Strategies to Improve Chimeric Antigen Receptor Therapies for Neuroblastoma

 and
and
Abstract
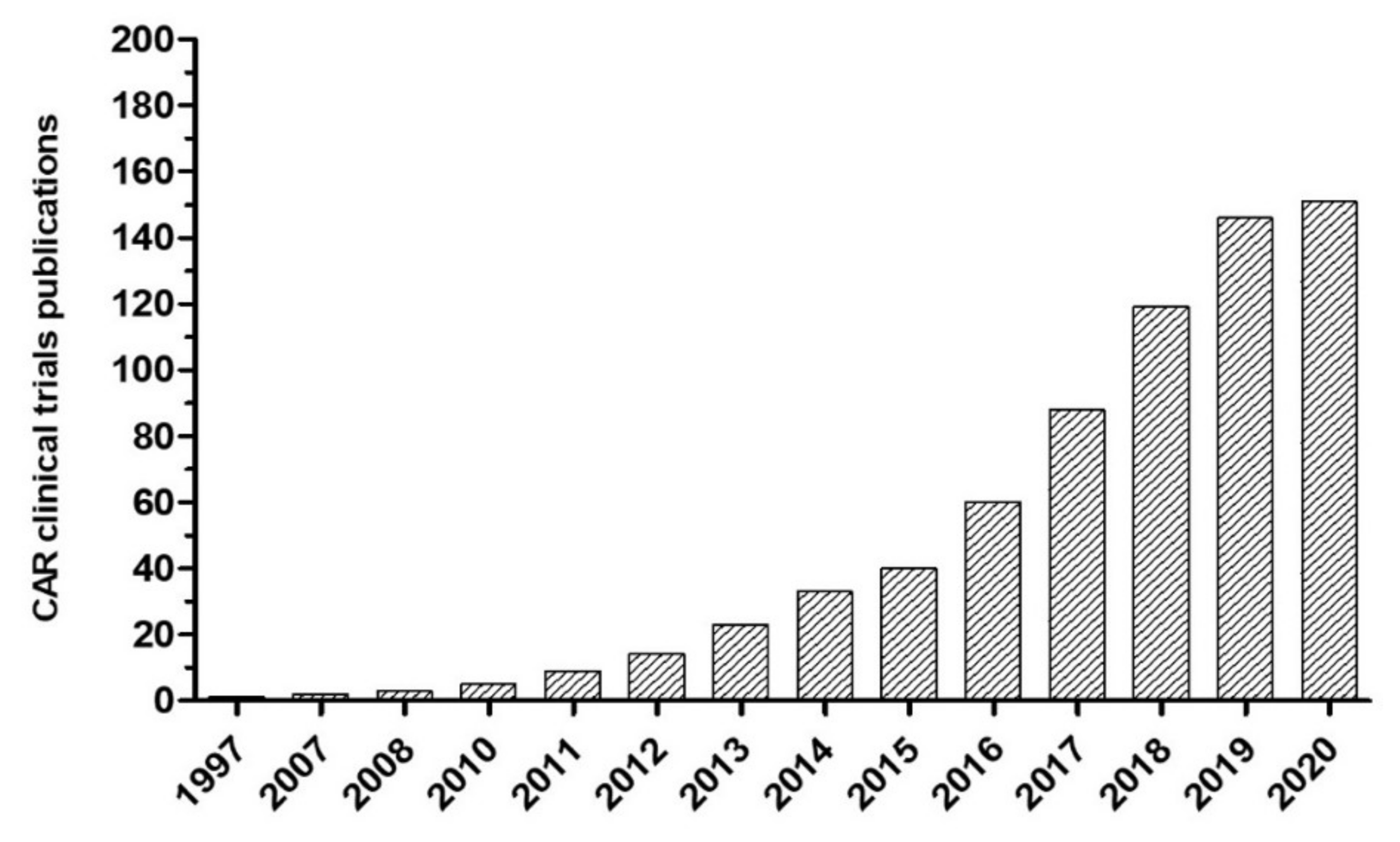
1. Introduction
2. CARs in Neuroblastoma
2.1. Summary of CAR Experience
2.2. Obstacles to Using CARs in Neuroblastoma
2.2.1. CAR T Cell Persistence and Exhaustion
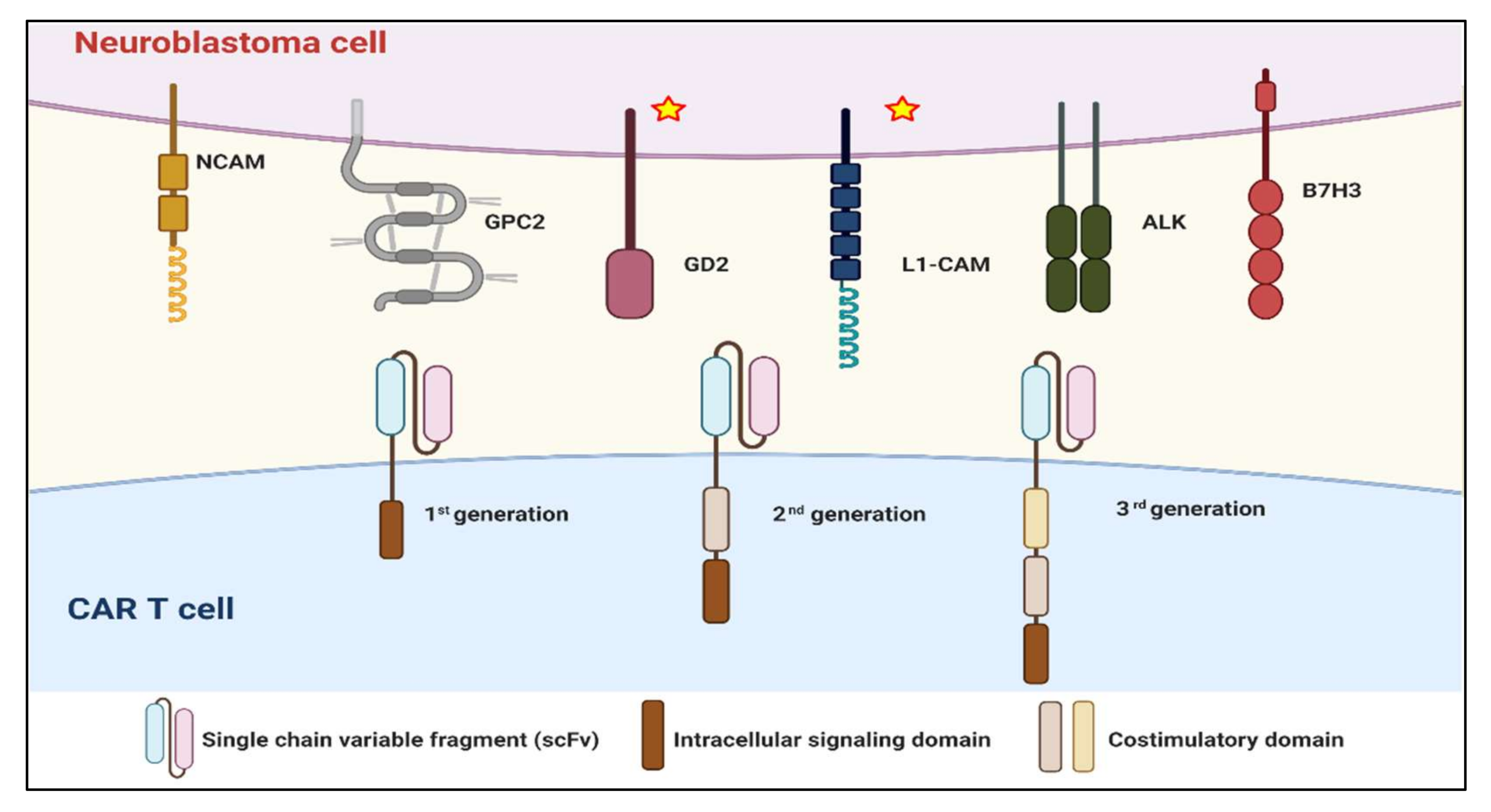
2.2.2. Target Selection and On-Target, Off-Tumor Effect
2.2.3. Tumor Microenvironment
2.2.4. CAR Trafficking
3. Strategies to Improve CARs in Neuroblastoma
3.1. Improving Effector Immune Cells
3.1.1. T lymphocytes
3.1.2. Natural Killer Cells
3.1.3. NKT Cells
3.1.4. γδT Cells
3.2. Modification of CAR Constructs
3.2.1. Hinge and Transmembrane Domain
3.2.2. Costimulatory Domains
3.2.3. Signaling-Transducing Domain
3.2.4. Other Generations of CAR Structures
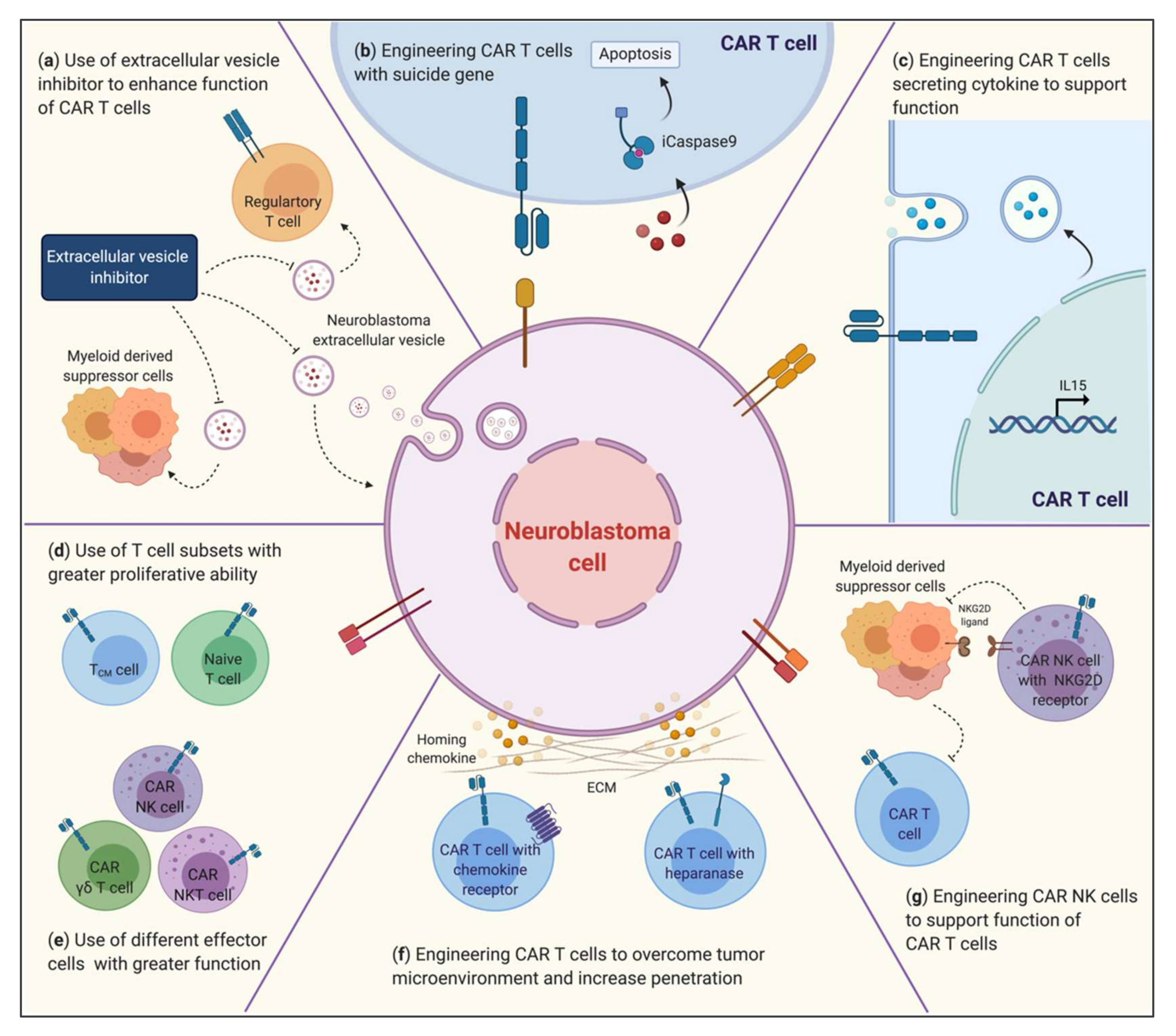
3.3. Overcoming the TME to Improve the Efficacy of CAR Immunotherapy
3.3.1. ECM: Trafficking and Infiltration of Effector Cells
3.3.2. Myeloid-Derived Suppressor Cells
3.3.3. Tumor Extracellular Vesicles
3.4. Combination of CARs
3.4.1. PD-1/PDL-1
3.4.2. Cytokines
4. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ahmed, A.A.; Zhang, L.; Reddivalla, N.; Hetherington, M. Neuroblastoma in children: Update on clinicopathologic and genetic prognostic factors. Pediatr. Hematol. Oncol. 2017, 34, 165–185. [Google Scholar] [CrossRef]
- Moreno, L.; Rubie, H.; Varo, A.; Le Deley, M.C.; Amoroso, L.; Chevance, A.; Garaventa, A.; Gambart, M.; Bautista, F.; Valteau-Couanet, D.; et al. Outcome of children with relapsed or refractory neuroblastoma: A meta-analysis of ITCC/SIOPEN European phase II clinical trials. Pediatr. Blood Cancer 2017, 64, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Altekruse, S.F.; Adamson, P.C.; Reaman, G.H.; Seibel, N.L. Declining childhood and adolescent cancer mortality. Cancer 2014, 120, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Ries, L.A.G.; Smith, M.A.; Gurney, J.G.; Linet, M.; Tamra, T.; Young, J.L.; Bunin, G.R.; Bernstein, L.; Key, C.R.; Lynch, C.F.; et al. (Eds.) Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975–1995; National Cancer Institute, SEER Program: Bethesda, MD, USA, 1999; NIH Pub. No. 99-4649. [Google Scholar]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef]
- Huibregtse, K.E.; Vo, K.T.; DuBois, S.G.; Fetzko, S.; Neuhaus, J.; Batra, V.; Maris, J.M.; Weiss, B.; Marachelian, A.; Yanik, G.A.; et al. Incidence and risk factors for secondary malignancy in patients with neuroblastoma after treatment with (131)I-metaiodobenzylguanidine. Eur. J. Cancer 2016, 66, 144–152. [Google Scholar] [CrossRef]
- Li, R.; Polishchuk, A.; DuBois, S.; Hawkins, R.; Lee, S.W.; Bagatell, R.; Shusterman, S.; Hill-Kayser, C.; Al-Sayegh, H.; Diller, L.; et al. Patterns of Relapse in High-Risk Neuroblastoma Patients Treated With and Without Total Body Irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2017, 97, 270–277. [Google Scholar] [CrossRef]
- Cheung, I.Y.; Kushner, B.H.; Modak, S.; Basu, E.M.; Roberts, S.S.; Cheung, N.V. Phase I trial of anti-GD2 monoclonal antibody hu3F8 plus GM-CSF: Impact of body weight, immunogenicity and anti-GD2 response on pharmacokinetics and survival. Oncoimmunology 2017, 6, e1358331. [Google Scholar] [CrossRef]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334. [Google Scholar] [CrossRef]
- Kushner, B.H.; Ostrovnaya, I.; Cheung, I.Y.; Kuk, D.; Kramer, K.; Modak, S.; Yataghene, K.; Cheung, N.K. Prolonged progression-free survival after consolidating second or later remissions of neuroblastoma with Anti-GD2 immunotherapy and isotretinoin: A prospective Phase II study. Oncoimmunology 2015, 4, e1016704. [Google Scholar] [CrossRef]
- Park, J.R.; Digiusto, D.L.; Slovak, M.; Wright, C.; Naranjo, A.; Wagner, J.; Meechoovet, H.B.; Bautista, C.; Chang, W.C.; Ostberg, J.R.; et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol. Ther. 2007, 15, 825–833. [Google Scholar] [CrossRef]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017, 129, 3322–3331. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Asgharzadeh, S.; Salo, J.A.; Ji, L.; Oberthuer, A.; Fischer, M.; Berthold, F.; Hadjidaniel, M.; Liu, C.W.; Metelitsa, L.S.; Pique-Regi, R.; et al. Clinical significance of tumor-associated inflammatory cells in metastatic neuroblastoma. J. Clin. Oncol. 2012, 30, 3525–3532. [Google Scholar] [CrossRef] [PubMed]
- Heczey, A.; Louis, C.U. Advances in chimeric antigen receptor immunotherapy for neuroblastoma. Discov. Med. 2013, 16, 287–294. [Google Scholar] [PubMed]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.; Gorochov, G.; Waks, T.; Eshhar, Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant. Proc. 1989, 21, 127–130. [Google Scholar]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Savoldo, B.; Dotti, G. Chimeric antigen receptors (CARs) from bench-to-bedside. Immunol. Lett. 2013, 155, 40–42. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.M.; Sotillo, E.; Majzner, R.G. CAR T Cell Therapy for Neuroblastoma. Front. Immunol. 2018, 9, 2380. [Google Scholar] [CrossRef]
- Mosse, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef]
- De Brouwer, S.; De Preter, K.; Kumps, C.; Zabrocki, P.; Porcu, M.; Westerhout, E.M.; Lakeman, A.; Vandesompele, J.; Hoebeeck, J.; Van Maerken, T.; et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin. Cancer Res. 2010, 16, 4353–4362. [Google Scholar] [CrossRef]
- Moog-Lutz, C.; Degoutin, J.; Gouzi, J.Y.; Frobert, Y.; Brunet-de Carvalho, N.; Bureau, J.; Creminon, C.; Vigny, M. Activation and inhibition of anaplastic lymphoma kinase receptor tyrosine kinase by monoclonal antibodies and absence of agonist activity of pleiotrophin. J. Biol. Chem. 2005, 280, 26039–26048. [Google Scholar] [CrossRef]
- Walker, A.J.; Majzner, R.G.; Zhang, L.; Wanhainen, K.; Long, A.H.; Nguyen, S.M.; Lopomo, P.; Vigny, M.; Fry, T.J.; Orentas, R.J.; et al. Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol. Ther. 2017, 25, 2189–2201. [Google Scholar] [CrossRef]
- Li, N.; Fu, H.; Hewitt, S.M.; Dimitrov, D.S.; Ho, M. Therapeutically targeting glypican-2 via single-domain antibody-based chimeric antigen receptors and immunotoxins in neuroblastoma. Proc. Natl. Acad. Sci. USA 2017, 114, E6623–E6631. [Google Scholar] [CrossRef]
