]
- Liu, J.; Ghneim, K.; Sok, D.; Bosche, W.J.; Li, Y.; Chipriano, E.; Berkemeier, B.; Oswald, K.; Borducchi, E.; Cabral, C.; et al. Antibody-Mediated Protection against Shiv Challenge Includes Systemic Clearance of Distal Virus. Science 2016, 353, 1045–1049. [Google Scholar] [CrossRef]
- Lu, M.; Ma, X.; Castillo-Menendez, L.R.; Gorman, J.; Alsahafi, N.; Ermel, U.; Terry, D.S.; Chambers, M.; Peng, D.; Zhang, B.; et al. Associating Hiv-1 Envelope Glycoprotein Structures with States on the Virus Observed by Smfret. Nature 2019, 568, 415–419. [Google Scholar] [CrossRef] [PubMed]
- White, T.A.; Bartesaghi, A.; Borgnia, M.J.; Meyerson, J.R.; De La Cruz, M.J.V.; Bess, J.W.; Nandwani, R.; Hoxie, J.A.; Lifson, J.D.; Milne, J.L.S.; et al. Molecular Architectures of Trimeric Siv and Hiv-1 Envelope Glycoproteins on Intact Viruses: Strain-Dependent Variation in Quaternary Structure. PLoS Pathog. 2010, 6, e1001249. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Rao, V.B.; Rao, M. Primary HIV-1 and Infectious Molecular Clones Are Differentially Susceptible to Broadly Neutralizing Antibodies. Vaccines 2020, 8, 782. https://doi.org/10.3390/vaccines8040782
Kim J, Rao VB, Rao M. Primary HIV-1 and Infectious Molecular Clones Are Differentially Susceptible to Broadly Neutralizing Antibodies. Vaccines. 2020; 8(4):782. https://doi.org/10.3390/vaccines8040782
Chicago/Turabian StyleKim, Jiae, Venigalla B. Rao, and Mangala Rao. 2020. "Primary HIV-1 and Infectious Molecular Clones Are Differentially Susceptible to Broadly Neutralizing Antibodies" Vaccines 8, no. 4: 782. https://doi.org/10.3390/vaccines8040782
APA StyleKim, J., Rao, V. B., & Rao, M. (2020). Primary HIV-1 and Infectious Molecular Clones Are Differentially Susceptible to Broadly Neutralizing Antibodies. Vaccines, 8(4), 782. https://doi.org/10.3390/vaccines8040782