Development and Applications of Viral Vectored Vaccines to Combat Zoonotic and Emerging Public Health Threats




Abstract
1. Introduction
2. General Concepts and Approaches to the Development of Viral Vectors
3. Expansive Repertoire and Selection of Currently Available Viral Vectors for Vaccine Development
3.1. Retrovirus and Lentivirus Vectors
3.2. Adenovirus Vectors
3.3. Poxvirus Vectors
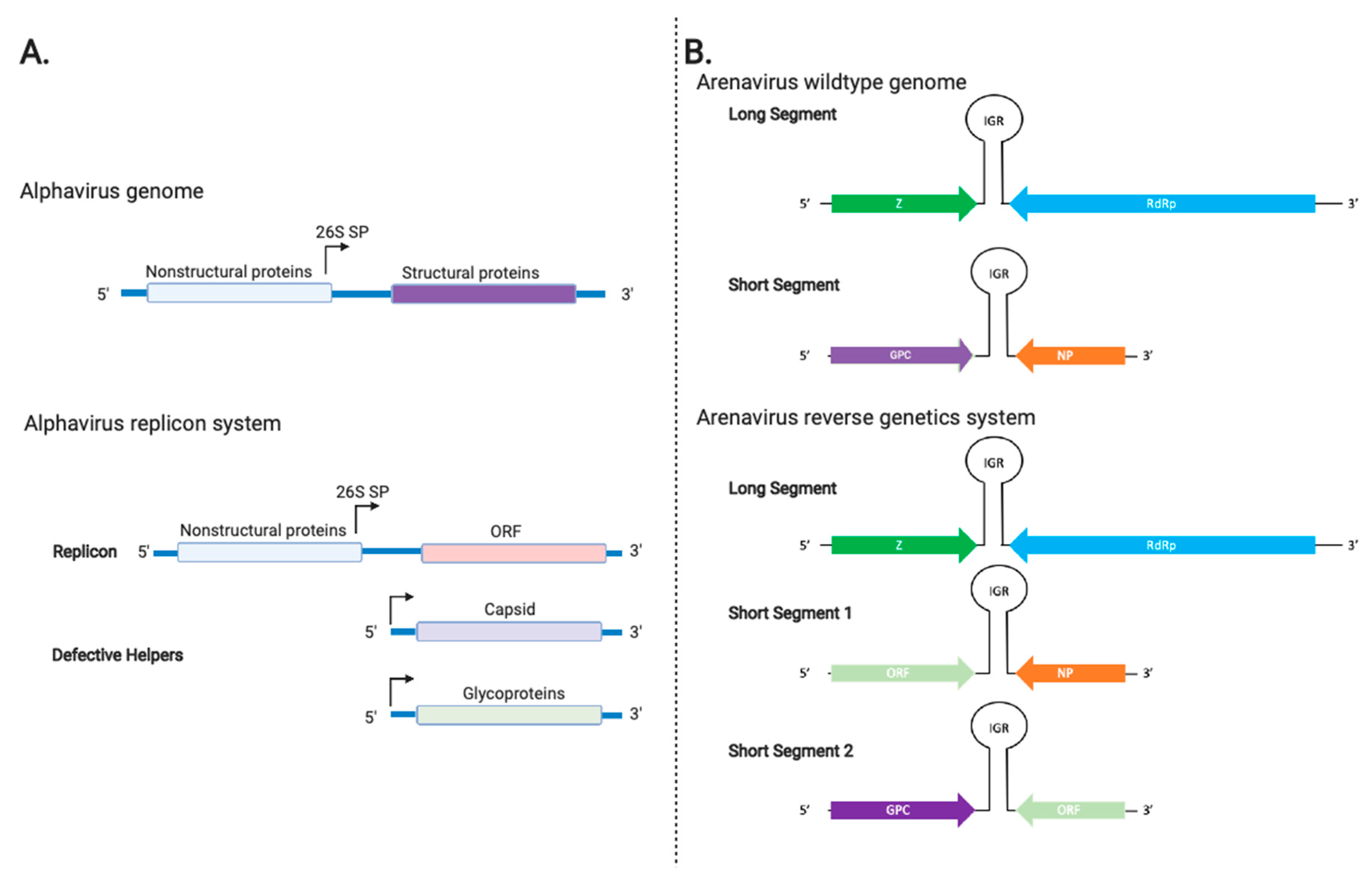
3.4. Alphavirus Vectors
3.5. Arenavirus Vectors
3.6. Herpesvirus Vectors
3.7. Flavivirus Vectors
3.8. Paramyxovirus Vectors
3.9. VSV and Rabies Virus Vectors
4. Viral Vaccine Vectors in Veterinary Medicine: A Story of Success
5. Examples of Application of Viral Vectored Vaccines for Zoonotic Infection of a High-Consequence Pathogen, SARS-CoV-2, the Causative Agent of COVID-19
6. Summary
Author Contributions
Funding
Conflicts of Interest
References
- FDA Vaccines Licensed for Use in the United States. 2020. Available online: https://www.fda.gov/vaccines-blood-biologics/vaccines/vaccines-licensed-use-united-states (accessed on 13 November 2020).
