Neo-Antigen mRNA Vaccines

 , , , and
, , , and
Abstract
1. Introduction
2. Neo-Antigens and Their Recognition by CD4+ or CD8+ T Cells
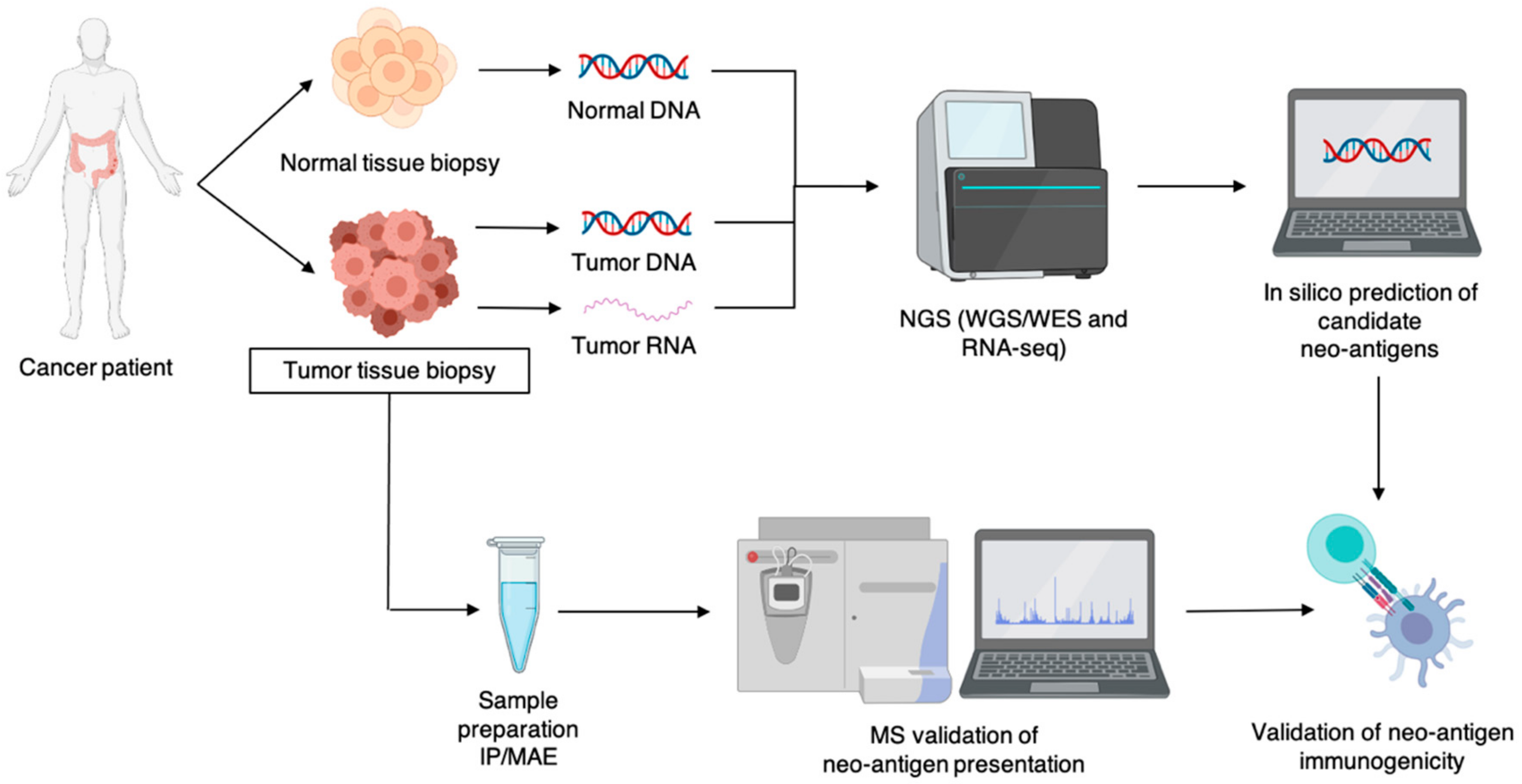
3. Identification and Validation of Neo-Antigens
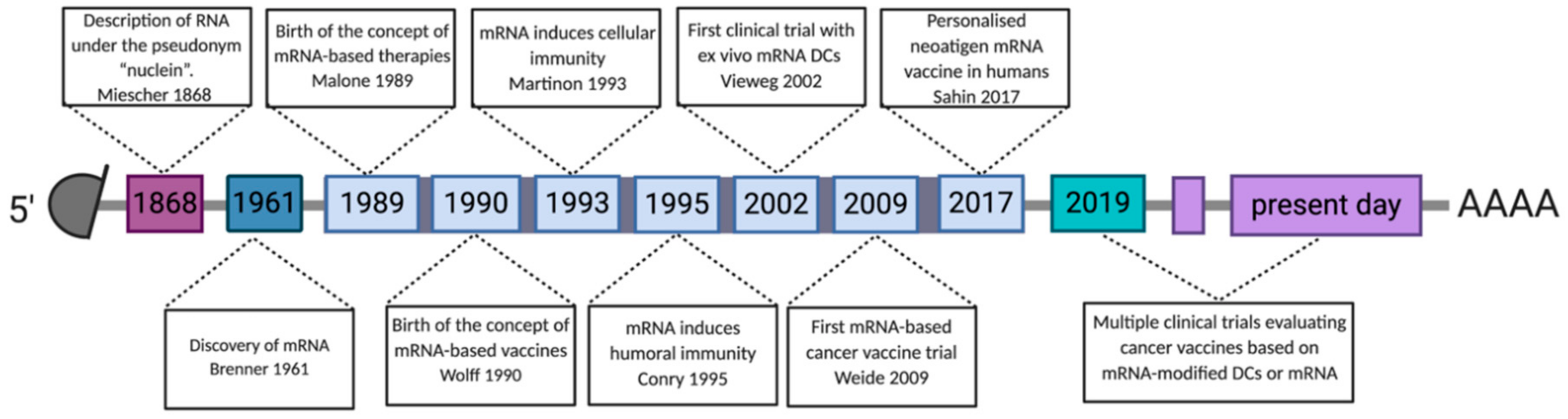
4. History of mRNA-Based Cancer Vaccines
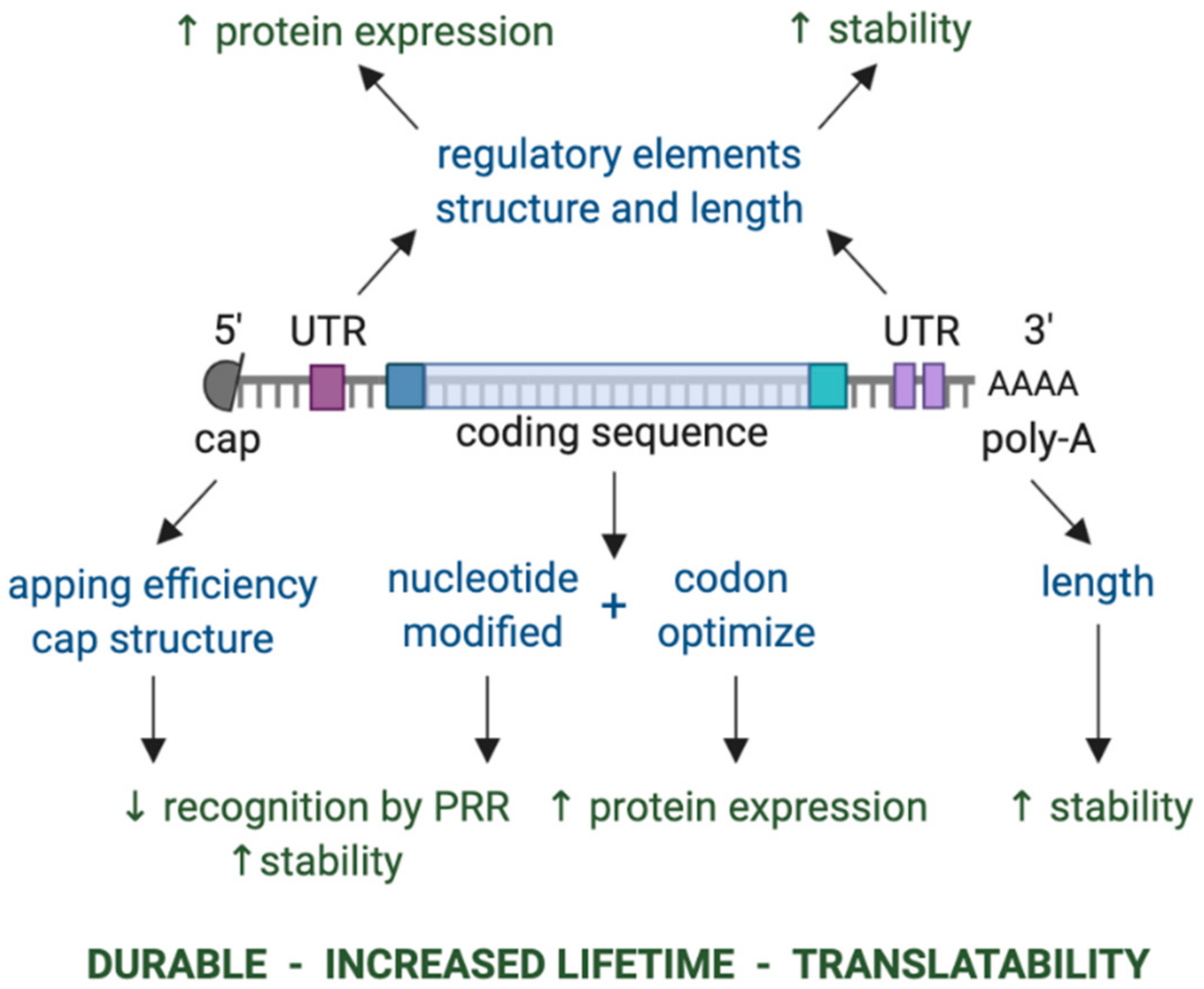
5. mRNA as a Platform for Neo-Antigen Vaccination
6. Studies with Neo-Antigen mRNA
7. Future Perspectives
7.1. Neo-Antigen Prediction and Prioritization
7.2. Neo-Antigen Immunogenicity Screening
7.3. Monitoring the Efficacy of Neo-Antigen Vaccines?
7.4. Combining with Other Immunotherapies
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3′ poly-A | three prime polyadenylic acid |
| 5′ cap | five prime cap |
| APC | antigen-presenting cell |
| CD40L | CD40 ligand |
| CRC | colorectal cancer |
| CTL | cytotoxic T lymphocyte |
| DC | dendritic cell |
| GFP | green fluorescent protein |
| GMP | good manufacturing practice |
| HLA | human leukocyte antigen |
| i.d. | intradermal |
| i.m. | intramuscular |
| i.n. | intranodal |
| i.v. | intravenous |
| IFN | interferon |
| INDEL | insertion and deletion |
| iNKT | invariant natural killer T cell |
| IP | immunoprecipitation |
| IVT | in vitro transcribed |
| LAMP | lysosomal-associated membrane protein |
| LNP | lipid nanoparticle |
| MAE | mild acid elution |
| MCR | MHC-TCR chimeric receptor |
| MHC | major histocompatibility complex |
| moDC | monocyte-derived dendritic cell |
| mRNA | messenger RNA |
| MS | mass spectrometry |
| NGS | next-generation sequencing |
| NSCL | non-small cell lung cancer |
| PBMC | peripheral blood mononuclear cell |
| PD-1 | programmed death-1 |
| PRR | pattern recognition receptor |
| PSA | prostate specific antigen |
| PTM | post-translational modification |
| RNases | ribonucleases |
| s.c. | subcutaneous |
| SABR | signaling and antigen-presenting bifunctional receptor |
| SNV | single nucleotide variants |
| TAA | tumor-associated antigen |
| TCR | T cell receptor |
| TH1 | T helper 1 cell |
| TLR | toll-like receptor |
| TNBC | triple negative breast cancer |
| UTR | untranslated region |
References
- Benteyn, D.; Heirman, C.; Bonehill, A.; Thielemans, K.; Breckpot, K. mRNA-based dendritic cell vaccines. Expert Rev. Vaccines 2015, 14, 161–176. [Google Scholar] [CrossRef]
- Bonehill, A.; Heirman, C.; Tuyaerts, S.; Michiels, A.; Breckpot, K.; Brasseur, F.; Zhang, Y.; Van Der Bruggen, P.; Thielemans, K. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J. Immunol. 2004, 172, 6649–6657. [Google Scholar] [CrossRef]
- Verbeke, R.; Lentacker, I.; Breckpot, K.; Janssens, J.; Van Calenbergh, S.; De Smedt, S.C.; Dewitte, H. Broadening the Message: A Nanovaccine Co-loaded with Messenger RNA and α-GalCer Induces Antitumor Immunity through Conventional and Natural Killer T Cells. ACS Nano 2019, 13, 1655–1669. [Google Scholar] [CrossRef]
- Hunder, N.N.; Wallen, H.; Cao, J.; Hendricks, D.W.; Reilly, J.Z.; Rodmyre, R.; Jungbluth, A.; Gnjatic, S.; Thompson, J.A.; Yee, C. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N. Engl. J. Med. 2008, 358, 2698–2703. [Google Scholar] [CrossRef]
- Quezada, S.A.; Simpson, T.R.; Peggs, K.S.; Merghoub, T.; Vider, J.; Fan, X.; Blasberg, R.; Yagita, H.; Muranski, P.; Antony, P.A.; et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J. Exp. Med. 2010, 207, 637–650. [Google Scholar] [CrossRef]
- Nair, S.; Dhodapkar, M.V. Natural Killer T Cells in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1178. [Google Scholar] [CrossRef]
- Perez-Diez, A.; Joncker, N.T.; Choi, K.; Chan, W.F.; Anderson, C.C.; Lantz, O.; Matzinger, P. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood 2007, 109, 5346–5354. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Van Nuffel, A.M.T.; Benteyn, D.; Corthals, J.; Aerts, C.; Heirman, C.; Van Riet, I.; Bonehill, A.; Thielemans, K.; Neyns, B. A phase IB study on intravenous synthetic mRNA electroporated dendritic cell immunotherapy in pretreated advanced melanoma patients. Ann. Oncol. 2013, 24, 2686–2693. [Google Scholar] [CrossRef]
- Chen, X.; Yang, J.; Wang, L.; Liu, B. Personalized neoantigen vaccination with synthetic long peptides: Recent advances and future perspectives. Theranostics 2020, 10, 6011–6023. [Google Scholar] [CrossRef]
- Buonaguro, L.; Tagliamonte, M. Selecting Target Antigens for Cancer Vaccine Development. Vaccines 2020, 8, 615. [Google Scholar] [CrossRef]
- Han, X.J.; Ma, X.L.; Yang, L.; Wei, Y.Q.; Peng, Y.; Wei, X.W. Progress in Neoantigen Targeted Cancer Immunotherapies. Front. Cell Dev. Biol. 2020, 8, 728. [Google Scholar] [CrossRef]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Löwer, M.; van de Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef]
- Heemskerk, B.; Kvistborg, P.; Schumacher, T.N. The cancer antigenome. EMBO J. 2013, 32, 194–203. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef]