- Baral, A.; Ye, H.X.; Jiang, P.C.; Yao, Y.; Mao, Y. B7-H3 and B7-H1 expression in cerebral spinal fluid and tumor tissue correlates with the malignancy grade of glioma patients. Oncol. Lett. 2014, 8, 1195–1201. [Google Scholar] [CrossRef]
- Zhou, Z.; Luther, N.; Ibrahim, G.M.; Hawkins, C.; Vibhakar, R.; Handler, M.H.; Souweidane, M.M. B7-H3, a potential therapeutic target, is expressed in diffuse intrinsic pontine glioma. J. Neurooncol. 2013, 111, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Hirabayashi, K.; Ahn, S.; Kren, N.P.; Montgomery, S.A.; Wang, X.; Tiruthani, K.; Mirlekar, B.; Michaud, D.; Greene, K.; et al. Antitumor Responses in the Absence of Toxicity in Solid Tumors by Targeting B7-H3 via Chimeric Antigen Receptor T Cells. Cancer Cell 2019, 35, 221–237 e228. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef] [PubMed]
- Crossland, D.L.; Denning, W.L.; Ang, S.; Olivares, S.; Mi, T.; Switzer, K.; Singh, H.; Huls, H.; Gold, K.S.; Glisson, B.S.; et al. Antitumor activity of CD56-chimeric antigen receptor T cells in neuroblastoma and SCLC models. Oncogene 2018, 37, 3686–3697. [Google Scholar] [CrossRef]
- Zeromski, J.; Nyczak, E.; Dyszkiewicz, W. Significance of cell adhesion molecules, CD56/NCAM in particular, in human tumor growth and spreading. Folia Histochem. Cytobiol. 2001, 39 (Suppl. 2), 36–37. [Google Scholar]
- Singh, N.; Kulikovskaya, I.; Barrett, D.M.; Binder-Scholl, G.; Jakobsen, B.; Martinez, D.; Pawel, B.; June, C.H.; Kalos, M.D.; Grupp, S.A. T cells targeting NY-ESO-1 demonstrate efficacy against disseminated neuroblastoma. Oncoimmunology 2016, 5, e1040216. [Google Scholar] [CrossRef]
- Spel, L.; Boelens, J.J.; van der Steen, D.M.; Blokland, N.J.; van Noesel, M.M.; Molenaar, J.J.; Heemskerk, M.H.; Boes, M.; Nierkens, S. Natural killer cells facilitate PRAME-specific T-cell reactivity against neuroblastoma. Oncotarget 2015, 6, 35770–35781. [Google Scholar] [CrossRef]
- Arlt, M.J.; Novak-Hofer, I.; Gast, D.; Gschwend, V.; Moldenhauer, G.; Grunberg, J.; Honer, M.; Schubiger, P.A.; Altevogt, P.; Kruger, A. Efficient inhibition of intra-peritoneal tumor growth and dissemination of human ovarian carcinoma cells in nude mice by anti-L1-cell adhesion molecule monoclonal antibody treatment. Cancer Res. 2006, 66, 936–943. [Google Scholar] [CrossRef]
- Wang, X.; Chang, W.C.; Wong, C.W.; Colcher, D.; Sherman, M.; Ostberg, J.R.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011, 118, 1255–1263. [Google Scholar] [CrossRef]
- Kunkele, A.; Taraseviciute, A.; Finn, L.S.; Johnson, A.J.; Berger, C.; Finney, O.; Chang, C.A.; Rolczynski, L.S.; Brown, C.; Mgebroff, S.; et al. Preclinical Assessment of CD171-Directed CAR T-cell Adoptive Therapy for Childhood Neuroblastoma: CE7 Epitope Target Safety and Product Manufacturing Feasibility. Clin. Cancer Res. 2017, 23, 466–477. [Google Scholar] [CrossRef]
- Rossig, C.; Bollard, C.M.; Nuchtern, J.G.; Merchant, D.A.; Brenner, M.K. Targeting of G(D2)-positive tumor cells by human T lymphocytes engineered to express chimeric T-cell receptor genes. Int. J. Cancer 2001, 94, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Cheung, N.K.V. Disialoganglioside GD2 as a therapeutic target for human diseases. Expert Opin. Ther. Tar. 2015, 19, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.K.; Cheung, I.Y.; Kramer, K.; Modak, S.; Kuk, D.; Pandit-Taskar, N.; Chamberlain, E.; Ostrovnaya, I.; Kushner, B.H. Key role for myeloid cells: Phase II results of anti-G(D2) antibody 3F8 plus granulocyte-macrophage colony-stimulating factor for chemoresistant osteomedullary neuroblastoma. Int. J. Cancer 2014, 135, 2199–2205. [Google Scholar] [CrossRef] [PubMed]
- Gilman, A.L.; Ozkaynak, M.F.; Matthay, K.K.; Krailo, M.; Yu, A.L.; Gan, J.; Sternberg, A.; Hank, J.A.; Seeger, R.; Reaman, G.H.; et al. Phase I study of ch14.18 with granulocyte-macrophage colony-stimulating factor and interleukin-2 in children with neuroblastoma after autologous bone marrow transplantation or stem-cell rescue: A report from the Children’s Oncology Group. J. Clin. Oncol. 2009, 27, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Ozkaynak, M.F.; Sondel, P.M.; Krailo, M.D.; Gan, J.; Javorsky, B.; Reisfeld, R.A.; Matthay, K.K.; Reaman, G.H.; Seeger, R.C. Phase I study of chimeric human/murine anti-ganglioside G(D2) monoclonal antibody (ch14.18) with granulocyte-macrophage colony-stimulating factor in children with neuroblastoma immediately after hematopoietic stem-cell transplantation: A Children’s Cancer Group Study. J. Clin. Oncol. 2000, 18, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Straathof, K.; Flutter, B.; Wallace, R.; Thomas, S.; Cheung, G.; Collura, A.; Gileadi, T.; Barton, J.; Wright, G.; Inglott, S.; et al. A Cancer Research UK phase I trial of anti-GD2 chimeric antigen receptor (CAR) transduced T-cells (1RG-CART) in patients with relapsed or refractory neuroblastoma. Cancer Res. 2018, 78. [Google Scholar] [CrossRef]
- Pule, M.A.; Straathof, K.C.; Dotti, G.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol. Ther. 2005, 12, 933–941. [Google Scholar] [CrossRef]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.F.; Mei, Z.Y.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.L.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Sun, J.; Huye, L.E.; Lapteva, N.; Mamonkin, M.; Hiregange, M.; Ballard, B.; Dakhova, O.; Raghavan, D.; Durett, A.G.; Perna, S.K.; et al. Early transduction produces highly functional chimeric antigen receptor-modified virus-specific T-cells with central memory markers: A Production Assistant for Cell Therapy (PACT) translational application. J. Immunother. Cancer 2015, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Huang, W.; Heczey, A.; Liu, D.; Guo, L.; Wood, M.; Jin, J.; Courtney, A.N.; Liu, B.; Di Pierro, E.J.; et al. NKT Cells Coexpressing a GD2-Specific Chimeric Antigen Receptor and IL15 Show Enhanced In Vivo Persistence and Antitumor Activity against Neuroblastoma. Clin. Cancer Res. 2019, 25, 7126–7138. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhao, W.; Yue, Z.; Qin, M.; Jin, M.; Chang, L.J.; Ma, X. 4SCAR-GD2-modified T-cell therapy in neuroblastoma with MYCN amplification: A case report with over 4-year follow-up data. Pediatr. Investig. 2020, 4, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Hattinger, C.M.; Patrizio, M.P.; Magagnoli, F.; Luppi, S.; Serra, M. An update on emerging drugs in osteosarcoma: Towards tailored therapies? Expert Opin. Emerg. Drugs 2019, 24, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Tashiro, H.; Omer, B.; Lapteva, N.; Ando, J.; Ngo, M.; Mehta, B.; Dotti, G.; Kinchington, P.R.; Leen, A.M.; et al. Vaccination Targeting Native Receptors to Enhance the Function and Proliferation of Chimeric Antigen Receptor (CAR)-Modified T Cells. Clin. Cancer Res. 2017, 23, 3499–3509. [Google Scholar] [CrossRef]