- Tannock, G.A.; Kim, H.; Xue, L. Why are vaccines against many human viral diseases still unavailable; an historic perspective? J. Med. Virol. 2020, 92, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.L.; MacKett, M.; Moss, B. Infectious vaccinia virus recombinants that express hepatitis B virus surface antigen. Nature 1983, 302, 490–495. [Google Scholar] [CrossRef]
- Moss, B.; Smith, G.L.; Gerin, J.L.; Purcell, R.H. Live recombinant vaccinia virus protects chimpanzees against hepatitis B. Nature 1984, 311, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Pastoret, P.P.; Brochier, B. The development and use of a vaccinia-rabies recombinant oral vaccine for the control of wildlife rabies; A link between Jenner and Pasteur. Epidemiol. Infect. 1996, 116, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Mackowiak, M.; Maki, J.; Motes-Kreimeyer, L.; Harbin, T.; Kampen, K. Van Vaccination of wildlife against rabies: Successful use of a vectored vaccine obtained by recombinant technology. Adv. Vet. Med. 1999, 41, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Escors, D.; Breckpot, K. Lentiviral vectors in gene therapy: Their current status and future potential. Arch. Immunol. Ther. Exp. (Warsz.) 2010, 58, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Rauch, S.; Jasny, E.; Schmidt, K.E.; Petsch, B. New vaccine technologies to combat outbreak situations. Front. Immunol. 2018, 9, 1963. [Google Scholar] [CrossRef]
- Barry, M. Single-cycle adenovirus vectors in the current vaccine landscape. Expert Rev. Vaccines 2018, 17, 163–173. [Google Scholar] [CrossRef]
- Vemula, S.V.; Mittal, S.K. Production of adenovirus vectors and their use as a delivery system for influenza vaccines. Expert Opin. Biol. Ther. 2010, 10, 1469–1487. [Google Scholar] [CrossRef]
- Shiver, J.W.; Emini, E.A. Recent advances in the development of HIV-1 vaccines using replication-incompetent adenovirus vectors. Annu. Rev. Med. 2004, 55, 355–372. [Google Scholar] [CrossRef]
- Wold, W.S.M.; Toth, K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 2013, 13, 421–433. [Google Scholar] [CrossRef]
- Crystal, R.G. Adenovirus: The first effective in vivo gene delivery vector. Hum. Gene Ther. 2014, 25, 3–11. [Google Scholar] [CrossRef]
- Sánchez-Sampedro, L.; Perdiguero, B.; Mejías-Pérez, E.; García-Arriaza, J.; Di Pilato, M.; Esteban, M. The evolution of poxvirus vaccines. Viruses 2015, 7, 1726–1803. [Google Scholar] [CrossRef] [PubMed]
- Volz, A.; Sutter, G. Modified Vaccinia Virus Ankara: History, Value in Basic Research, and Current Perspectives for Vaccine Development. In Advances in Virus Research; Academic Press Inc.: Cambridge, MA, USA, 2017; Volume 97, pp. 187–243. [Google Scholar]
- Robert-Guroff, M. Replicating and non-replicating viral vectors for vaccine development. Curr. Opin. Biotechnol. 2007, 18, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.; Weinberg, R.; Languet, B.; Desmettre, P.; Paoletti, E. Recombinant fowlpox virus inducing protective immunity in non-avian species. Vaccine 1988, 6, 497–503. [Google Scholar] [CrossRef]
- Wang, M.; Jokinen, J.; Tretyakova, I.; Pushko, P.; Lukashevich, I.S. Alphavirus vector-based replicon particles expressing multivalent cross-protective Lassa virus glycoproteins. Vaccine 2018, 36, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Liljeström, P.; Garoff, H. A New Generation of Animal Cell Expression Vectors Based on the Semliki Forest Virus Replicon. Nat. Biotechnol. 1991, 9, 1356–1361. [Google Scholar] [CrossRef] [PubMed]
- Emonet, S.F.; Garidou, L.; McGavern, D.B.; De la Torre, C.; De La Torre, J.C. Generation of recombinant lymphocytic choriomeningitis viruses with trisegmented genomes stably expressing two additional genes of interest. Proc. Natl. Acad. Sci. USA 2009, 106, 3473–3478. [Google Scholar] [CrossRef] [PubMed]
- Emonet, S.E.; Urata, S.; de la Torre, J.C. Arenavirus reverse genetics: New approaches for the investigation of arenavirus biology and development of antiviral strategies. Virology 2011, 411, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Dhanwani, R.; Ly, H.; Liang, Y. Recombinant Tri-Segmented Pichinde Virus as a Novel Live Viral Vaccine Platform. Methods Mol. Biol. 2017, 1581, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Slavuljica, I.; Busche, A.; Babić, M.; Mitrović, M.; Gašparović, I.; Cekinović, D.; Markova Car, E.; Pernjak Pugel, E.; Ciković, A.; Lisnić, V.J.; et al. Recombinant mouse cytomegalovirus expressing a ligand for the NKG2D receptor is attenuated and has improved vaccine properties. J. Clin. Investig. 2010, 120, 4532–4545. [Google Scholar] [CrossRef] [PubMed]
- Hiršl, L.; Brizić, I.; Jenuš, T.; Lisnić, V.J.; Reichel, J.J.; Jurković, S.; Krmpotić, A.; Jonjić, S. Murine CMV Expressing the High Affinity NKG2D Ligand MULT-1: A Model for the Development of Cytomegalovirus-Based Vaccines. Front. Immunol. 2018, 9, 991. [Google Scholar] [CrossRef] [PubMed]