- Strønen, E.; Toebes, M.; Kelderman, S.; van Buuren, M.M.; Yang, W.; van Rooij, N.; Donia, M.; Böschen, M.L.; Lund-Johansen, F.; Olweus, J.; et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 2016, 352, 1337–1341. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. ‘Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef]
- Linette, G.P.; Carreno, B.M. Neoantigen Vaccines Pass the Immunogenicity Test. Trends Mol. Med. 2017, 23, 869–871. [Google Scholar] [CrossRef]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Dörrie, J.; Schaft, N.; Schuler, G.; Schuler-Thurner, B. Therapeutic Cancer Vaccination with Ex Vivo RNA-Transfected Dendritic Cells-An Update. Pharmaceutics 2020, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Thess, A.; Grund, S.; Mui, B.L.; Hope, M.J.; Baumhof, P.; Fotin-Mleczek, M.; Schlake, T. Sequence-engineered mRNA Without Chemical Nucleoside Modifications Enables an Effective Protein Therapy in Large Animals. Mol. Ther. 2015, 23, 1456–1464. [Google Scholar] [CrossRef]
- Grudzien-Nogalska, E.; Kowalska, J.; Su, W.; Kuhn, A.N.; Slepenkov, S.V.; Darzynkiewicz, E.; Sahin, U.; Jemielity, J.; Rhoads, R.E. Synthetic mRNAs with superior translation and stability properties. Methods Mol. Biol. 2013, 969, 55–72. [Google Scholar] [CrossRef]
- Asrani, K.H.; Farelli, J.D.; Stahley, M.R.; Miller, R.L.; Cheng, C.J.; Subramanian, R.R.; Brown, J.M. Optimization of mRNA untranslated regions for improved expression of therapeutic mRNA. RNA Biol. 2018, 15, 756–762. [Google Scholar] [CrossRef]
- Trepotec, Z.; Geiger, J.; Plank, C.; Aneja, M.K.; Rudolph, C. Segmented poly(A) tails significantly reduce recombination of plasmid DNA without affecting mRNA translation efficiency or half-life. RNA 2019, 25, 507–518. [Google Scholar] [CrossRef]
- Nelson, J.; Sorensen, E.W.; Mintri, S.; Rabideau, A.E.; Zheng, W.; Besin, G.; Khatwani, N.; Su, S.V.; Miracco, E.J.; Issa, W.J.; et al. Impact of mRNA chemistry and manufacturing process on innate immune activation. Sci. Adv. 2020, 6, eaaz6893. [Google Scholar] [CrossRef]
- Barret, L.W.; Fletcher, S.; Wilton, S.D. Untranslated Gene Regions and Other Non-Coding Elements; Springer: Basel, Switzerland, 2013. [Google Scholar]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef] [PubMed]
- Tuyaerts, S.; Noppe, S.; Corthals, J.; Breckpot, K.; Heirman, C.; De Greef, C.; Van Riet, I.; Thielemans, K. Generation of large numbers of dendritic cells in a closed system using Cell Factories (TM). J. Immunol. Methods 2002, 264, 135–151. [Google Scholar] [CrossRef]
- Van Lint, S.; Thielemans, K.; Breckpot, K. mRNA: Delivering an antitumor message? Immunotherapy 2011, 3, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, S.; Renmans, D.; Broos, K.; Dewitte, H.; Lentacker, I.; Heirman, C.; Breckpot, K.; Thielemans, K. The ReNAissanCe of mRNA-based cancer therapy. Expert Rev. Vaccines 2015, 14, 235–251. [Google Scholar] [CrossRef]
- Dewitte, H.; Verbeke, R.; Breckpot, K.; De Smedt, S.C.; Lentacker, I. Nanoparticle design to induce tumor immunity and challenge the suppressive tumor microenvironment. Nano Today 2014, 9, 743–758. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Zhang, X.; Qi, Y.; Zhang, Q.; Liu, W. Application of mass spectrometry-based MHC immunopeptidome profiling in neoantigen identification for tumor immunotherapy. Biomed. Pharmacother. 2019, 120, 109542. [Google Scholar] [CrossRef]
- Lennerz, V.; Fatho, M.; Gentilini, C.; Frye, R.A.; Lifke, A.; Ferel, D.; Wölfel, C.; Huber, C.; Wölfel, T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl. Acad. Sci. USA 2005, 102, 16013–16018. [Google Scholar] [CrossRef]
- Pritchard, A.L.; Burel, J.G.; Neller, M.A.; Hayward, N.K.; Lopez, J.A.; Fatho, M.; Lennerz, V.; Wölfel, T.; Schmidt, C.W. Exome Sequencing to Predict Neoantigens in Melanoma. In Cancer Immunol. Res.; 2015; Volume 3, pp. 992–998. [Google Scholar]
- Chang, T.C.; Carter, R.A.; Li, Y.; Wang, H.; Edmonson, M.N.; Chen, X.; Arnold, P.; Geiger, T.L.; Wu, G.; Peng, J.; et al. The neoepitope landscape in pediatric cancers. Genome Med. 2017, 9, 78. [Google Scholar]
- Skipper, J.C.; Hendrickson, R.C.; Gulden, P.H.; Brichard, V.; Van Pel, A.; Chen, Y.; Shabanowitz, J.; Wolfel, T.; Slingluff, C.L., Jr.; Boon, T.; et al. An HLA-A2-restricted tyrosinase antigen on melanoma cells results from posttranslational modification and suggests a novel pathway for processing of membrane proteins. J. Exp. Med. 1996, 183, 527–534. [Google Scholar] [CrossRef]
- Monteuuis, G.; Schmitz, U.; Petrova, V.; Kearney, P.S.; Rasko, J.E.J. Holding on to junk bonds: Intron retention in cancer and therapy. In Cancer Research; American Association for Cancer Research: Philadelphia, PA, USA, 2020. [Google Scholar]
- Lord, J.M.; Davey, J.; Frigerio, L.; Roberts, L.M. Endoplasmic reticulum-associated protein degradation. Semin. Cell Dev. Biol. 2000, 11, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Goldberg, A.L. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu. Rev. Immunol. 1999, 17, 739–779. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, N.; Van den Eynde, B.J. Insights into the processing of MHC class I ligands gained from the study of human tumor epitopes. Cell. Mol. Life Sci. 2011, 68, 1503–1520. [Google Scholar] [CrossRef]