- Robbins, P.F.; Dudley, M.E.; Wunderlich, J.; El-Gamil, M.; Li, Y.F.; Zhou, J.; Huang, J.; Powell, D.J., Jr.; Rosenberg, S.A. Cutting edge: Persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J. Immunol. 2004, 173, 7125–7130. [Google Scholar] [CrossRef]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef]
- Himoudi, N.; Yan, M.Y.; Papanastasiou, A.; Anderson, J. MYCN as a target for cancer immunotherapy. Cancer Immunol. Immun. 2008, 57, 693–700. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-transduced T Cells Targeting EGFRvIII in Patients With Glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Thistlethwaite, F.C.; Gilham, D.E.; Guest, R.D.; Rothwell, D.G.; Pillai, M.; Burt, D.J.; Byatte, A.J.; Kirillova, N.; Valle, J.W.; Sharma, S.K.; et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother. 2017, 66, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; DeRenzo, C.C.; Zhang, H.M.; Mata, M.; Gerken, C.; Shree, A.; Yi, Z.Z.; Brawley, V.; Dakhova, O.; Wu, M.F.; et al. Expansion of HER2-CAR T cells after lymphodepletion and clinical responses in patients with advanced sarcoma. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Navai, S.A.; Derenzo, C.; Joseph, S.; Sanber, K.; Byrd, T.; Zhang, H.M.; Mata, M.; Gerken, C.; Shree, A.; Mathew, P.R.; et al. Administration of HER2-CAR T cells after lymphodepletion safely improves T cell expansion and induces clinical responses in patients with advanced sarcomas. Cancer Res. 2019, 79. [Google Scholar] [CrossRef]
- Mina, M.; Boldrini, R.; Citti, A.; Romania, P.; D’Alicandro, V.; De Ioris, M.; Castellano, A.; Furlanello, C.; Locatelli, F.; Fruci, D. Tumor-infiltrating T lymphocytes improve clinical outcome of therapy-resistant neuroblastoma. Oncoimmunology 2015, 4, e1019981. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Simon, J.S.; Grosso, J.F.; Martinez, D.; Pawel, B.R.; Santi, M.; Merchant, M.S.; Geoerger, B.; Hezam, I.; Marty, V.; et al. Assessment of programmed death-ligand 1 expression and tumor-associated immune cells in pediatric cancer tissues. Cancer 2017, 123, 3807–3815. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Moon, E.K.; Wang, L.C.; Dolfi, D.V.; Wilson, C.B.; Ranganathan, R.; Sun, J.; Kapoor, V.; Scholler, J.; Pure, E.; Milone, M.C.; et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin. Cancer Res. 2014, 20, 4262–4273. [Google Scholar] [CrossRef]
- Dwivedi, V.P.; Tousif, S.; Bhattacharya, D.; Prasad, D.V.; Van Kaer, L.; Das, J.; Das, G. Transforming growth factor-beta protein inversely regulates in vivo differentiation of interleukin-17 (IL-17)-producing CD4+ and CD8+ T cells. J. Biol. Chem. 2012, 287, 2943–2947. [Google Scholar] [CrossRef]
- Seo, N.; Hayakawa, S.; Takigawa, M.; Tokura, Y. Interleukin-10 expressed at early tumour sites induces subsequent generation of CD4(+) T-regulatory cells and systemic collapse of antitumour immunity. Immunology 2001, 103, 449–457. [Google Scholar] [CrossRef]
- Soldati, R.; Berger, E.; Zenclussen, A.C.; Jorch, G.; Lode, H.N.; Salatino, M.; Rabinovich, G.A.; Fest, S. Neuroblastoma triggers an immunoevasive program involving galectin-1-dependent modulation of T cell and dendritic cell compartments. Int. J. Cancer 2012, 131, 1131–1141. [Google Scholar] [CrossRef]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-beta Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [PubMed]
- Vanichapol, T.; Chiangjong, W.; Panachan, J.; Anurathapan, U.; Chutipongtanate, S.; Hongeng, S. Secretory High-Mobility Group Box 1 Protein Affects Regulatory T Cell Differentiation in Neuroblastoma Microenvironment In Vitro. J. Oncol. 2018, 2018, 7946021. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P.; Plymoth, A. Targeting the hallmarks of cancer: Towards a rational approach to next-generation cancer therapy. Curr. Opin. Oncol. 2013, 25, 50–51. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W.P. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef]
- Craddock, J.A.; Lu, A.; Bear, A.; Pule, M.; Brenner, M.K.; Rooney, C.M.; Foster, A.E. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 2010, 33, 780–788. [Google Scholar] [CrossRef]
- Di Stasi, A.; De Angelis, B.; Rooney, C.M.; Zhang, L.; Mahendravada, A.; Foster, A.E.; Heslop, H.E.; Brenner, M.K.; Dotti, G.; Savoldo, B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009, 113, 6392–6402. [Google Scholar] [CrossRef]
- Mohammed, S.; Sukumaran, S.; Bajgain, P.; Watanabe, N.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Fisher, W.E.; Leen, A.M.; Vera, J.F. Improving Chimeric Antigen Receptor-Modified T Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Pancreatic Cancer. Mol. Ther. 2017, 25, 249–258. [Google Scholar] [CrossRef]
- Davila, M.L.; Papapetrou, E.P. CARs Move To the Fast Lane. Mol. Ther. 2014, 22, 477–478. [Google Scholar] [CrossRef][Green Version]
- Prasongtanakij, S.; Anurathapan, U.; Vanichapol, T.; Jittorntrum, B.; Atjanasuppat, K.; Pongpitcha, P.; Pakakasama, S.; Songdej, D.; Sirachainan, N.; Paisooksantivatana, K.; et al. Production and characterization of haploidentical CD19 CAR T cells: Validated to induce a continuous complete remission in a patient with relapsed refractory B-cell ALL. Asia Pac. J. Clin. Oncol. 2020. [Google Scholar] [CrossRef]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Shusterman, S.; London, W.B.; Gillies, S.D.; Hank, J.A.; Voss, S.D.; Seeger, R.C.; Reynolds, C.P.; Kimball, J.; Albertini, M.R.; Wagner, B.; et al. Antitumor activity of hu14.18-IL2 in patients with relapsed/refractory neuroblastoma: A Children’s Oncology Group (COG) phase II study. J. Clin. Oncol. 2010, 28, 4969–4975. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.S.; McGree, B.; Noyes, S.; Plummer, S.; Wong, C.; Chen, Y.B.; Palmer, E.; Albertson, T.; Ferry, J.A.; Arrillaga-Romany, I.C. Anti-CD19 CAR T Cells in CNS Diffuse Large-B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Heczey, A.; Liu, D.; Tian, G.; Courtney, A.N.; Wei, J.; Marinova, E.; Gao, X.; Guo, L.; Yvon, E.; Hicks, J.; et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 2014, 124, 2824–2833. [Google Scholar] [CrossRef]