- Lauer, K.B.; Borrow, R.; Blanchard, T.J. Multivalent and Multipathogen Viral Vector Vaccines. Clin. Vaccine Immunol. 2017, 24, e00298-16. [Google Scholar] [CrossRef]
- Kenneson, A.; Cannon, M.J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol. 2007, 17, 253–276. [Google Scholar] [CrossRef]
- de Castro Ferreira, C.; Campi-Azevedo, A.C.; Peruhype-Magalhāes, V.; Costa-Pereira, C.; de Albuquerque, C.P.; Muniz, L.F.; de Souza, T.Y.; Oliveira, A.C.V.; Martins-Filho, O.A.; da Mota, L.M.H. The 17D-204 and 17DD yellow fever vaccines: An overview of major similarities and subtle differences. Expert Rev. Vaccines 2018, 17, 79–90. [Google Scholar] [CrossRef]
- Ahmed, R.; Akondy, R.S. Insights into human CD8+ T-cell memory using the yellow fever and smallpox vaccines. Immunol. Cell Biol. 2011, 89, 340–345. [Google Scholar] [CrossRef]
- DiNapoli, J.M.; Yang, L.; Samal, S.K.; Murphy, B.R.; Collins, P.L.; Bukreyev, A. Respiratory tract immunization of non-human primates with a Newcastle disease virus-vectored vaccine candidate against Ebola virus elicits a neutralizing antibody response. Vaccine 2010, 29, 17–25. [Google Scholar] [CrossRef]
- Basavarajappa, M.K.; Kumar, S.; Khattar, S.K.; Gebreluul, G.T.; Paldurai, A.; Samal, S.K. A recombinant Newcastle disease virus (NDV) expressing infectious laryngotracheitis virus (ILTV) surface glycoprotein D protects against highly virulent ILTV and NDV challenges in chickens. Vaccine 2014, 32, 3555–3563. [Google Scholar] [CrossRef]
- Wertz, G.W.; Perepelitsa, V.P.; Ball, L.A. Gene rearrangement attenuates expression and lethality of a nonsegmented negative strand RNA virus. Proc. Natl. Acad. Sci. USA 1998, 95, 3501–3506. [Google Scholar] [CrossRef]
- Ball, L.A.; Pringle, C.R.; Flanagan, B.; Perepelitsa, V.P.; Wertz, G.W. Phenotypic Consequences of Rearranging the P, M, and G Genes of Vesicular Stomatitis Virus. J. Virol. 1999, 73, 4705–4712. [Google Scholar] [CrossRef] [PubMed]
- Gomme, E.A.; Faul, E.J.; Flomenberg, P.; McGettigan, J.P.; Schnell, M.J. Characterization of a Single-Cycle Rabies Virus-Based Vaccine Vector. J. Virol. 2010. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Etessami, R.; Conzelmann, K.-K.; Fadai-Ghotbi, B.; Natelson, B.; Tsiang, H.; Ceccaldi, P.-E. Spread and pathogenic characteristics of a G-deficient rabies virus recombinant: An in vitro and in vivo study. J. Gen. Virol. 2000, 81, 2147–2153. [Google Scholar] [CrossRef] [PubMed]
- Shashkova, E.V.; May, S.M.; Barry, M.A. Characterization of human adenovirus serotypes 5, 6, 11, and 35 as anticancer agents. Virology 2009, 394, 311–320. [Google Scholar] [CrossRef]
- Florescu, D.F.; Schaenman, J.M.; on behalf of the AST Infectious Diseases Community of Practice. Adenovirus in solid organ transplant recipients: Guidelines from the American Society of Transplantation Infectious Diseases Community of Practice. Clin. Transplant. 2019, 33, e13527. [Google Scholar] [CrossRef]
- Peng, B.; Wang, L.R.; Gomez-Roman, V.R.; Davis-Warren, A.; Montefiori, D.C.; Kalyanaraman, V.S.; Venzon, D.; Zhao, J.; Kan, E.; Rowell, T.J.; et al. Replicating Rather than Nonreplicating Adenovirus-Human Immunodeficiency Virus Recombinant Vaccines Are Better at Eliciting Potent Cellular Immunity and Priming High-Titer Antibodies. J. Virol. 2005, 79, 10200–10209. [Google Scholar] [CrossRef]
- Chea, L.S.; Wyatt, L.S.; Gangadhara, S.; Moss, B.; Amara, R.R. Novel Modified Vaccinia Virus Ankara Vector Expressing Anti-apoptotic Gene B13R Delays Apoptosis and Enhances Humoral Responses. J. Virol. 2019, 93, e01648-18. [Google Scholar] [CrossRef]
- Dudek, T.; Knipe, D.M. Replication-defective viruses as vaccines and vaccine vectors. Virology 2006, 344, 230–239. [Google Scholar] [CrossRef]
- Henao-Restrepo, A.M.; Camacho, A.; Longini, I.M.; Watson, C.H.; Edmunds, W.J.; Egger, M.; Carroll, M.W.; Dean, N.E.; Diatta, I.; Doumbia, M.; et al. Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease: Final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ça Suffit!). Lancet 2017, 389, 505–518. [Google Scholar] [CrossRef]
- First FDA-Approved Vaccine for the Prevention of Ebola Virus Disease, Marking a Critical Milestone in Public Health Preparedness and Response | FDA. Available online: https://www.fda.gov/news-events/press-announcements/first-fda-approved-vaccine-prevention-ebola-virus-disease-marking-critical-milestone-public-health (accessed on 1 September 2020).
- ERVEBO® [Ebola Zaire Vaccine (rVSVΔG-ZEBOV-GP) live] Awarded Prequalification Status by the World Health Organization (WHO)—Merck.com. Available online: https://www.merck.com/news/ervebo-ebola-zaire-vaccine-rvsvδg-zebov-gp-live-awarded-prequalification-status-by-the-world-health-organization-who/ (accessed on 10 November 2020).