- Raghavan, M.; Del Cid, N.; Rizvi, S.M.; Peters, L.R. MHC class I assembly: Out and about. Trends Immunol. 2008, 29, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Lu, Y.C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef]
- Kambayashi, T.; Laufer, T.M. Atypical MHC class II-expressing antigen-presenting cells: Can anything replace a dendritic cell? Nat. Rev. Immunol. 2014, 14, 719–730. [Google Scholar] [CrossRef]
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Löwer, M.; Diekmann, J.; Boegel, S.; Schrörs, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef]
- Overwijk, W.W. Human CD4(+) T cells spontaneously detect somatic mutations in cancer cells. Nat. Med. 2015, 21, 12–14. [Google Scholar] [CrossRef]
- Kim, H.J.; Cantor, H. CD4 T-cell subsets and tumor immunity: The helpful and the not-so-helpful. Cancer Immunol. Res. 2014, 2, 91–98. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Bonehill, A.; Heirman, C.; Thielemans, K. Genetic approaches for the induction of a CD4+ T cell response in cancer immunotherapy. J. Gene Med. 2005, 7, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Bonehill, A.; Heirman, C.; Tuyaerts, S.; Michiels, A.; Zhang, Y.; van der Bruggen, P.; Thielemans, K. Efficient presentation of known HLA class II-restricted MAGE-A3 epitopes by dendritic cells electroporated with messenger RNA encoding an invariant chain with genetic exchange of class II-associated invariant chain peptide. Cancer Res. 2003, 63, 5587–5594. [Google Scholar] [PubMed]
- Leng, Q.; Tarbe, M.; Long, Q.; Wang, F. Pre-existing heterologous T-cell immunity and neoantigen immunogenicity. Clin. Transl. Immunol. 2020, 9, e01111. [Google Scholar] [CrossRef]
- Loftus, D.J.; Castelli, C.; Clay, T.M.; Squarcina, P.; Marincola, F.M.; Nishimura, M.I.; Parmiani, G.; Appella, E.; Rivoltini, L. Identification of epitope mimics recognized by CTL reactive to the melanoma/melanocyte-derived peptide MART-1(27-35). J. Exp. Med. 1996, 184, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Vujanovic, L.; Shi, J.; Kirkwood, J.M.; Storkus, W.J.; Butterfield, L.H. Molecular mimicry of MAGE-A6 and. Oncoimmunology 2014, 3, e954501. [Google Scholar] [CrossRef] [PubMed]
- Vujanovic, L.; Mandic, M.; Olson, W.C.; Kirkwood, J.M.; Storkus, W.J. A mycoplasma peptide elicits heteroclitic CD4+ T cell responses against tumor antigen MAGE-A6. Clin. Cancer Res. 2007, 13, 6796–6806. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Kesmir, C.; van Buuren, M.M. Biomarkers in cancer immunotherapy. Cancer Cell 2015, 27, 12–14. [Google Scholar] [CrossRef]
- Wells, D.K.; van Buuren, M.M.; Dang, K.K.; Hubbard-Lucey, V.M.; Sheehan, K.C.F.; Campbell, K.M.; Lamb, A.; Ward, J.P.; Sidney, J.; Blazquez, A.B.; et al. Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell 2020, 183, 818–834.e13. [Google Scholar] [CrossRef] [PubMed]
- De Plaen, E.; Lurquin, C.; Van Pel, A.; Mariamé, B.; Szikora, J.P.; Wölfel, T.; Sibille, C.; Chomez, P.; Boon, T. Immunogenic (tum-) variants of mouse tumor P815: Cloning of the gene of tum- antigen P91A and identification of the tum- mutation. Proc. Natl. Acad. Sci. USA 1988, 85, 2274–2278. [Google Scholar] [CrossRef] [PubMed]
- Lurquin, C.; Van Pel, A.; Mariamé, B.; De Plaen, E.; Szikora, J.P.; Janssens, C.; Reddehase, M.J.; Lejeune, J.; Boon, T. Structure of the gene of tum- transplantation antigen P91A: The mutated exon encodes a peptide recognized with Ld by cytolytic T cells. Cell 1989, 58, 293–303. [Google Scholar] [CrossRef]
- Monach, P.A.; Meredith, S.C.; Siegel, C.T.; Schreiber, H. A unique tumor antigen produced by a single amino acid substitution. Immunity 1995, 2, 45–59. [Google Scholar] [CrossRef]
- Wölfel, T.; Hauer, M.; Schneider, J.; Serrano, M.; Wölfel, C.; Klehmann-Hieb, E.; De Plaen, E.; Hankeln, T.; Meyer zum Büschenfelde, K.H.; Beach, D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 1995, 269, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.F.; Wang, X.; Atwood, A.C.; Topalian, S.L.; Rosenberg, S.A. Cloning genes encoding MHC class II-restricted antigens: Mutated CDC27 as a tumor antigen. Science 1999, 284, 1351–1354. [Google Scholar] [CrossRef]
- Coulie, P.G.; Lehmann, F.; Lethé, B.; Herman, J.; Lurquin, C.; Andrawiss, M.; Boon, T. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc. Natl. Acad. Sci. USA 1995, 92, 7976–7980. [Google Scholar] [CrossRef]
- Roberts, N.D.; Kortschak, R.D.; Parker, W.T.; Schreiber, A.W.; Branford, S.; Scott, H.S.; Glonek, G.; Adelson, D.L. A comparative analysis of algorithms for somatic SNV detection in cancer. Bioinformatics 2013, 29, 2223–2230. [Google Scholar] [CrossRef]
- Horak, P.; Fröhling, S.; Glimm, H. Integrating next-generation sequencing into clinical oncology: Strategies, promises and pitfalls. ESMO Open 2016, 1, e000094. [Google Scholar] [CrossRef]