- Harrer, D.C.; Simon, B.; Fujii, S.I.; Shimizu, K.; Uslu, U.; Schuler, G.; Gerer, K.F.; Hoyer, S.; Dorrie, J.; Schaft, N. RNA-transfection of gamma/delta T cells with a chimeric antigen receptor or an alpha/beta T-cell receptor: A safer alternative to genetically engineered alpha/beta T cells for the immunotherapy of melanoma. BMC Cancer 2017, 17, 551. [Google Scholar] [CrossRef]
- Daher, M.; Rezvani, K. Next generation natural killer cells for cancer immunotherapy: The promise of genetic engineering. Curr. Opin. Immunol. 2018, 51, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Hudecek, M.; Pender, B.; Robinson, E.; Hawkins, R.; Chaney, C.; Cherian, S.; Chen, X.; et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci. Transl. Med. 2016, 8, 355ra116. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Schmueck-Henneresse, M.; Omer, B.; Shum, T.; Tashiro, H.; Mamonkin, M.; Lapteva, N.; Sharma, S.; Rollins, L.; Dotti, G.; Reinke, P.; et al. Comprehensive Approach for Identifying the T Cell Subset Origin of CD3 and CD28 Antibody-Activated Chimeric Antigen Receptor-Modified T Cells. J. Immunol. 2017, 199, 348–362. [Google Scholar] [CrossRef]
- Graef, P.; Buchholz, V.R.; Stemberger, C.; Flossdorf, M.; Henkel, L.; Schiemann, M.; Drexler, I.; Hofer, T.; Riddell, S.R.; Busch, D.H. Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8(+) central memory T cells. Immunity 2014, 41, 116–126. [Google Scholar] [CrossRef]
- Tian, G.; Courtney, A.N.; Jena, B.; Heczey, A.; Liu, D.; Marinova, E.; Guo, L.; Xu, X.; Torikai, H.; Mo, Q.; et al. CD62L+ NKT cells have prolonged persistence and antitumor activity in vivo. J. Clin. Investig. 2016, 126, 2341–2355. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J., Jr.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, 1109. [Google Scholar] [CrossRef]
- Pietra, G.; Vitale, C.; Pende, D.; Bertaina, A.; Moretta, F.; Falco, M.; Vacca, P.; Montaldo, E.; Cantoni, C.; Mingari, M.C.; et al. Human natural killer cells: News in the therapy of solid tumors and high-risk leukemias. Cancer Immunol. Immunother. 2016, 65, 465–476. [Google Scholar] [CrossRef]
- Rezvani, K.; Rouce, R.; Liu, E.; Shpall, E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol. Ther. 2017, 25, 1769–1781. [Google Scholar] [CrossRef]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef]
- Parihar, R.; Rivas, C.; Huynh, M.; Omer, B.; Lapteva, N.; Metelitsa, L.S.; Gottschalk, S.M.; Rooney, C.M. NK Cells Expressing a Chimeric Activating Receptor Eliminate MDSCs and Rescue Impaired CAR-T Cell Activity against Solid Tumors. Cancer Immunol. Res. 2019, 7, 363–375. [Google Scholar] [CrossRef]
- Jamali, A.; Hadjati, J.; Madjd, Z.; Mirzaei, H.R.; Thalheimer, F.B.; Agarwal, S.; Bonig, H.; Ullrich, E.; Hartmann, J. Highly Efficient Generation of Transgenically Augmented CAR NK Cells Overexpressing CXCR4. Front. Immunol. 2020, 11, 2028. [Google Scholar] [CrossRef]
- Tseng, H.C.; Xiong, W.; Badeti, S.; Yang, Y.; Ma, M.; Liu, T.; Ramos, C.A.; Dotti, G.; Fritzky, L.; Jiang, J.G.; et al. Efficacy of anti-CD147 chimeric antigen receptors targeting hepatocellular carcinoma. Nat. Commun. 2020, 11, 4810. [Google Scholar] [CrossRef]
- Morgan, M.A.; Buning, H.; Sauer, M.; Schambach, A. Use of Cell and Genome Modification Technologies to Generate Improved “Off-the-Shelf” CAR T and CAR NK Cells. Front. Immunol. 2020, 11, 1965. [Google Scholar] [CrossRef]
- Rotolo, R.; Leuci, V.; Donini, C.; Cykowska, A.; Gammaitoni, L.; Medico, G.; Valabrega, G.; Aglietta, M.; Sangiolo, D. CAR-Based Strategies beyond T Lymphocytes: Integrative Opportunities for Cancer Adoptive Immunotherapy. Int. J. Mol. Sci. 2019, 20, 2839. [Google Scholar] [CrossRef] [PubMed]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural Killer Cells for Immunotherapy-Advantages of the NK-92 Cell Line over Blood NK Cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.Y.; Tay, J.C.K.; Wang, S. CXCR1 Expression to Improve Anti-Cancer Efficacy of Intravenously Injected CAR-NK Cells in Mice with Peritoneal Xenografts. Mol. Ther. Oncolytics 2020, 16, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhi, L.; Yin, M.; Guo, C.; Zhang, H.; Lu, C.; Zhu, W. A novel bispecific chimeric PD1-DAP10/NKG2D receptor augments NK92-cell therapy efficacy for human gastric cancer SGC-7901 cell. Biochem. Biophys. Res. Commun. 2020, 523, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Montagner, I.M.; Penna, A.; Fracasso, G.; Carpanese, D.; Dalla Pieta, A.; Barbieri, V.; Zuccolotto, G.; Rosato, A. Anti-PSMA CAR-engineered NK-92 Cells: An Off-the-shelf Cell Therapy for Prostate Cancer. Cells 2020, 9, 1382. [Google Scholar] [CrossRef] [PubMed]
- Gang, M.; Marin, N.D.; Wong, P.; Neal, C.C.; Marsala, L.; Foster, M.; Schappe, T.; Meng, W.; Tran, J.; Schaettler, M.; et al. CAR-modified memory-like NK cells exhibit potent responses to NK-resistant lymphomas. Blood 2020, 136, 2308–2318. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Dasgupta, S.; Bhattacharya-Chatterjee, M.; O’Malley, B.W., Jr.; Chatterjee, S.K. Inhibition of NK cell activity through TGF-beta 1 by down-regulation of NKG2D in a murine model of head and neck cancer. J. Immunol. 2005, 175, 5541–5550. [Google Scholar] [CrossRef]
- Porcelli, S.; Yockey, C.E.; Brenner, M.B.; Balk, S.P. Analysis of T cell antigen receptor (TCR) expression by human peripheral blood CD4-8- alpha/beta T cells demonstrates preferential use of several V beta genes and an invariant TCR alpha chain. J. Exp. Med. 1993, 178, 1–16. [Google Scholar] [CrossRef]
- Bendelac, A. CD1: Presenting unusual antigens to unusual T lymphocytes. Science 1995, 269, 185–186. [Google Scholar] [CrossRef]