- Alharbi, N.K. Poxviral promoters for improving the immunogenicity of MVA delivered vaccines. Hum. Vaccin. Immunother. 2019, 15, 203–209. [Google Scholar] [CrossRef]
- Crosby, C.M.; Nehete, P.; Sastry, K.J.; Barry, M.A. Amplified and Persistent Immune Responses Generated by Single-Cycle Replicating Adenovirus Vaccines. J. Virol. 2015, 89, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Joglekar, A.V.; Sandoval, S. Pseudotyped Lentiviral Vectors: One Vector, Many Guises. Hum. Gene Ther. Methods 2017, 28, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Andrake, M.D.; Skalka, A.M. Retroviral Integrase: Then and Now. Annu. Rev. Virol. 2015, 2, 241–264. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Lee, J.-T.; Lee, K.; Kim, S.; Kim, J.Y.; Yoon, K.; Kim, S. Development of murine leukemia virus-based retroviral vectors with a minimum possibility of cis-activation. Gene Ther. 2011, 18, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A. Severe combined immunodeficiencies (SCID). Clin. Exp. Immunol. 2000, 122, 143–149. [Google Scholar] [CrossRef]
- Ram, Z.; Culver, K.W.; Oshiro, E.M.; Viola, J.J.; DeVroom, H.L.; Otto, E.; Long, Z.; Chiang, Y.; McGarrity, G.J.; Muul, L.M.; et al. Therapy of malignant brain tumors by intratumoral implantation of retroviral vector-producing cells. Nat. Med. 1997, 3, 1354–1361. [Google Scholar] [CrossRef]
- Schroers, R.; Sinha, I.; Segall, H.; Schmidt-Wolf, I.G.; Rooney, C.M.; Brenner, M.K.; Sutton, R.E.; Chen, S.Y. Transduction of human PBMC-derived dendritic cells and macrophages by an HIV-1-based lentiviral vector system. Mol. Ther. 2000, 1, 171–179. [Google Scholar] [CrossRef]
- Kobinger, G.P.; Weiner, D.J.; Yu, Q.-C.; Wilson, J.M. Filovirus-pseudotyped lentiviral vector can efficiently and stably transduce airway epithelia in vivo. Nat. Biotechnol. 2001, 19, 225–230. [Google Scholar] [CrossRef]
- Watson, D.J.; Kobinger, G.P.; Passini, M.A.; Wilson, J.M.; Wolfe, J.H. Targeted transduction patterns in the mouse brain by lentivirus vectors pseudotyped with VSV, Ebola, Mokola, LCMV, or MuLV envelope proteins. Mol. Ther. 2002, 5, 528–537. [Google Scholar] [CrossRef]
- Frecha, C.; Costa, C.; Nègre, D.; Gauthier, E.; Russell, S.J.; Cosset, F.-L.; Verhoeyen, E. Stable transduction of quiescent T cells without induction of cycle progression by a novel lentiviral vector pseudotyped with measles virus glycoproteins. Blood 2008, 112, 4843–4852. [Google Scholar] [CrossRef]
- Crosby, C.M.; Barry, M.A. IIIa deleted adenovirus as a single-cycle genome replicating vector. Virology 2014, 462–463, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Anguiano-Zarate, S.S.; Matchett, W.E.; Nehete, P.N.; Sastry, J.K.; Marzi, A.; Barry, M.A. A replicating single-cycle adenovirus vaccine against Ebola virus. J. Infect. Dis. 2018, 218, 1883–1889. [Google Scholar] [CrossRef] [PubMed]
- Crosby, C.M.; Matchett, W.E.; Anguiano-Zarate, S.S.; Parks, C.A.; Weaver, E.A.; Pease, L.R.; Webby, R.J.; Barry, M.A. Replicating Single-Cycle Adenovirus Vectors Generate Amplified Influenza Vaccine Responses. J. Virol. 2017, 91, e00720-16. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.; Hörig, H.; Kaufman, H.L. Poxviruses as vectors for cancer immunotherapy. Curr. Opin. Drug Discov. Devel. 2003, 6, 161–168. [Google Scholar]
- Taylor, J.; Trimarchi, C.; Weinberg, R.; Languet, B.; Guillermin, F.; Desmettre, P.; Paoletti, E. Efficacy studies on a canarypox-rabies recombinant virus. Vaccine 1991, 9, 190–193. [Google Scholar] [CrossRef]
- Teigler, J.E.; Phogat, S.; Franchini, G.; Hirsch, V.M.; Michael, N.L.; Barouch, D.H. The canarypox virus vector ALVAC induces distinct cytokine responses compared to the vaccinia virus-based vectors MVA and NYVAC in rhesus monkeys. J. Virol. 2014, 88, 1809–1814. [Google Scholar] [CrossRef]
- Mastrangelo, M.J.; Eisenlohr, L.C.; Gomella, L.; Lattime, E.C. Poxvirus vectors: Orphaned and underappreciated. J. Clin. Investig. 2000, 105, 1031–1034. [Google Scholar] [CrossRef]
- Guillaume-Vasselin, V.; Lemaitre, L.; Dhondt, K.P.; Tedeschi, L.; Poulard, A.; Charreyre, C.; Horvat, B. Protection from Hendra virus infection with Canarypox recombinant vaccine. npj Vaccines 2016, 1, 16003. [Google Scholar] [CrossRef]
- Gómez, C.E.; Perdiguero, B.; Garcia-Arriaza, J.; Esteban, M. Poxvirus vectors as HIV/AIDS vaccines in humans. Hum. Vaccin. Immunother. 2012, 8, 1192–1207. [Google Scholar] [CrossRef]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; De Souza, M.; Adams, E.; et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar] [CrossRef]