- Karasaki, T.; Nagayama, K.; Kuwano, H.; Nitadori, J.I.; Sato, M.; Anraku, M.; Hosoi, A.; Matsushita, H.; Takazawa, M.; Ohara, O.; et al. Prediction and prioritization of neoantigens: Integration of RNA sequencing data with whole-exome sequencing. Cancer Sci. 2017, 108, 170–177. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Bräunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016, 7, 13404. [Google Scholar] [CrossRef] [PubMed]
- Westerink, J.; Visseren, F.L.J.; Spiering, W. Diagnostic Clinical Genome and Exome Sequencing. N. Engl. J. Med. 2014, 371, 1169. [Google Scholar] [PubMed]
- Zhou, C.; Zhu, C.; Liu, Q. Toward in silico Identification of Tumor Neoantigens in Immunotherapy. Trends Mol. Med. 2019, 25, 980–992. [Google Scholar] [CrossRef] [PubMed]
- De Mattos-Arruda, L.; Blanco-Heredia, J.; Aguilar-Gurrieri, C.; Carrillo, J.; Blanco, J. New emerging targets in cancer immunotherapy: The role of neoantigens. ESMO Open 2020, 4. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Coukos, G. Mass spectrometry-based antigen discovery for cancer immunotherapy. Curr. Opin. Immunol. 2016, 41, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Gros, A.; Parkhurst, M.R.; Tran, E.; Pasetto, A.; Robbins, P.F.; Ilyas, S.; Prickett, T.D.; Gartner, J.J.; Crystal, J.S.; Roberts, I.M.; et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 2016, 22, 433–438. [Google Scholar] [CrossRef]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef]
- Yossef, R.; Tran, E.; Deniger, D.C.; Gros, A.; Pasetto, A.; Parkhurst, M.R.; Gartner, J.J.; Prickett, T.D.; Cafri, G.; Robbins, P.F.; et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Kote, S.; Pirog, A.; Bedran, G.; Alfaro, J.; Dapic, I. Mass Spectrometry-Based Identification of MHC-Associated Peptides. Cancers 2020, 12, 535. [Google Scholar] [CrossRef]
- Chen, R.; Fulton, K.M.; Twine, S.M.; Li, J. Identification of MHC peptides using mass spectrometry for neoantigen discovery and vaccine development. Mass Spectrom. Rev. 2019. [Google Scholar] [CrossRef]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Kalaora, S.; Barnea, E.; Merhavi-Shoham, E.; Qutob, N.; Teer, J.K.; Shimony, N.; Schachter, J.; Rosenberg, S.A.; Besser, M.J.; Admon, A.; et al. Use of HLA peptidomics and whole exome sequencing to identify human immunogenic neo-antigens. Oncotarget 2016, 7, 5110–5117. [Google Scholar] [CrossRef] [PubMed]
- Ritz, D.; Gloger, A.; Weide, B.; Garbe, C.; Neri, D.; Fugmann, T. High-sensitivity HLA class I peptidome analysis enables a precise definition of peptide motifs and the identification of peptides from cell lines and patients’ sera. Proteomics 2016, 16, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Bassani-Sternberg, M.; Pletscher-Frankild, S.; Jensen, L.J.; Mann, M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol. Cell. Proteomics 2015, 14, 658–673. [Google Scholar] [CrossRef]
- Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Haabeth, O.A.; Chen, B.; Swaminathan, K.; Rawson, K.; Liu, C.L.; Steiner, D.; Lund, P.; et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature 2017, 543, 723–727. [Google Scholar] [CrossRef]
- Bräunlein, E.; Krackhardt, A.M. Identification and Characterization of Neoantigens As Well As Respective Immune Responses in Cancer Patients. Front. Immunol. 2017, 8, 1702. [Google Scholar] [CrossRef]
- Lanoix, J.; Durette, C.; Courcelles, M.; Cossette, É.; Comtois-Marotte, S.; Hardy, M.P.; Côté, C.; Perreault, C.; Thibault, P. Comparison of the MHC I Immunopeptidome Repertoire of B-Cell Lymphoblasts Using Two Isolation Methods. Proteomics 2018, 18, e1700251. [Google Scholar] [CrossRef]
- Chen, F.; Zou, Z.; Du, J.; Su, S.; Shao, J.; Meng, F.; Yang, J.; Xu, Q.; Ding, N.; Yang, Y.; et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. J. Clin. Investig. 2019, 129, 2056–2070. [Google Scholar] [CrossRef]
- Perumal, D.; Imai, N.; Laganà, A.; Finnigan, J.; Melnekoff, D.; Leshchenko, V.V.; Solovyov, A.; Madduri, D.; Chari, A.; Cho, H.J.; et al. Mutation-derived Neoantigen-specific T-cell Responses in Multiple Myeloma. Clin. Cancer Res. 2020, 26, 450–464. [Google Scholar] [CrossRef]
- Joglekar, A.V.; Leonard, M.T.; Jeppson, J.D.; Swift, M.; Li, G.; Wong, S.; Peng, S.; Zaretsky, J.M.; Heath, J.R.; Ribas, A.; et al. T cell antigen discovery via signaling and antigen-presenting bifunctional receptors. Nat. Methods 2019, 16, 191–198. [Google Scholar] [CrossRef]
- Kisielow, J.; Obermair, F.J.; Kopf, M. Deciphering CD4. Nat. Immunol. 2019, 20, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.N.; Kishton, R.J.; Restifo, N.P. Developing neoantigen-targeted T cell-based treatments for solid tumors. Nat. Med. 2019, 25, 1488–1499. [Google Scholar] [CrossRef] [PubMed]
- D’Ippolito, E.; Wagner, K.I.; Busch, D.H. Needle in a Haystack: The Naïve Repertoire as a Source of T Cell Receptors for Adoptive Therapy with Engineered T Cells. Int. J. Mol. Sci. 2020, 21, 8324. [Google Scholar] [CrossRef] [PubMed]
- Miescher, F. Die Histochemischen und Physiologischen Arbeiten; Leipzig: Vogel, Japan, 1897; 704p, Available online: https://rmda.kulib.kyoto-u.ac.jp/en/item/rb00029652#?c=0&m=0&s=0&cv=0&r=0&xywh=-6240%2C-223%2C18287%2C4457.