- Halder, R.C.; Aguilera, C.; Maricic, I.; Kumar, V. Type II NKT cell-mediated anergy induction in type I NKT cells prevents inflammatory liver disease. J. Clin. Investig. 2007, 117, 2302–2312. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.H.; Deng, H.; Matthews, P.; Krasovsky, J.; Ragupathi, G.; Spisek, R.; Mazumder, A.; Vesole, D.H.; Jagannath, S.; Dhodapkar, M.V. Inflammation-associated lysophospholipids as ligands for CD1d-restricted T cells in human cancer. Blood 2008, 112, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Dhodapkar, M.V.; Geller, M.D.; Chang, D.H.; Shimizu, K.; Fujii, S.; Dhodapkar, K.M.; Krasovsky, J. A reversible defect in natural killer T cell function characterizes the progression of premalignant to malignant multiple myeloma. J. Exp. Med. 2003, 197, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Rotolo, A.; Caputo, V.S.; Holubova, M.; Baxan, N.; Dubois, O.; Chaudhry, M.S.; Xiao, X.; Goudevenou, K.; Pitcher, D.S.; Petevi, K.; et al. Enhanced Anti-lymphoma Activity of CAR19-iNKT Cells Underpinned by Dual CD19 and CD1d Targeting. Cancer Cell 2018, 34, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Simon, B.; Wiesinger, M.; Marz, J.; Wistuba-Hamprecht, K.; Weide, B.; Schuler-Thurner, B.; Schuler, G.; Dorrie, J.; Uslu, U. The Generation of CAR-Transfected Natural Killer T Cells for the Immunotherapy of Melanoma. Int. J. Mol. Sci. 2018, 19, 2365. [Google Scholar] [CrossRef] [PubMed]
- Metelitsa, L.S. Anti-tumor potential of type-I NKT cells against CD1d-positive and CD1d-negative tumors in humans. Clin. Immunol. 2011, 140, 119–129. [Google Scholar] [CrossRef]
- Vivier, E.; Ugolini, S.; Blaise, D.; Chabannon, C.; Brossay, L. Targeting natural killer cells and natural killer T cells in cancer. Nat. Rev. Immunol. 2012, 12, 239–252. [Google Scholar] [CrossRef]
- Olle Hurtado, M.; Wolbert, J.; Fisher, J.; Flutter, B.; Stafford, S.; Barton, J.; Jain, N.; Barone, G.; Majani, Y.; Anderson, J. Tumor infiltrating lymphocytes expanded from pediatric neuroblastoma display heterogeneity of phenotype and function. PLoS ONE 2019, 14, e0216373. [Google Scholar] [CrossRef]
- Heczey, A.; Courtney, A.N.; Montalbano, A.; Robinson, S.; Liu, K.; Li, M.; Ghatwai, N.; Dakhova, O.; Liu, B.; Raveh-Sadka, T.; et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: An interim analysis. Nat. Med. 2020, 26, 1686–1690. [Google Scholar] [CrossRef]
- Fisher, J.; Kramer, A.M.; Gustafsson, K.; Anderson, J. Non-V delta 2 gamma delta T lymphocytes as effectors of cancer immunotherapy. Oncoimmunology 2015, 4, e973808. [Google Scholar] [CrossRef]
- Fisher, J.P.; Heuijerjans, J.; Yan, M.; Gustafsson, K.; Anderson, J. gammadelta T cells for cancer immunotherapy: A systematic review of clinical trials. Oncoimmunology 2014, 3, e27572. [Google Scholar] [CrossRef] [PubMed]
- Himoudi, N.; Morgenstern, D.A.; Yan, M.; Vernay, B.; Saraiva, L.; Wu, Y.; Cohen, C.J.; Gustafsson, K.; Anderson, J. Human gammadelta T lymphocytes are licensed for professional antigen presentation by interaction with opsonized target cells. J. Immunol. 2012, 188, 1708–1716. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Meyer, C.; Bonneville, M. gammadelta T cells: First line of defense and beyond. Annu. Rev. Immunol. 2014, 32, 121–155. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, B.; Ravens, S.; Silva-Santos, B. High-throughput analysis of the human thymic Vdelta1(+) T cell receptor repertoire. Sci. Data 2019, 6, 115. [Google Scholar] [CrossRef]
- Vantourout, P.; Hayday, A. Six-of-the-best: Unique contributions of gammadelta T cells to immunology. Nat. Rev. Immunol. 2013, 13, 88–100. [Google Scholar] [CrossRef]
- Fisher, J.P.; Yan, M.; Heuijerjans, J.; Carter, L.; Abolhassani, A.; Frosch, J.; Wallace, R.; Flutter, B.; Capsomidis, A.; Hubank, M.; et al. Neuroblastoma killing properties of Vdelta2 and Vdelta2-negative gammadeltaT cells following expansion by artificial antigen-presenting cells. Clin. Cancer Res. 2014, 20, 5720–5732. [Google Scholar] [CrossRef]
- Miyagawa, F.; Tanaka, Y.; Yamashita, S.; Minato, N. Essential requirement of antigen presentation by monocyte lineage cells for the activation of primary human gamma delta T cells by aminobisphosphonate antigen. J. Immunol. 2001, 166, 5508–5514. [Google Scholar] [CrossRef]
- Deseke, M.; Prinz, I. Ligand recognition by the gammadelta TCR and discrimination between homeostasis and stress conditions. Cell. Mol. Immunol. 2020, 17, 914–924. [Google Scholar] [CrossRef]
- Brandes, M.; Willimann, K.; Moser, B. Professional antigen-presentation function by human gammadelta T Cells. Science 2005, 309, 264–268. [Google Scholar] [CrossRef]
- Hamid, O.; Carvajal, R.D. Anti-programmed death-1 and anti-programmed death-ligand 1 antibodies in cancer therapy. Expert Opin. Biol. Ther. 2013, 13, 847–861. [Google Scholar] [CrossRef]
- Deniger, D.C.; Switzer, K.; Mi, T.; Maiti, S.; Hurton, L.; Singh, H.; Huls, H.; Olivares, S.; Lee, D.A.; Champlin, R.E.; et al. Bispecific T-cells expressing polyclonal repertoire of endogenous gammadelta T-cell receptors and introduced CD19-specific chimeric antigen receptor. Mol. Ther. 2013, 21, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Rozenbaum, M.; Meir, A.; Aharony, Y.; Itzhaki, O.; Schachter, J.; Bank, I.; Jacoby, E.; Besser, M.J. Gamma-Delta CAR-T Cells Show CAR-Directed and Independent Activity Against Leukemia. Front. Immunol. 2020, 11, 1347. [Google Scholar] [CrossRef] [PubMed]
- Capsomidis, A.; Benthall, G.; Van Acker, H.H.; Fisher, J.; Kramer, A.M.; Abeln, Z.; Majani, Y.; Gileadi, T.; Wallace, R.; Gustafsson, K.; et al. Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation. Mol. Ther. 2018, 26, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.; Abramowski, P.; Wisidagamage Don, N.D.; Flutter, B.; Capsomidis, A.; Cheung, G.W.; Gustafsson, K.; Anderson, J. Avoidance of On-Target Off-Tumor Activation Using a Co-stimulation-Only Chimeric Antigen Receptor. Mol. Ther. 2017, 25, 1234–1247. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef]
- Jensen, M.C.; Riddell, S.R. Designing chimeric antigen receptors to effectively and safely target tumors. Curr. Opin. Immunol. 2015, 33, 9–15. [Google Scholar] [CrossRef]
- Alabanza, L.; Pegues, M.; Geldres, C.; Shi, V.; Wiltzius, J.J.W.; Sievers, S.A.; Yang, S.; Kochenderfer, J.N. Function of Novel Anti-CD19 Chimeric Antigen Receptors with Human Variable Regions Is Affected by Hinge and Transmembrane Domains. Mol. Ther. 2017, 25, 2452–2465. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Suck, G.; Odendahl, M.; Nowakowska, P.; Seidl, C.; Wels, W.S.; Klingemann, H.G.; Tonn, T. NK-92: An ‘off-the-shelf therapeutic’ for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol. Immunother. 2016, 65, 485–492. [Google Scholar] [CrossRef]