- Kelleher, A.D.; Puls, R.L.; Bebbington, M.; Boyle, D.; Ffrench, R.; Kent, S.J.; Kippax, S.; Purcell, D.F.J.; Thomson, S.; Wand, H.; et al. A randomized, placebo-controlled phase I trial of DNA prime, recombinant fowlpox virus boost prophylactic vaccine for HIV-1. AIDS 2006, 20, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Dale, C.J.; De Rose, R.; Wilson, K.M.; Croom, H.A.; Thomson, S.; Coupar, B.E.H.; Ramsay, A.; Purcell, D.F.J.; Ffrench, R.; Law, M.; et al. Evaluation in macaques of HIV-1 DNA vaccines containing primate CpG motifs and fowlpoxvirus vaccines co-expressing IFNγ or IL-12. Vaccine 2004, 23, 188–197. [Google Scholar] [CrossRef] [PubMed]
- De Rose, R.; Batten, C.J.; Smith, M.Z.; Fernandez, C.S.; Peut, V.; Thomson, S.; Ramshaw, I.A.; Coupar, B.E.H.; Boyle, D.B.; Venturi, V.; et al. Comparative efficacy of subtype AE simian-human immunodeficiency virus priming and boosting vaccines in pigtail macaques. J. Virol. 2007, 81, 292–300. [Google Scholar] [CrossRef][Green Version]
- De Rose, R.; Chea, S.; Dale, C.J.; Reece, J.; Fernandez, C.S.; Wilson, K.M.; Thomson, S.; Ramshaw, I.A.; Coupar, B.E.H.; Boyle, D.B.; et al. Subtype AE HIV-1 DNA and recombinant Fowlpoxvirus vaccines encoding five shared HIV-1 genes: Safety and T cell immunogenicity in macaques. Vaccine 2005, 23, 1949–1956. [Google Scholar] [CrossRef] [PubMed]
- Lousberg, E.L.; Diener, K.R.; Fraser, C.K.; Phipps, S.; Foster, P.S.; Chen, W.; Uematsu, S.; Akira, S.; Robertson, S.A.; Brown, M.P.; et al. Antigen-specific T-cell responses to a recombinant fowlpox virus are dependent on MyD88 and interleukin-18 and independent of Toll-like receptor 7 (TLR7)- and TLR9-mediated innate immune recognition. J. Virol. 2011, 85, 3385–3396. [Google Scholar] [CrossRef] [PubMed]
- Rayner, J.O.; Dryga, S.A.; Kamrud, K.I. Alphavirus vectors and vaccination. Rev. Med. Virol. 2002, 12, 279–296. [Google Scholar] [CrossRef]
- Schlesinger, S. Alphavirus vectors: Development and potential therapeutic applications. Expert Opin. Biol. Ther. 2001, 1, 177–191. [Google Scholar] [CrossRef]
- Xiong, C.; Levis, R.; Shen, P.; Schlesinger, S.; Rice, C.M.; Huang, H.V. Sindbis virus: An efficient, broad host range vector for gene expression in animal cells. Science 1989, 243, 1188–1191. [Google Scholar] [CrossRef]
- Davis, N.L.; Willis, L.V.; Smitht, J.F.; Johnston, R.E. In vitro synthesis of infectious venezuelan equine encephalitis virus RNA from a cDNA clone: Analysis of a viable deletion mutant. Virology 1989, 171, 189–204. [Google Scholar] [CrossRef]
- Pushko, P.; Parker, M.; Ludwig, G.V.; Davis, N.L.; Johnston, R.E.; Smith, J.F. Replicon-helper systems from attenuated Venezuelan equine encephalitis virus: Expression of heterologous genes in vitro and immunization against heterologous pathogens in vivo. Virology 1997, 239, 389–401. [Google Scholar] [CrossRef]
- Frolov, I.; Frolova, E.; Schlesinger, S. Sindbis virus replicons and Sindbis virus: Assembly of chimeras and of particles deficient in virus RNA. J. Virol. 1997, 71, 2819–2829. [Google Scholar] [CrossRef] [PubMed]
- Smerdou, C.; Liljeström, P. Two-Helper RNA System for Production of Recombinant Semliki Forest Virus Particles. J. Virol. 1999, 73, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Koutsoumpli, G.; van de Wall, S.; Daemen, T. An alphavirus-based therapeutic cancer vaccine: From design to clinical trial. Cancer Immunol. Immunother. 2019, 68, 849–859. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, G.H.; Johnston, R.E. Role of dendritic cell targeting in Venezuelan equine encephalitis virus pathogenesis. J. Virol. 2000, 74, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Huckriede, A.; Bungener, L.; Holtrop, M.; de Vries, J.; Waarts, B.-L.; Daemen, T.; Wilschut, J. Induction of cytotoxic T lymphocyte activity by immunization with recombinant Semliki Forest virus: Indications for cross-priming. Vaccine 2004, 22, 1104–1113. [Google Scholar] [CrossRef]
- Näslund, T.I.; Kostic, L.; Nordström, E.K.L.; Chen, M.; Liljeström, P. Role of innate signalling pathways in the immunogenicity of alphaviral replicon-based vaccines. Virol. J. 2011, 8, 36. [Google Scholar] [CrossRef]
- Brand, D.; Lemiale, F.; Turbica, I.; Buzelay, L.; Brunet, S.; Barin, F. Comparative analysis of humoral immune responses to HIV type 1 envelope glycoproteins in mice immunized with a DNA vaccine, recombinant semliki forest virus RNA, or recombinant semliki forest virus particles. AIDS Res. Hum. Retroviruses 1998, 14, 1369–1377. [Google Scholar] [CrossRef]
- Colombage, G.; Hall, R.; Pavy, M.; Lobigs, M. DNA-Based and Alphavirus-Vectored Immunisation with PrM and E Proteins Elicits Long-Lived and Protective Immunity against the Flavivirus, Murray Valley Encephalitis Virus. Virology 1998, 250, 151–163. [Google Scholar] [CrossRef]
- Berglund, P.; Fleeton, M.N.; Smerdou, C.; Liljeström, P. Immunization with recombinant Semliki Forest virus induces protection against influenza challenge in mice. Vaccine 1999, 17, 497–507. [Google Scholar] [CrossRef]