- Brenner, S.; Jacob, F.; Meselson, M. An unstable intermediate carrying information from genes to ribosomes for protein synthesis. Nature 1961, 190, 576–581. [Google Scholar] [CrossRef]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magné, R.; Gomard, E.; Guillet, J.G.; Lévy, J.P.; Meulien, P. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur. J. Immunol. 1993, 23, 1719–1722. [Google Scholar] [CrossRef]
- Conry, R.M.; LoBuglio, A.F.; Wright, M.; Sumerel, L.; Pike, M.J.; Johanning, F.; Benjamin, R.; Lu, D.; Curiel, D.T. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995, 55, 1397–1400. [Google Scholar]
- Heiser, A.; Coleman, D.; Dannull, J.; Yancey, D.; Maurice, M.A.; Lallas, C.D.; Dahm, P.; Niedzwiecki, D.; Gilboa, E.; Vieweg, J. Autologous dendritic cells transfected with prostate-specific antigen RNA stimulate CTL responses against metastatic prostate tumors. J. Clin. Investig. 2002, 109, 409–417. [Google Scholar] [CrossRef]
- Schuurhuis, D.H.; Verdijk, P.; Schreibelt, G.; Aarntzen, E.H.; Scharenborg, N.; de Boer, A.; van de Rakt, M.W.; Kerkhoff, M.; Gerritsen, M.J.; Eijckeler, F.; et al. In situ expression of tumor antigens by messenger RNA-electroporated dendritic cells in lymph nodes of melanoma patients. Cancer Res. 2009, 69, 2927–2934. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; van Baren, N.; Lucas, S.; Kvistborg, P.; Thielemans, K.; Neyns, B. Phase II Study of Autologous Monocyte-Derived mRNA Electroporated Dendritic Cells (TriMixDC-MEL) Plus Ipilimumab in Patients With Pretreated Advanced Melanoma. J. Clin. Oncol. 2016, 34, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Wilgenhof, S.; Corthals, J.; Van Nuffel, A.M.; Benteyn, D.; Heirman, C.; Bonehill, A.; Thielemans, K.; Neyns, B. Long-term clinical outcome of melanoma patients treated with messenger RNA-electroporated dendritic cell therapy following complete resection of metastases. Cancer Immunol. Immunother. 2015, 64, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Aarntzen, E.H.; Schreibelt, G.; Bol, K.; Lesterhuis, W.J.; Croockewit, A.J.; de Wilt, J.H.; van Rossum, M.M.; Blokx, W.A.; Jacobs, J.F.; Duiveman-de Boer, T.; et al. Vaccination with mRNA-electroporated dendritic cells induces robust tumor antigen-specific CD4+ and CD8+ T cells responses in stage III and IV melanoma patients. Clin. Cancer Res. 2012, 18, 5460–5470. [Google Scholar] [CrossRef] [PubMed]
- Bol, K.F.; Figdor, C.G.; Aarntzen, E.H.; Welzen, M.E.; van Rossum, M.M.; Blokx, W.A.; van de Rakt, M.W.; Scharenborg, N.M.; de Boer, A.J.; Pots, J.M.; et al. Intranodal vaccination with mRNA-optimized dendritic cells in metastatic melanoma patients. Oncoimmunology 2015, 4, e1019197. [Google Scholar] [CrossRef]
- Dannull, J.; Haley, N.R.; Archer, G.; Nair, S.; Boczkowski, D.; Harper, M.; De Rosa, N.; Pickett, N.; Mosca, P.J.; Burchette, J.; et al. Melanoma immunotherapy using mature DCs expressing the constitutive proteasome. J. Clin. Investig. 2013, 123, 3135–3145. [Google Scholar] [CrossRef] [PubMed]
- Coosemans, A.; Vanderstraeten, A.; Tuyaerts, S.; Verschuere, T.; Moerman, P.; Berneman, Z.N.; Vergote, I.; Amant, F.; VAN Gool, S.W. Wilms’ Tumor Gene 1 (WT1)--loaded dendritic cell immunotherapy in patients with uterine tumors: A phase I/II clinical trial. Anticancer Res. 2013, 33, 5495–5500. [Google Scholar] [PubMed]
- Coosemans, A.; Vanderstraeten, A.; Tuyaerts, S.; Verschuere, T.; Moerman, P.; Berneman, Z.; Vergote, I.; Amant, F.; Van Gool, S.W. Immunological response after WT1 mRNA-loaded dendritic cell immunotherapy in ovarian carcinoma and carcinosarcoma. Anticancer Res. 2013, 33, 3855–3859. [Google Scholar]
- Hernando, J.J.; Park, T.W.; Fischer, H.P.; Zivanovic, O.; Braun, M.; Pölcher, M.; Grünn, U.; Leutner, C.; Pötzsch, B.; Kuhn, W. Vaccination with dendritic cells transfected with mRNA-encoded folate-receptor-alpha for relapsed metastatic ovarian cancer. Lancet Oncol. 2007, 8, 451–454. [Google Scholar] [CrossRef]
- Shindo, Y.; Hazama, S.; Maeda, Y.; Matsui, H.; Iida, M.; Suzuki, N.; Yoshimura, K.; Ueno, T.; Yoshino, S.; Sakai, K.; et al. Adoptive immunotherapy with MUC1-mRNA transfected dendritic cells and cytotoxic lymphocytes plus gemcitabine for unresectable pancreatic cancer. J. Transl. Med. 2014, 12, 175. [Google Scholar] [CrossRef]
- Morse, M.A.; Nair, S.K.; Boczkowski, D.; Tyler, D.; Hurwitz, H.I.; Proia, A.; Clay, T.M.; Schlom, J.; Gilboa, E.; Lyerly, H.K. The feasibility and safety of immunotherapy with dendritic cells loaded with CEA mRNA following neoadjuvant chemoradiotherapy and resection of pancreatic cancer. Int. J. Gastrointest. Cancer 2002, 32, 1–6. [Google Scholar] [CrossRef]
- Lesterhuis, W.J.; De Vries, I.J.; Schreibelt, G.; Schuurhuis, D.H.; Aarntzen, E.H.; De Boer, A.; Scharenborg, N.M.; Van De Rakt, M.; Hesselink, E.J.; Figdor, C.G.; et al. Immunogenicity of dendritic cells pulsed with CEA peptide or transfected with CEA mRNA for vaccination of colorectal cancer patients. Anticancer Res. 2010, 30, 5091–5097. [Google Scholar] [PubMed]
- Wang, D.; Zhang, B.; Gao, H.; Ding, G.; Wu, Q.; Zhang, J.; Liao, L.; Chen, H. Clinical research of genetically modified dendritic cells in combination with cytokine-induced killer cell treatment in advanced renal cancer. BMC Cancer 2014, 14, 251. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Yoshimura, K.; Matsui, H.; Shindo, Y.; Tamesa, T.; Tokumitsu, Y.; Hashimoto, N.; Tokuhisa, Y.; Sakamoto, K.; Sakai, K.; et al. Dendritic cells transfected with heat-shock protein 70 messenger RNA for patients with hepatitis C virus-related hepatocellular carcinoma: A phase 1 dose escalation clinical trial. Cancer Immunol. Immunother. 2015, 64, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015, 519, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Batich, K.A.; Reap, E.A.; Archer, G.E.; Sanchez-Perez, L.; Nair, S.K.; Schmittling, R.J.; Norberg, P.; Xie, W.; Herndon, J.E.; Healy, P.; et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clin. Cancer Res. 2017, 23, 1898–1909. [Google Scholar] [CrossRef] [PubMed]