- Bryceson, Y.T.; Chiang, S.C.; Darmanin, S.; Fauriat, C.; Schlums, H.; Theorell, J.; Wood, S.M. Molecular mechanisms of natural killer cell activation. J. Innate Immun. 2011, 3, 216–226. [Google Scholar] [CrossRef]
- Salter, A.I.; Pont, M.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells: CD19 and the road beyond. Blood 2018, 131, 2621–2629. [Google Scholar] [CrossRef] [PubMed]
- Lo Presti, E.; Pizzolato, G.; Corsale, A.M.; Caccamo, N.; Sireci, G.; Dieli, F.; Meraviglia, S. gammadelta T Cells and Tumor Microenvironment: From Immunosurveillance to Tumor Evasion. Front. Immunol. 2018, 9, 1395. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.R.; Mirzaei, H.; Lee, S.Y.; Hadjati, J.; Till, B.G. Prospects for chimeric antigen receptor (CAR) gammadelta T cells: A potential game changer for adoptive T cell cancer immunotherapy. Cancer Lett. 2016, 380, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Oelsner, S.; Friede, M.E.; Zhang, C.; Wagner, J.; Badura, S.; Bader, P.; Ullrich, E.; Ottmann, O.G.; Klingemann, H.; Tonn, T.; et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy 2017, 19, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zeng, J.; Liu, T.; Xu, Q.; Song, X.; Zeng, J. DNAM1 and 2B4 Costimulatory Domains Enhance the Cytotoxicity of Anti-GPC3 Chimeric Antigen Receptor-Modified Natural Killer Cells Against Hepatocellular Cancer Cells in vitro. Cancer Manag. Res. 2020, 12, 3247–3255. [Google Scholar] [CrossRef]
- Fernandez, L.; Metais, J.Y.; Escudero, A.; Vela, M.; Valentin, J.; Vallcorba, I.; Leivas, A.; Torres, J.; Valeri, A.; Patino-Garcia, A.; et al. Memory T Cells Expressing an NKG2D-CAR Efficiently Target Osteosarcoma Cells. Clin. Cancer Res. 2017, 23, 5824–5835. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.Y.; Tay, J.C.K.; Li, Z.; Wang, J.; Zhu, J.; Wang, S. T Cells Expressing NKG2D CAR with a DAP12 Signaling Domain Stimulate Lower Cytokine Production While Effective in Tumor Eradication. Mol. Ther. 2020. [Google Scholar] [CrossRef]
- Tan, A.H.J.; Vinanica, N.; Campana, D. Chimeric antigen receptor-T cells with cytokine neutralizing capacity. Blood Adv. 2020, 4, 1419–1431. [Google Scholar] [CrossRef]
- Moretta, A.; Biassoni, R.; Bottino, C.; Pende, D.; Vitale, M.; Poggi, A.; Mingari, M.C.; Moretta, L. Major histocompatibility complex class I-specific receptors on human natural killer and T lymphocytes. Immunol. Rev. 1997, 155, 105–117. [Google Scholar] [CrossRef]
- Wang, E.; Wang, L.C.; Tsai, C.Y.; Bhoj, V.; Gershenson, Z.; Moon, E.; Newick, K.; Sun, J.; Lo, A.; Baradet, T.; et al. Generation of Potent T-cell Immunotherapy for Cancer Using DAP12-Based, Multichain, Chimeric Immunoreceptors. Cancer Immunol. Res. 2015, 3, 815–826. [Google Scholar] [CrossRef]
- Topfer, K.; Cartellieri, M.; Michen, S.; Wiedemuth, R.; Muller, N.; Lindemann, D.; Bachmann, M.; Fussel, M.; Schackert, G.; Temme, A. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J. Immunol. 2015, 194, 3201–3212. [Google Scholar] [CrossRef] [PubMed]
- Vanichapol, T.; Chutipongtanate, S.; Anurathapan, U.; Hongeng, S. Immune Escape Mechanisms and Future Prospects for Immunotherapy in Neuroblastoma. Biomed. Res. Int. 2018, 2018, 1812535. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.A.; Sweis, R.F.; Bao, R.; Luke, J.J. T Cell-Inflamed versus Non-T Cell-Inflamed Tumors: A Conceptual Framework for Cancer Immunotherapy Drug Development and Combination Therapy Selection. Cancer Immunol. Res. 2018, 6, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef]
- Whilding, L.M.; Halim, L.; Draper, B.; Parente-Pereira, A.C.; Zabinski, T.; Davies, D.M.; Maher, J. CAR T-Cells Targeting the Integrin alphavbeta6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy against Several Solid Malignancies. Cancers 2019, 11, 674. [Google Scholar] [CrossRef]
- Liu, G.; Rui, W.; Zheng, H.; Huang, D.; Yu, F.; Zhang, Y.; Dong, J.; Zhao, X.; Lin, X. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur. J. Immunol. 2020, 50, 712–724. [Google Scholar] [CrossRef]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef]
- Liu, L.B.; Xie, F.; Chang, K.K.; Li, M.Q.; Meng, Y.H.; Wang, X.H.; Li, H.; Li, D.J.; Yu, J.J. Hypoxia promotes the proliferation of cervical carcinoma cells through stimulating the secretion of IL-8. Int. J. Clin. Exp. Pathol. 2014, 7, 575–583. [Google Scholar]
- Kim, E.S.; Serur, A.; Huang, J.; Manley, C.A.; McCrudden, K.W.; Frischer, J.S.; Soffer, S.Z.; Ring, L.; New, T.; Zabski, S.; et al. Potent VEGF blockade causes regression of coopted vessels in a model of neuroblastoma. Proc. Natl. Acad. Sci. USA 2002, 99, 11399–11404. [Google Scholar] [CrossRef]
- Ara, T.; Song, L.; Shimada, H.; Keshelava, N.; Russell, H.V.; Metelitsa, L.S.; Groshen, S.G.; Seeger, R.C.; DeClerck, Y.A. Interleukin-6 in the bone marrow microenvironment promotes the growth and survival of neuroblastoma cells. Cancer Res. 2009, 69, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Brignole, C.; Marimpietri, D.; Pastorino, F.; Di Paolo, D.; Pagnan, G.; Loi, M.; Piccardi, F.; Cilli, M.; Tradori-Cappai, A.; Arrigoni, G.; et al. Anti-IL-10R antibody improves the therapeutic efficacy of targeted liposomal oligonucleotides. J. Control. Release 2009, 138, 122–127. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59. [Google Scholar] [CrossRef]
- Noguchi, T.; Ward, J.P.; Gubin, M.M.; Arthur, C.D.; Lee, S.H.; Hundal, J.; Selby, M.J.; Graziano, R.F.; Mardis, E.R.; Korman, A.J.; et al. Temporally Distinct PD-L1 Expression by Tumor and Host Cells Contributes to Immune Escape. Cancer Immunol. Res. 2017, 5, 106–117. [Google Scholar] [CrossRef]
- Whittle, S.B.; Smith, V.; Doherty, E.; Zhao, S.; McCarty, S.; Zage, P.E. Overview and recent advances in the treatment of neuroblastoma. Expert. Rev. Anticancer Ther. 2017, 17, 369–386. [Google Scholar] [CrossRef]
- Pinto, N.R.; Applebaum, M.A.; Volchenboum, S.L.; Matthay, K.K.; London, W.B.; Ambros, P.F.; Nakagawara, A.; Berthold, F.; Schleiermacher, G.; Park, J.R.; et al. Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J. Clin. Oncol. 2015, 33, 3008–3017. [Google Scholar] [CrossRef]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Antonyak, M.A.; Cerione, R.A. Microvesicles as mediators of intercellular communication in cancer. Methods Mol. Biol. 2014, 1165, 147–173. [Google Scholar] [CrossRef] [PubMed]