- Tsuji, M.; Bergmann, C.C.; Takita-Sonoda, Y.; Murata, K.; Rodrigues, E.G.; Nussenzweig, R.S.; Zavala, F. Recombinant Sindbis viruses expressing a cytotoxic T-lymphocyte epitope of a malaria parasite or of influenza virus elicit protection against the corresponding pathogen in mice. J. Virol. 1998, 72, 6907–6910. [Google Scholar] [CrossRef]
- Kamrud, K.I.; Hooper, J.W.; Elgh, F.; Schmaljohn, C.S. Comparison of the Protective Efficacy of Naked DNA, DNA-based Sindbis Replicon, and Packaged Sindbis Replicon Vectors Expressing Hantavirus Structural Genes in Hamsters. Virology 1999, 263, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Riezebos-Brilman, A.; Regts, J.; Freyschmidt, E.-J.; Dontje, B.; Wilschut, J.; Daemen, T. Induction of human papilloma virus E6/E7-specific cytotoxic T-lymphocyte activity in immune-tolerant, E6/E7-transgenic mice. Gene Ther. 2005, 12, 1410–1414. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.A.; Bray, M.; Bakken, R.; Hart, M.K. Vaccine Potential of Ebola Virus VP24, VP30, VP35, and VP40 Proteins. Virology 2001, 286, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.A.; Hart, M.K. Protection from Ebola virus mediated by cytotoxic T lymphocytes specific for the viral nucleoprotein. J. Virol. 2001, 75, 2660–2664. [Google Scholar] [CrossRef] [PubMed]
- Pushko, P.; Bray, M.; Ludwig, G.V.; Parker, M.; Schmaljohn, A.; Sanchez, A.; Jahrling, P.B.; Smith, J.F. Recombinant RNA replicons derived from attenuated Venezuelan equine encephalitis virus protect guinea pigs and mice from Ebola hemorrhagic fever virus. Vaccine 2000, 19, 142–153. [Google Scholar] [CrossRef]
- Hevey, M.; Negley, D.; Pushko, P.; Smith, J.; Schmaljohn, A. Marburg Virus Vaccines Based upon Alphavirus Replicons Protect Guinea Pigs and Nonhuman Primates. Virology 1998, 251, 28–37. [Google Scholar] [CrossRef]
- Davis, N.L.; Caley, I.J.; Brown, K.W.; Betts, M.R.; Irlbeck, D.M.; McGrath, K.M.; Connell, M.J.; Montefiori, D.C.; Frelinger, J.A.; Swanstrom, R.; et al. Vaccination of Macaques against Pathogenic Simian Immunodeficiency Virus with Venezuelan Equine Encephalitis Virus Replicon Particles. J. Virol. 2000, 74, 371–378. [Google Scholar] [CrossRef]
- Lee, J.S.; Pushko, P.; Parker, M.D.; Dertzbaugh, M.T.; Smith, L.A.; Smith, J.F. Candidate vaccine against botulinum neurotoxin serotype A derived from a Venezuelan equine encephalitis virus vector system. Infect. Immun. 2001, 69, 5709–5715. [Google Scholar] [CrossRef]
- Zhou, X.; Berglund, P.; Zhao, H.; Liljeström, P.; Jondal, M. Generation of cytotoxic and humoral immune responses by nonreplicative recombinant Semliki Forest virus. Proc. Natl. Acad. Sci. USA 1995, 92, 3009–3013. [Google Scholar] [CrossRef]
- Johnston, L.J.; Halliday, G.M.; King, N.J. Phenotypic changes in Langerhans’ cells after infection with arboviruses: A role in the immune response to epidermally acquired viral infection? J. Virol. 1996, 70, 4761–4766. [Google Scholar] [CrossRef]
- Lundstrom, K. Plasmid DNA-based Alphavirus Vaccines. Vaccines 2019, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D.I.; Reap, E.A.; Katen, K.; Watson, A.; Smith, K.; Norberg, P.; Olmsted, R.A.; Hoeper, A.; Morris, J.; Negri, S.; et al. Randomized, double-blind, Phase 1 trial of an alphavirus replicon vaccine for cytomegalovirus in CMV seronegative adult volunteers. Vaccine 2009, 28, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Hobeika, A.C.; Osada, T.; Berglund, P.; Hubby, B.; Negri, S.; Niedzwiecki, D.; Devi, G.R.; Burnett, B.K.; Clay, T.M.; et al. An alphavirus vector overcomes the presence of neutralizing antibodies and elevated numbers of Tregs to induce immune responses in humans with advanced cancer. J. Clin. Investig. 2010, 120, 3234–3241. [Google Scholar] [CrossRef] [PubMed]
- Wecker, M.; Gilbert, P.; Russell, N.; Hural, J.; Allen, M.; Pensiero, M.; Chulay, J.; Chiu, Y.-L.; Karim, S.S.A.; Burke, D.S.; et al. Phase I safety and immunogenicity evaluations of an alphavirus replicon HIV-1 subtype C gag vaccine in healthy HIV-1-uninfected adults. Clin. Vaccine Immunol. 2012, 19, 1651–1660. [Google Scholar] [CrossRef]
- McLay, L.; Liang, Y.; Ly, H. Comparative analysis of disease pathogenesis and molecular mechanisms of New World and Old World arenavirus infections. J. Gen. Virol. 2014, 95, 1–15. [Google Scholar] [CrossRef]
- Bonthius, D.J. Lymphocytic choriomeningitis virus: An underrecognized cause of neurologic disease in the fetus, child, and adult. Semin. Pediatr. Neurol. 2012, 19, 89–95. [Google Scholar] [CrossRef]
- Fischer, S.A.; Graham, M.B.; Kuehnert, M.J.; Kotton, C.N.; Srinivasan, A.; Marty, F.M.; Comer, J.A.; Guarner, J.; Paddock, C.D.; DeMeo, D.L.; et al. Transmission of lymphocytic choriomeningitis virus by organ transplantation. N. Engl. J. Med. 2006, 354, 2235–2249. [Google Scholar] [CrossRef]