- Reap, E.A.; Suryadevara, C.M.; Batich, K.A.; Sanchez-Perez, L.; Archer, G.E.; Schmittling, R.J.; Norberg, P.K.; Herndon, J.E.; Healy, P.; Congdon, K.L.; et al. Dendritic Cells Enhance Polyfunctionality of Adoptively Transferred T Cells That Target Cytomegalovirus in Glioblastoma. Cancer Res. 2018, 78, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Van de Velde, A.L.; Smits, E.L.; Van Tendeloo, V.F.; Juliusson, G.; Cools, N.; Nijs, G.; Stein, B.; Lion, E.; Van Driessche, A.; et al. Dendritic cell vaccination as postremission treatment to prevent or delay relapse in acute myeloid leukemia. Blood 2017, 130, 1713–1721. [Google Scholar] [CrossRef]
- Van Tendeloo, V.F.; Van de Velde, A.; Van Driessche, A.; Cools, N.; Anguille, S.; Ladell, K.; Gostick, E.; Vermeulen, K.; Pieters, K.; Nijs, G.; et al. Induction of complete and molecular remissions in acute myeloid leukemia by Wilms’ tumor 1 antigen-targeted dendritic cell vaccination. Proc. Natl. Acad. Sci. USA 2010, 107, 13824–13829. [Google Scholar] [CrossRef]
- Khoury, H.J.; Collins, R.H.; Blum, W.; Stiff, P.S.; Elias, L.; Lebkowski, J.S.; Reddy, A.; Nishimoto, K.P.; Sen, D.; Wirth, E.D.; et al. Immune responses and long-term disease recurrence status after telomerase-based dendritic cell immunotherapy in patients with acute myeloid leukemia. Cancer 2017, 123, 3061–3072. [Google Scholar] [CrossRef]
- Hobo, W.; Strobbe, L.; Maas, F.; Fredrix, H.; Greupink-Draaisma, A.; Esendam, B.; de Witte, T.; Preijers, F.; Levenga, H.; van Rees, B.; et al. Immunogenicity of dendritic cells pulsed with MAGE3, Survivin and B-cell maturation antigen mRNA for vaccination of multiple myeloma patients. Cancer Immunol. Immunother. 2013, 62, 1381–1392. [Google Scholar] [CrossRef]
- Van Lint, S.; Wilgenhof, S.; Heirman, C.; Corthals, J.; Breckpot, K.; Bonehill, A.; Neyns, B.; Thielemans, K. Optimized dendritic cell-based immunotherapy for melanoma: The TriMix-formula. Cancer Immunol. Immunother. 2014, 63, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Weide, B.; Pascolo, S.; Scheel, B.; Derhovanessian, E.; Pflugfelder, A.; Eigentler, T.K.; Pawelec, G.; Hoerr, I.; Rammensee, H.G.; Garbe, C. Direct injection of protamine-protected mRNA: Results of a phase 1/2 vaccination trial in metastatic melanoma patients. J. Immunother. 2009, 32, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Yang, K.; Li, R.; Zhang, L. mRNA Vaccine Era-Mechanisms, Drug Platform and Clinical Prospection. Int. J. Mol. Sci. 2020, 21, 6582. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Dewitte, H. Three decades of mRNA development. Nano Today 2019, 28, 100766. [Google Scholar] [CrossRef]
- Boczkowski, D.; Nair, S.K.; Snyder, D.; Gilboa, E. Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J. Exp. Med. 1996, 184, 465–472. [Google Scholar] [CrossRef]
- Kreiter, S.; Selmi, A.; Diken, M.; Koslowski, M.; Britten, C.M.; Huber, C.; Türeci, O.; Sahin, U. Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. 2010, 70, 9031–9040. [Google Scholar] [CrossRef]
- Van Lint, S.; Goyvaerts, C.; Maenhout, S.; Goethals, L.; Disy, A.; Benteyn, D.; Pen, J.; Bonehill, A.; Heirman, C.; Breckpot, K.; et al. Preclinical Evaluation of TriMix and Antigen mRNA-Based Antitumor Therapy. Cancer Res. 2012, 72, 1661–1671. [Google Scholar] [CrossRef]
- Van Lint, S.; Renmans, D.; Broos, K.; Goethals, L.; Maenhout, S.; Benteyn, D.; Goyvaerts, C.; Du Four, S.; Van der Jeught, K.; Bialkowski, L.; et al. Intratumoral Delivery of TriMix mRNA Results in T-cell Activation by Cross-Presenting Dendritic Cells. Cancer Immunol. Res. 2016. [Google Scholar] [CrossRef]
- Broos, K.; Van der Jeught, K.; Puttemans, J.; Goyvaerts, C.; Heirman, C.; Dewitte, H.; Verbeke, R.; Lentacker, I.; Thielemans, K.; Breckpot, K. Particle-mediated Intravenous Delivery of Antigen mRNA Results in Strong Antigen-specific T-cell Responses Despite the Induction of Type I Interferon. Mol. Ther. Nucleic Acids 2016, 5, e326. [Google Scholar] [CrossRef]
- Van der Jeught, K.; De Koker, S.; Bialkowski, L.; Heirman, C.; Tjok Joe, P.; Perche, F.; Maenhout, S.; Bevers, S.; Broos, K.; Deswarte, K.; et al. Dendritic Cell Targeting mRNA Lipopolyplexes Combine Strong Antitumor T-Cell Immunity with Improved Inflammatory Safety. ACS Nano 2018, 12, 9815–9829. [Google Scholar] [CrossRef]
- Verbeke, R.; Lentacker, I.; Wayteck, L.; Breckpot, K.; Van Bockstal, M.; Descamps, B.; Vanhove, C.; De Smedt, S.C.; Dewitte, H. Co-delivery of nucleoside-modified mRNA and TLR agonists for cancer immunotherapy: Restoring the immunogenicity of immunosilent mRNA. J. Control. Release off. J. Control. Release Soc. 2017. [Google Scholar] [CrossRef]
- Udhayakumar, V.K.; De Beuckelaer, A.; McCaffrey, J.; McCrudden, C.M.; Kirschman, J.o.L.; Vanover, D.; Van Hoecke, L.; Roose, K.; Deswarte, K.; De Geest, B.G.; et al. Arginine-Rich Peptide-Based mRNA Nanocomplexes Efficiently Instigate Cytotoxic T Cell Immunity Dependent on the Amphipathic Organization of the Peptide. Adv. Healthc. Mater. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Michiels, A.; Tuyaerts, S.; Bonehill, A.; Corthals, J.; Breckpot, K.; Heirman, C.; Van Meirvenne, S.; Dullaers, M.; Allard, S.; Brasseur, F.; et al. Electroporation of immature and mature dendritic cells: Implications for dendritic cell-based vaccines. Gene Ther. 2005, 12, 772–782. [Google Scholar] [CrossRef] [PubMed]
- Van Tendeloo, V.F.; Ponsaerts, P.; Lardon, F.; Nijs, G.; Lenjou, M.; Van Broeckhoven, C.; Van Bockstaele, D.R.; Berneman, Z.N. Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: Superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood 2001, 98, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Dewitte, H.; Van Lint, S.; Heirman, C.; Thielemans, K.; De Smedt, S.C.; Breckpot, K.; Lentacker, I. The potential of antigen and TriMix sonoporation using mRNA-loaded microbubbles for ultrasound-triggered cancer immunotherapy. J. Control. Release 2014, 194, 28–36. [Google Scholar] [CrossRef]
- Devoldere, J.; Dewitte, H.; De Smedt, S.C.; Remaut, K. Evading innate immunity in nonviral mRNA delivery: Don’t shoot the messenger. Drug Discov. Today 2016, 21, 11–25. [Google Scholar] [CrossRef]
- Andries, O.; De Filette, M.; De Smedt, S.C.; Demeester, J.; Van Poucke, M.; Peelman, L.; Sanders, N.N. Innate immune response and programmed cell death following carrier-mediated delivery of unmodified mRNA to respiratory cells. J. Control. Release 2013, 167, 157–166. [Google Scholar] [CrossRef]