- Blavier, L.; Yang, R.M.; DeClerck, Y.A. The Tumor Microenvironment in Neuroblastoma: New Players, New Mechanisms of Interaction and New Perspectives. Cancers 2020, 12, 2912. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Fonseka, P.; Liem, M.; Ozcitti, C.; Adda, C.G.; Ang, C.S.; Mathivanan, S. Exosomes from N-Myc amplified neuroblastoma cells induce migration and confer chemoresistance to non-N-Myc amplified cells: Implications of intra-tumour heterogeneity. J. Extracell Vesicles 2019, 8, 1597614. [Google Scholar] [CrossRef] [PubMed]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef]
- Ali, S.; Toews, K.; Schwiebert, S.; Klaus, A.; Winkler, A.; Grunewald, L.; Oevermann, L.; Deubzer, H.E.; Tuns, A.; Jensen, M.C.; et al. Tumor-Derived Extracellular Vesicles Impair CD171-Specific CD4(+) CAR T Cell Efficacy. Front. Immunol. 2020, 11, 531. [Google Scholar] [CrossRef]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Ji, Q.; Zhou, L.; Sui, H.; Yang, L.; Wu, X.; Song, Q.; Jia, R.; Li, R.; Sun, J.; Wang, Z.; et al. Primary tumors release ITGBL1-rich extracellular vesicles to promote distal metastatic tumor growth through fibroblast-niche formation. Nat. Commun. 2020, 11, 1211. [Google Scholar] [CrossRef]
- Nakata, R.; Shimada, H.; Fernandez, G.E.; Fanter, R.; Fabbri, M.; Malvar, J.; Zimmermann, P.; DeClerck, Y.A. Contribution of neuroblastoma-derived exosomes to the production of pro-tumorigenic signals by bone marrow mesenchymal stromal cells. J. Extracell Vesicles 2017, 6, 1332941. [Google Scholar] [CrossRef]
- Challagundla, K.B.; Wise, P.M.; Neviani, P.; Chava, H.; Murtadha, M.; Xu, T.; Kennedy, R.; Ivan, C.; Zhang, X.; Vannini, I.; et al. Exosome-mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef] [PubMed]
- Zuo, S.; Sho, M.; Sawai, T.; Kanehiro, H.; Maeda, K.; Yoshida, M.; Tsukada, R.; Nomura, M.; Okuyama, H. Potential role of the PD-L1 expression and tumor-infiltrating lymphocytes on neuroblastoma. Pediatr. Surg. Int. 2020, 36, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Dondero, A.; Pastorino, F.; Della Chiesa, M.; Corrias, M.V.; Morandi, F.; Pistoia, V.; Olive, D.; Bellora, F.; Locatelli, F.; Castellano, A.; et al. PD-L1 expression in metastatic neuroblastoma as an additional mechanism for limiting immune surveillance. Oncoimmunology 2016, 5, e1064578. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef]
- Li, S.; Siriwon, N.; Zhang, X.; Yang, S.; Jin, T.; He, F.; Kim, Y.J.; Mac, J.; Lu, Z.; Wang, S.; et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin. Cancer Res. 2017, 23, 6982–6992. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef]
- Qin, L.; Zhao, R.; Chen, D.; Wei, X.; Wu, Q.; Long, Y.; Jiang, Z.; Li, Y.; Wu, H.; Zhang, X.; et al. Chimeric antigen receptor T cells targeting PD-L1 suppress tumor growth. Biomark. Res. 2020, 8, 19. [Google Scholar] [CrossRef]
- Gargett, T.; Yu, W.; Dotti, G.; Yvon, E.S.; Christo, S.N.; Hayball, J.D.; Lewis, I.D.; Brenner, M.K.; Brown, M.P. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol. Ther. 2016, 24, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Toews, K.; Grunewald, L.; Schwiebert, S.; Klaus, A.; Winkler, A.; Ali, S.; Zirngibl, F.; Astrahantseff, K.; Wagner, D.L.; Henssen, A.G.; et al. Central memory phenotype drives success of checkpoint inhibition in combination with CAR T cells. Mol. Carcinog. 2020, 59, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, P.; Hanaei, S.; Rezaei, N. State of the art in immunotherapy of neuroblastoma. Immunotherapy 2019, 11, 831–850. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Carranza-Rua, O.; Alfaro, C.; Onate, C.; Martin-Algarra, S.; Perez, G.; Landazuri, S.F.; Gonzalez, A.; Gross, S.; Rodriguez, I.; et al. Serum interleukin-8 reflects tumor burden and treatment response across malignancies of multiple tissue origins. Clin. Cancer Res. 2014, 20, 5697–5707. [Google Scholar] [CrossRef] [PubMed]
- Kuwada, Y.; Sasaki, T.; Morinaka, K.; Kitadai, Y.; Mukaida, N.; Chayama, K. Potential involvement of IL-8 and its receptors in the invasiveness of pancreatic cancer cells. Int. J. Oncol. 2003, 22, 765–771. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, C.; Landoni, E.; Metelitsa, L.; Dotti, G.; Savoldo, B. Eradication of Neuroblastoma by T Cells Redirected with an Optimized GD2-Specific Chimeric Antigen Receptor and Interleukin-15. Clin. Cancer Res. 2019, 25, 2915–2924. [Google Scholar] [CrossRef]
- Alvarez-Rueda, N.; Desselle, A.; Cochonneau, D.; Chaumette, T.; Clemenceau, B.; Leprieur, S.; Bougras, G.; Supiot, S.; Mussini, J.M.; Barbet, J.; et al. A monoclonal antibody to O-acetyl-GD2 ganglioside and not to GD2 shows potent anti-tumor activity without peripheral nervous system cross-reactivity. PLoS ONE 2011, 6, e25220. [Google Scholar] [CrossRef]
- Kramer, K.; Khan, O.; Haque, S. Central Nervous System Neuroblastoma Metastases Pseudoprogression Following Intraventricular Anti-B7h3 Radioimmunotherapy. Neuro-Oncology 2019, 21, 95. [Google Scholar] [CrossRef]
- Yang, L.H.; Ma, X.L.; Liu, Y.C.; Zhao, W.; Yu, L.H.; Qin, M.Q.; Zhu, G.H.; Wang, K.; Shi, X.D.; Zhang, Z.X.; et al. Chimeric Antigen Receptor 4SCAR-GD2-Modified T Cells Targeting High-Risk and Recurrent Neuroblastoma: A Phase II Multi-Center Trial in China. Blood 2017, 130. [Google Scholar] [CrossRef]
- Zhao, W.; Dong, L.; Wang, H.; Zheng, X.; Zhang, C.; Wang, K.; Zhu, G.; Wang, B.; Qin, M.; Liu, Y.; et al. Chimeric Antigen Receptor 4SCAR-GD2/CD56-Modified T Cells in the Refractory and Recurrent Paediatric Solid Tumors. Pediatric Blood Cancer 2016, 63, S38. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Clinicaltrials.Gov Identifier | Type of Study | Status | Title | Interventions | Target/ScFv/Signaling Domain | Date | Key Result | Reference |
|---|---|---|---|---|---|---|---|---|
| N/A | Phase I N = 6 | Completed | N/A | Anti- L1-CAM CAR T cells | L1-CAM/CE7R/ CD3ζ | N/A | Partial response in one patient | [12] |