- Lledó, L.; Gegúndez, M.I.; Saz, J.V.; Bahamontes, N.; Beltrán, M. Lymphocytic choriomeningitis virus infection in a province of Spain: Analysis of sera from the general population and wild rodents. J. Med. Virol. 2003, 70, 273–275. [Google Scholar] [CrossRef]
- Buchmeier, M.; Adam, E.; Rawls, W.E. Serological evidence of infection by Pichinde virus among laboratory workers. Infect. Immun. 1974, 9, 821–823. [Google Scholar] [CrossRef]
- Dhanwani, R.; Ly, H. Arenaviral vaccine vectors to combat infectious diseases. Oncotarget 2016, 7, 44875. [Google Scholar] [CrossRef]
- Cheng, B.Y.H.; Ortiz-Riaño, E.; de la Torre, J.C.; Martínez-Sobrido, L. Arenavirus Genome Rearrangement for the Development of Live Attenuated Vaccines. J. Virol. 2015, 89, 7373–7384. [Google Scholar] [CrossRef] [PubMed]
- Flatz, L.; Hegazy, A.N.; Bergthaler, A.; Verschoor, A.; Claus, C.; Fernandez, M.; Gattinoni, L.; Johnson, S.; Kreppel, F.; Kochanek, S.; et al. Development of replication-defective lymphocytic choriomeningitis virus vectors for the induction of potent CD8+ T cell immunity. Nat. Med. 2010, 16, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Penaloza MacMaster, P.; Shields, J.L.; Alayo, Q.A.; Cabral, C.; Jimenez, J.; Mondesir, J.; Chandrashekar, A.; Cabral, J.M.; Lim, M.; Iampietro, M.J.; et al. Development of novel replication-defective lymphocytic choriomeningitis virus vectors expressing SIV antigens. Vaccine 2017, 35, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dhanwani, R.; Zhou, Y.; Huang, Q.; Verma, V.; Dileepan, M.; Ly, H.; Liang, Y. A Novel Live Pichinde Virus-Based Vaccine Vector Induces Enhanced Humoral and Cellular Immunity after a Booster Dose. J. Virol. 2015, 90, 2551–2560. [Google Scholar] [CrossRef] [PubMed]
- Sommerstein, R.; Flatz, L.; Remy, M.M.; Malinge, P.; Magistrelli, G.; Fischer, N.; Sahin, M.; Bergthaler, A.; Igonet, S.; ter Meulen, J.; et al. Arenavirus Glycan Shield Promotes Neutralizing Antibody Evasion and Protracted Infection. PLOS Pathog. 2015, 11, e1005276. [Google Scholar] [CrossRef]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef]
- Méndez, A.C.; Rodríguez-Rojas, C.; Val, M. Del Vaccine vectors: The bright side of cytomegalovirus. Med. Microbiol. Immunol. 2019, 208, 349–363. [Google Scholar] [CrossRef]
- Wang, D.; Freed, D.C.; He, X.; Li, F.; Tang, A.; Cox, K.S.; Dubey, S.A.; Cole, S.; Medi, M.B.; Liu, Y.; et al. A replication-defective human cytomegalovirus vaccine for prevention of congenital infection. Sci. Transl. Med. 2016, 8, 362ra145 LP-362ra145. [Google Scholar] [CrossRef]
- Hansen, S.G.; Vieville, C.; Whizin, N.; Coyne-Johnson, L.; Siess, D.C.; Drummond, D.D.; Legasse, A.W.; Axthelm, M.K.; Oswald, K.; Trubey, C.M.; et al. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat. Med. 2009, 15, 293–299. [Google Scholar] [CrossRef]
- Hansen, S.G.; Ford, J.C.; Lewis, M.S.; Ventura, A.B.; Hughes, C.M.; Coyne-Johnson, L.; Whizin, N.; Oswald, K.; Shoemaker, R.; Swanson, T.; et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 2011, 473, 523–527. [Google Scholar] [CrossRef]
- Hansen, S.G.; Jr, M.P.; Ventura, A.B.; Hughes, C.M.; Gilbride, R.M.; Ford, J.C.; Oswald, K.; Shoemaker, R.; Li, Y.; Lewis, M.S.; et al. Immune clearance of highly pathogenic SIV infection. Nature 2013, 502, 100–104. [Google Scholar] [CrossRef]
- Tsuda, Y.; Parkins, C.J.; Caposio, P.; Feldmann, F.; Botto, S.; Ball, S.; Messaoudi, I.; Cicin-Sain, L.; Feldmann, H.; Jarvis, M.A. A cytomegalovirus-based vaccine provides long-lasting protection against lethal Ebola virus challenge after a single dose. Vaccine 2015, 33, 2261–2266. [Google Scholar] [CrossRef] [PubMed]
- Marzi, A.; Murphy, A.A.; Feldmann, F.; Parkins, C.J.; Haddock, E.; Hanley, P.W.; Emery, M.J.; Engelmann, F.; Messaoudi, I.; Feldmann, H.; et al. Cytomegalovirus-based vaccine expressing Ebola virus glycoprotein protects nonhuman primates from Ebola virus infection. Sci. Rep. 2016, 6, 21674. [Google Scholar] [CrossRef] [PubMed]
- Tierney, R.; Nakai, T.; Parkins, C.J.; Caposio, P.; Fairweather, N.F.; Sesardic, D.; Jarvis, M.A. A single-dose cytomegalovirus-based vaccine encoding tetanus toxin fragment C induces sustained levels of protective tetanus toxin antibodies in mice. Vaccine 2012, 30, 3047–3052. [Google Scholar] [CrossRef] [PubMed]
- Beverley, P.C.L.; Ruzsics, Z.; Hey, A.; Hutchings, C.; Boos, S.; Bolinger, B.; Marchi, E.; O’Hara, G.; Klenerman, P.; Koszinowski, U.H.; et al. A Novel Murine Cytomegalovirus Vaccine Vector Protects against Mycobacterium tuberculosis. J. Immunol. 2014, 193, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Zak, D.E.; Xu, G.; Ford, J.C.; Marshall, E.E.; Malouli, D.; Gilbride, R.M.; Hughes, C.M.; Ventura, A.B.; Ainslie, E.; et al. Prevention of tuberculosis in rhesus macaques by a cytomegalovirus-based vaccine. Nat. Med. 2018, 24, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Gotuzzo, E.; Yactayo, S.; Córdova, E. Efficacy and duration of immunity after yellow fever vaccination: Systematic review on the need for a booster every 10 years. Am. J. Trop. Med. Hyg. 2013, 89, 434–444. [Google Scholar] [CrossRef]