- Karikó, K.; Ni, H.; Capodici, J.; Lamphier, M.; Weissman, D. mRNA is an endogenous ligand for Toll-like receptor 3. J. Biol. Chem. 2004, 279, 12542–12550. [Google Scholar] [CrossRef]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5’-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef]
- Züst, R.; Cervantes-Barragan, L.; Habjan, M.; Maier, R.; Neuman, B.W.; Ziebuhr, J.; Szretter, K.J.; Baker, S.C.; Barchet, W.; Diamond, M.S.; et al. Ribose 2’-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011, 12, 137–143. [Google Scholar] [CrossRef] [PubMed]
- De Beuckelaer, A.; Grooten, J.; De Koker, S. Type I Interferons Modulate CD8. Trends Mol. Med. 2017, 23, 216–226. [Google Scholar] [CrossRef] [PubMed]
- De Beuckelaer, A.; Pollard, C.; Van Lint, S.; Roose, K.; Van Hoecke, L.; Naessens, T.; Udhayakumar, V.K.; Smet, M.; Sanders, N.; Lienenklaus, S.; et al. Type I Interferons Interfere with the Capacity of mRNA Lipoplex Vaccines to Elicit Cytolytic T Cell Responses. Mol. Ther. 2016, 24, 2012–2020. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Kallen, K.J.; Heidenreich, R.; Schnee, M.; Petsch, B.; Schlake, T.; Thess, A.; Baumhof, P.; Scheel, B.; Koch, S.D.; Fotin-Mleczek, M. A novel, disruptive vaccination technology: Self-adjuvanted RNActive(®) vaccines. Hum. Vaccines Immunother. 2013, 9, 2263–2276. [Google Scholar] [CrossRef] [PubMed]
- Aerts-Toegaert, C.; Heirman, C.; Tuyaerts, S.; Corthals, J.; Aerts, J.L.; Bonehill, A.; Thielemans, K.; Breckpot, K. CD83 expression on dendritic cells and T cells: Correlation with effective immune responses. Eur. J. Immunol. 2007, 37, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Scheidegger, D.; Palmer-Lehmann, K.; Lane, P.; Lanzavecchia, A.; Alber, G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J. Exp. Med. 1996, 184, 747–752. [Google Scholar] [CrossRef]
- Ma, X.; Chow, J.M.; Gri, G.; Carra, G.; Gerosa, F.; Wolf, S.F.; Dzialo, R.; Trinchieri, G. The interleukin 12 p40 gene promoter is primed by interferon gamma in monocytic cells. J. Exp. Med. 1996, 183, 147–157. [Google Scholar] [CrossRef]
- Kormann, M.S.; Hasenpusch, G.; Aneja, M.K.; Nica, G.; Flemmer, A.W.; Herber-Jonat, S.; Huppmann, M.; Mays, L.E.; Illenyi, M.; Schams, A.; et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat. Biotechnol. 2011, 29, 154–157. [Google Scholar] [CrossRef]
- Martin, S.A.; Moss, B. Modification of RNA by mRNA guanylyltransferase and mRNA (guanine-7-)methyltransferase from vaccinia virions. J. Biol. Chem. 1975, 250, 9330–9335. [Google Scholar]
- Stepinski, J.; Waddell, C.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Synthesis and properties of mRNAs containing the novel “anti-reverse” cap analogs 7-methyl(3’-O-methyl)GpppG and 7-methyl (3’-deoxy)GpppG. RNA 2001, 7, 1486–1495. [Google Scholar] [PubMed]
- Grudzien-Nogalska, E.; Stepinski, J.; Jemielity, J.; Zuberek, J.; Stolarski, R.; Rhoads, R.E.; Darzynkiewicz, E. Synthesis of anti-reverse cap analogs (ARCAs) and their applications in mRNA translation and stability. Methods Enzymol. 2007, 431, 203–227. [Google Scholar] [CrossRef] [PubMed]
- Grudzien-Nogalska, E.; Jemielity, J.; Kowalska, J.; Darzynkiewicz, E.; Rhoads, R.E. Phosphorothioate cap analogs stabilize mRNA and increase translational efficiency in mammalian cells. RNA 2007, 13, 1745–1755. [Google Scholar] [CrossRef] [PubMed]
- Geall, A.J.; Mandl, C.W.; Ulmer, J.B. RNA: The new revolution in nucleic acid vaccines. Semin. Immunol. 2013, 25, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Baiersdörfer, M.; Boros, G.; Muramatsu, H.; Mahiny, A.; Vlatkovic, I.; Sahin, U.; Karikó, K. A Facile Method for the Removal of dsRNA Contaminant from In Vitro-Transcribed mRNA. Mol. Ther. Nucleic Acids 2019, 15, 26–35. [Google Scholar] [CrossRef]
- Foster, J.B.; Choudhari, N.; Perazzelli, J.; Storm, J.; Hofmann, T.J.; Jain, P.; Storm, P.B.; Pardi, N.; Weissman, D.; Waanders, A.J.; et al. Purification of mRNA Encoding Chimeric Antigen Receptor Is Critical for Generation of a Robust T-Cell Response. Hum. Gene Ther. 2019, 30, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Krupp, G. Unusual promoter-independent transcription reactions with bacteriophage RNA polymerases. Nucleic Acids Res. 1989, 17, 3023–3036. [Google Scholar] [CrossRef]
- Cazenave, C.; Uhlenbeck, O.C. RNA template-directed RNA synthesis by T7 RNA polymerase. Proc. Natl. Acad. Sci. USA 1994, 91, 6972–6976. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Van Nuffel, A.M.; Corthals, J.; Heirman, C.; Tuyaerts, S.; Benteyn, D.; De Coninck, A.; Van Riet, I.; Verfaillie, G.; Vandeloo, J.; et al. Therapeutic vaccination with an autologous mRNA electroporated dendritic cell vaccine in patients with advanced melanoma. J. Immunother. 2011, 34, 448–456. [Google Scholar] [CrossRef]
- Jansen, Y.; Kruse, V.; Corthals, J.; Schats, K.; van Dam, P.J.; Seremet, T.; Heirman, C.; Brochez, L.; Kockx, M.; Thielemans, K.; et al. A randomized controlled phase II clinical trial on mRNA electroporated autologous monocyte-derived dendritic cells (TriMixDC-MEL) as adjuvant treatment for stage III/IV melanoma patients who are disease-free following the resection of macrometastases. Cancer Immunol. Immunother. 2020. [Google Scholar] [CrossRef]
- De Keersmaecker, B.; Claerhout, S.; Carrasco, J.; Bar, I.; Corthals, J.; Wilgenhof, S.; Neyns, B.; Thielemans, K. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: Link between T-cell activation and clinical responses in advanced melanoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Heesen, L.; Frenzel, K.; Bolte, S.; Bukur, V.; Diken, M.; Derhovanessian, E.; Kreiter, S.; Kuhn, A.N.; Kühlcke, K.; Löwer, M.; et al. Mutanome engineered RNA immuno-therapy (MERIT) for patients with triple negative breast cancer (TNBC). In Proceedings of the AACR Annual Meeting, Atlanta, GA, USA, 29 March–3 April 2019; p. CT221. [Google Scholar]
- Roudko, V.; Greenbaum, B.; Bhardwaj, N. Computational Prediction and Validation of Tumor-Associated Neoantigens. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Richters, M.M.; Xia, H.M.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 2019, 11, 21. [Google Scholar] [CrossRef]