| NCT02761915 | Phase I N = 27 | Recruiting | A UK cancer research trial of anti-GD2 T cells (1RG-CART) | Anti-GD2 CAR T cells | GD2/KM8138/ CD28.CD3ζ | Study start: February 2016 Primary completion: February 2023 | On target activity in bone and bone marrow were detected | [47] |
| NCT01822652 | Phase I N = 11 | Active, not recruiting | Third generation GD-2 chimeric antigen receptor and icaspase suicide safety switch, neuroblastoma, GRAIN | iC9-GD2 CAR T cells | GD2/14g2a/ CD28.OX40.CD3ζ | Study start: August 2013 Primary completion: December 2015 | Modest early antitumor responses were detected | [49] |
| NCT00085930 | Phase I N = 19 | Active, not recruiting | Blood T-cells and EBV specific CTLs expressing GD-2 specific chimeric T Cell receptors to neuroblastoma patients | EBV-specific CTLs | GD2/14g2a/ CD3ζ | Study start: April 2003 Primary completion: January 2010 | Complete response in one patient | [18,50] |
| NCT01460901 | Phase I N = 5 | Completed | Study of donor-derived, multi-virus-specific, cytotoxic T-lymphocytes for relapsed/refractory neuroblastoma (STALLONe) | GD2 CAR modified Tri-virus-specific cytotoxic t-cells | GD2/14g2a/CD3ζ | Study start: October 2012 Primary completion: January 2015 | Partial response in 30% of patients | [51] |
| NCT03294954 | Phase I N = 24 | Recruiting | GD2-specific CAR and interleukin-15 expressing autologous NKT cells to treat children with neuroblastoma (GINAKIT2) | GINAKIT Cells | GD2/14g2a/ CD28.CD3ζ | Study start: 18 January 2018 Primary completion: 1 September 2021 | N/A | [52] |
| NCT02765243 | Phase II N = 34 | Recruiting | Anti-GD2 fourth generation CART cells targeting refractory and/ or recurrent neuroblastoma | Anti-GD2 CAR T cells | GD2/N/A/CD28.4-1BB.CD27.CD3ζ | Study start: 23 March 2016 Primary completion: 12 May 2019 | Partial response in 15% of patients | [53] |
| NCT03618381 | Phase I N = 36 | Recruiting | EGFR806 CAR T cell immunotherapy for recurrent/refractory solid tumors in children and young adults | Anti-EGFR806-EGFRt CAR T cells | EGFR/N/A/4-1BB.CD3ζ | Study start: 18 June 2019 Primary completion: June 2021 | N/A | [54] |
| NCT01953900 | Phase I N = 26 | Active, not recruiting | iC9-GD2-CAR-VZV-CTLs/refractory or metastatic GD2-positive sarcoma and neuroblastoma | Anti-GD2 CAR T cells | GD2/14g2a/ CD28.OX40.CD3ζ | Study start: April 2014 Primary completion: April 2021 | N/A | [54,55] |
| NCT03635632 | Phase I N = 94 | Recruiting | C7R-GD2.CAR T cells for patients with relapsed or refractory neuroblastoma and other GD2 Positive cancers (GAIL-N) | C7R-GD2 CAR T cells | GD2/14g2a/ CD28.OX40.CD3ζ | Study start: 23 April 2019 Primary completion: June 2022 | N/A | Unpublished |
| NCT03373097 | Phase I/II N = 42 | Recruiting | Anti-GD2 CAR T cells in pediatric patients affected by high risk and/or relapsed/refractory neuroblastoma or other GD2-positive solid tumors | Anti-GD2 CAR T cells | GD2/14g2a/ CD28.4-1BB.CD3ζ | Study start: January 2018 Primary completion: December 2024 | N/A | Unpublished |
| NCT03721068 | Phase I N = 18 | Recruiting | Study of CAR T cells targeting the GD2 with IL-15+icaspase9 for relapsed/refractory neuroblastoma | iC9-GD2 CAR T cells | GD2/N/A | Study start: 19 February 2019 Primary completion: 19 June 2024 | N/A | Unpublished |
| NCT04637503 | Phase I/II N = 100 | Recruiting | 4SCAR-T therapy targeting GD2, PSMA and CD276 for treating neuroblastoma | 4SCAR T cell | GD2, PSMA, CD276/N/A | Study start: 18 November 2020 Primary completion: 19 November 2020 | N/A | Unpublished |
| NCT04539366 | Phase I N = 67 | Not yet recruiting | Testing a new immune cell therapy, GD2-targeted modified T-cells (GD2CART), in children, adolescents, and young adults with relapsed/refractory osteosarcoma and neuroblastoma, the GD2-CAR persistence trial | Anti-GD2 CAR T cells | GD2/N/A/ CD28.OX40.CD3ζ | Study start: 1 January 2021 Primary completion: 1 August 2024 | N/A | Unpublished |
| NCT04483778 | Phase I N = 68 | Recruiting | B7H3 CAR T cell immunotherapy for recurrent/refractory solid tumors in children and young adults | Anti B7H3-EGFRt-DHFR CAR T cells | B7H3/N/A/4-1BB.CD3ζ | Study start: 13 July 2020 Primary completion: December 2025 | N/A | Unpublished |
| NCT02107963 | Phase I N = 15 | Completed | A Phase I trial of T cells expressing an anti-GD2 chimeric antigen receptor in children and young adults with GD2+ solid tumors | Anti-GD2 CAR T cells | GD2/14g2a/OX40.CD28.CD3ζ | Study start: 28 February 2014 Primary completion: 15 August 2016 | N/A | Unpublished |
| Effector Cell | Ex vivo Expansion | Type of Target Cells | CAR Design | Combination | ||||
|---|---|---|---|---|---|---|---|---|
| Localized | CTC | Costimulating | Signaling | Cytokine | Effector Cell | Signaling Inhibitor | ||
| T | *** | * | *** | Dim expression target: CD28 High expression target: 4-1BB | Normally: CD3ζ Decrease CRS: DAP12 | IL-2, IL-7, IL-15 | NKG2D.CAR NK | anti-PD-1/PD-L1, anti-IL-10, IL-6 inhibitor |
| NK | * | *** | ** | CD28, 4-1BB, DNAM1 and 2B4 | CD3ζ and DAP12 | IL-2, IL-7, IL-21 | NB-targeted CAR T | anti-PD-1/PD-L1, anti-TGFβ, anti-IL-10, IL-6 inhibitor |
| NKT | ** | *** | * | CD28 | CD3ζ and DAP12 | IL-2, IL-7, IL-15 | N/A | anti-PD-1/PD-L1, anti-IL-10, IL-6 inhibitor |
| γδT | *** | *** | * | Both CD28 and 4-1BB | CD3ζ [no data for DAP12] | IL-2, IL-7, IL-15 | N/A | anti-PD-1/PD-L1, anti-IL-10, IL-6 inhibitor |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sawaisorn, P.; Atjanasuppat, K.; Anurathapan, U.; Chutipongtanate, S.; Hongeng, S. Strategies to Improve Chimeric Antigen Receptor Therapies for Neuroblastoma. Vaccines 2020, 8, 753. https://doi.org/10.3390/vaccines8040753
Sawaisorn P, Atjanasuppat K, Anurathapan U, Chutipongtanate S, Hongeng S. Strategies to Improve Chimeric Antigen Receptor Therapies for Neuroblastoma. Vaccines. 2020; 8(4):753. https://doi.org/10.3390/vaccines8040753
Chicago/Turabian StyleSawaisorn, Piamsiri, Korakot Atjanasuppat, Usanarat Anurathapan, Somchai Chutipongtanate, and Suradej Hongeng. 2020. "Strategies to Improve Chimeric Antigen Receptor Therapies for Neuroblastoma" Vaccines 8, no. 4: 753. https://doi.org/10.3390/vaccines8040753
APA StyleSawaisorn, P., Atjanasuppat, K., Anurathapan, U., Chutipongtanate, S., & Hongeng, S. (2020). Strategies to Improve Chimeric Antigen Receptor Therapies for Neuroblastoma. Vaccines, 8(4), 753. https://doi.org/10.3390/vaccines8040753