- Bulik-Sullivan, B.; Busby, J.; Palmer, C.D.; Davis, M.J.; Murphy, T.; Clark, A.; Busby, M.; Duke, F.; Yang, A.; Young, L.; et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat. Biotechnol. 2019, 37, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Jou, J.; Harrington, K.J.; Zocca, M.B.; Ehrnrooth, E.; Cohen, E.E.W. The Changing Landscape of Therapeutic Cancer Vaccines—Novel Platforms and Neoantigen Identification. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Koster, J.; Plasterk, R.H.A. A library of Neo Open Reading Frame peptides (NOPs) as a sustainable resource of common neoantigens in up to 50% of cancer patients. Sci. Rep. 2019, 9, 6577. [Google Scholar] [CrossRef] [PubMed]
- Roudko, V.; Bozkus, C.C.; Orfanelli, T.; McClain, C.B.; Carr, C.; O’Donnell, T.; Chakraborty, L.; Samstein, R.; Huang, K.L.; Blank, S.V.; et al. Shared Immunogenic Poly-Epitope Frameshift Mutations in Microsatellite Unstable Tumors. Cell 2020, 183, 1634–1649.e1617. [Google Scholar] [CrossRef]
- Danilova, L.; Anagnostou, V.; Caushi, J.X.; Sidhom, J.W.; Guo, H.; Chan, H.Y.; Suri, P.; Tam, A.; Zhang, J.; Asmar, M.E.; et al. The Mutation-Associated Neoantigen Functional Expansion of Specific T Cells (MANAFEST) Assay: A Sensitive Platform for Monitoring Antitumor Immunity. Cancer Immunol. Res. 2018, 6, 888–899. [Google Scholar] [CrossRef]
- Broos, K.; Lecocq, Q.; Keersmaecker, B.; Raes, G.; Corthals, J.; Lion, E.; Thielemans, K.; Devoogdt, N.; Keyaerts, M.; Breckpot, K. Single Domain Antibody-Mediated Blockade of Programmed Death-Ligand 1 on Dendritic Cells Enhances CD8 T-cell Activation and Cytokine Production. Vaccines 2019, 7, 85. [Google Scholar] [CrossRef]
- Versteven, M.; Van den Bergh, J.M.J.; Broos, K.; Fujiki, F.; Campillo-Davo, D.; De Reu, H.; Morimoto, S.; Lecocq, Q.; Keyaerts, M.; Berneman, Z.; et al. A versatile T cell-based assay to assess therapeutic antigen-specific PD-1-targeted approaches. Oncotarget 2018, 9, 27797–27808. [Google Scholar] [CrossRef] [PubMed]
- Kellner, C.; Otte, A.; Cappuzzello, E.; Klausz, K.; Peipp, M. Modulating Cytotoxic Effector Functions by Fc Engineering to Improve Cancer Therapy. Transfus. Med. Hemother. 2017, 44, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Van den Ende, T.; van den Boorn, H.G.; Hoonhout, N.M.; van Etten-Jamaludin, F.S.; Meijer, S.L.; Derks, S.; de Gruijl, T.D.; Bijlsma, M.F.; van Oijen, M.G.H.; van Laarhoven, H.W.M. Priming the tumor immune microenvironment with chemo(radio)therapy: A systematic review across tumor types. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188386. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.L.; Schlom, J.; Hamilton, D.H. Combination therapies utilizing neoepitope-targeted vaccines. Cancer Immunol. Immunother. 2020. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Company or Consortium | Start Date | Country | Core Business | Website |
|---|---|---|---|---|
| TriLink | 1996 | USA | Contract development and manufacturing of mRNA medicines | https://www.trilinkbiotech.com/ |
| Curevac | 2000 | Germany | mRNA medicines, including cancer vaccines | https://www.curevac.com/ |
| BioNtech | 2008 | Germany | mRNA medicines, including neo-antigen cancer vaccines | https://biontech.de/ |
| ModeRNA | 2010 | USA | mRNA medicines for a wide range of diseases and conditions, including cancer | https://www.modernatx.com/ |
| eTheRNA | 2013 | Belgium | mRNA medicines to treat infectious diseases and cancer, including neo-antigen vaccines | https://www.etherna.be/ |
| Kernal Biologics | 2016 | USA | mRNA medicines to treat infectious diseases and cancer | https://www.kernalbio.com/ |
| Stemirna Therapeutics | 2016 | China | mRNA medicines to treat infectious diseases and cancer, including neo-antigen vaccines | http://www.stemirna.com/ |
| Persomed | 2020 | Belgium | DC vaccines based on moDCs modified with neo-antigen and DC activating mRNA | https://www.persomed.be/ |
| Cancer Type | Study | Phase | Formulation | Route | Other Therapy | Response |
|---|---|---|---|---|---|---|
| TNBC | NCT02316457 | Phase I | LNP | i.v. | NA | Study extended |
| Melanoma | NCT02035956 | Phase I | Naked mRNA | i.n. | RBL001/RBL002 | Not published |
| NSCLC | NCT03164772 | Phase I/II | LNP | i.d. | Durvalumab, Tremelumumab | Study ongoing |
| Solid tumors | NCT03289962 | Phase I | Naked mRNA | i.v. | Atezolizumab | Study ongoing |
| Solid tumors | NCT03313778 | Phase I | LNP | i.d. | Pembrolizumab | Study ongoing |
| Solid tumors | NCT03480152 | Phase I/II | Naked mRNA | i.m. | NA | MTD not reached |
| Solid tumors, lymphoma | NCT03468244 | NN | Naked mRNA | s.c. | NA | Study ongoing |
| Metastatic melanoma | NCT03815058 | Phase II | LNP | i.v. | Pembrolizumab | Study ongoing |
| High-risk of recurrence melanoma | NCT03897881 | Phase II | NN | NN | Pembrolizumab | Study ongoing |
| Esophageal cancer, NSCLC | NCT03908671 | NN | LNP | s.c. | NA | Study ongoing |
| Pancreas cancer | NCT04161755 | Phase I | NN | NN | Atezolizumab, chemotherapy | Study Ongoing |
| NSCLC | NCT04267237 | Phase I | LNP | i.v. | Atezolizumab | Study ongoing |
| KRAS-mutant pancreas, colon and lung cancer | NCT03948673 | Phase I | NN | i.m. | Pembrolizumab | Study ongoing |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esprit, A.; de Mey, W.; Bahadur Shahi, R.; Thielemans, K.; Franceschini, L.; Breckpot, K. Neo-Antigen mRNA Vaccines. Vaccines 2020, 8, 776. https://doi.org/10.3390/vaccines8040776
Esprit A, de Mey W, Bahadur Shahi R, Thielemans K, Franceschini L, Breckpot K. Neo-Antigen mRNA Vaccines. Vaccines. 2020; 8(4):776. https://doi.org/10.3390/vaccines8040776
Chicago/Turabian StyleEsprit, Arthur, Wout de Mey, Rajendra Bahadur Shahi, Kris Thielemans, Lorenzo Franceschini, and Karine Breckpot. 2020. "Neo-Antigen mRNA Vaccines" Vaccines 8, no. 4: 776. https://doi.org/10.3390/vaccines8040776
APA StyleEsprit, A., de Mey, W., Bahadur Shahi, R., Thielemans, K., Franceschini, L., & Breckpot, K. (2020). Neo-Antigen mRNA Vaccines. Vaccines, 8(4), 776. https://doi.org/10.3390/vaccines8040776