- Niedrig, M.; Lademann, M.; Emmerich, P.; Lafrenz, M. Assessment of IgG antibodies against yellow fever virus after vaccination with 17D by different assays: Neutralization test, haemagglutination inhibition test, immunofluorescence assay and ELISA. Trop. Med. Int. Health 1999, 4, 867–871. [Google Scholar] [CrossRef]
- Akondy, R.S.; Monson, N.D.; Miller, J.D.; Edupuganti, S.; Teuwen, D.; Wu, H.; Quyyumi, F.; Garg, S.; Altman, J.D.; Del Rio, C.; et al. The Yellow Fever Virus Vaccine Induces a Broad and Polyfunctional Human Memory CD8 T Cell Response. J. Immunol. 2009, 183, 7919–7930. [Google Scholar] [CrossRef]
- Chambers, T.J.; Hahn, C.S.; Galler, R.; Rice, C.M. Flavivirus Genome Organization, Expression, and Replication. Annu. Rev. Microbiol. 1990, 44, 649–688. [Google Scholar] [CrossRef]
- Chambers, T.J.; Nestorowicz, A.; Mason, P.W.; Rice, C.M. Yellow fever/Japanese encephalitis chimeric viruses: Construction and biological properties. J. Virol. 1999, 73, 3095–3101. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.; Miller, C.; Catalan, J.; Myers, G.A.; Ratterree, M.S.; Trent, D.W.; Monath, T.P. ChimeriVax-West Nile virus live-attenuated vaccine: Preclinical evaluation of safety, immunogenicity, and efficacy. J. Virol. 2004, 78, 12497–12507. [Google Scholar] [CrossRef] [PubMed]
- Guirakhoo, F.; Arroyo, J.; Pugachev, K.V.; Miller, C.; Zhang, Z.X.; Weltzin, R.; Georgakopoulos, K.; Catalan, J.; Ocran, S.; Soike, K.; et al. Construction, safety, and immunogenicity in nonhuman primates of a chimeric yellow fever-dengue virus tetravalent vaccine. J. Virol. 2001, 75, 7290–7304. [Google Scholar] [CrossRef] [PubMed]
- Sabchareon, A.; Wallace, D.; Sirivichayakul, C.; Limkittikul, K.; Chanthavanich, P.; Suvannadabba, S.; Jiwariyavej, V.; Dulyachai, W.; Pengsaa, K.; Wartel, T.A.; et al. Protective efficacy of the recombinant, live-attenuated, CYD tetravalent dengue vaccine in Thai schoolchildren: A randomised, controlled phase 2b trial. Lancet 2012, 380, 1559–1567. [Google Scholar] [CrossRef]
- Dayan, G.H.; Garbes, P.; Noriega, F.; de Sadovsky, A.D.I.; Rodrigues, P.M.; Giuberti, C.; Dietze, R. Immunogenicity and safety of a recombinant tetravalent dengue vaccine in children and adolescents ages 9-16 years in Brazil. Am. J. Trop. Med. Hyg. 2013, 89, 1058–1065. [Google Scholar] [CrossRef]
- Dayan, G.H.; Thakur, M.; Boaz, M.; Johnson, C. Safety and immunogenicity of three tetravalent dengue vaccine formulations in healthy adults in the USA. Vaccine 2013, 31, 5047–5054. [Google Scholar] [CrossRef]
- Guirakhoo, F.; Kitchener, S.; Morrison, D.; Forrat, R.; McCarthy, K.; Nichols, R.; Yoksan, S.; Duan, X.; Ermak, T.H.; Kanesa-Thasan, N.; et al. Live attenuated chimeric yellow fever dengue type 2 (ChimeriVax-DEN2) vaccine: Phase I clinical trial for safety and immunogenicity: Effect of yellow fever pre-immunity in induction of cross neutralizing antibody responses to all 4 dengue serotypes. Hum. Vaccin. 2006, 2, 60–67. [Google Scholar] [CrossRef]
- Qiao, M.; Shaw, D.; Forrat, R.; Wartel-Tram, A.; Lang, J. Priming effect of dengue and yellow fever vaccination on the immunogenicity, infectivity, and safety of a tetravalent dengue vaccine in humans. Am. J. Trop. Med. Hyg. 2011, 85, 724–731. [Google Scholar] [CrossRef]
- Monath, T.P.; McCarthy, K.; Bedford, P.; Johnson, C.T.; Nichols, R.; Yoksan, S.; Marchesani, R.; Knauber, M.; Wells, K.H.; Arroyo, J.; et al. Clinical proof of principle for ChimeriVaxTM: Recombinant live, attenuated vaccines against flavivirus infections. Vaccine 2002, 20, 1004–1018. [Google Scholar] [CrossRef]
- Chokephaibulkit, K.; Sirivichayakul, C.; Thisyakorn, U.; Sabchareon, A.; Pancharoen, C.; Bouckenooghe, A.; Gailhardou, S.; Boaz, M.; Feroldi, E. Safety and immunogenicity of a single administration of live-attenuated japanese encephalitis vaccine in previously primed 2- to 5-year-olds and naive 12- to 24-month-olds: Multicenter randomized controlled trial. Pediatr. Infect. Dis. J. 2010, 29, 1111–1117. [Google Scholar] [CrossRef]
- Monath, T.P.; Guirakhoo, F.; Nichols, R.; Yoksan, S.; Schrader, R.; Murphy, C.; Blum, P.; Woodward, S.; McCarthy, K.; Mathis, D.; et al. Chimeric Live, Attenuated Vaccine against Japanese Encephalitis (ChimeriVax-JE): Phase 2 Clinical Trials for Safety and Immunogenicity, Effect of Vaccine Dose and Schedule, and Memory Response to Challenge with Inactivated Japanese Encephalitis Antigen. J. Infect. Dis. 2003, 188, 1213–1230. [Google Scholar] [CrossRef] [PubMed]
- De Filette, M.; Ulbert, S.; Diamond, M.; Sanders, N.N. Recent progress in West Nile virus diagnosis and vaccination. Vet. Res. 2012, 43, 16. [Google Scholar] [CrossRef] [PubMed]
- Giel-Moloney, M.; Goncalvez, A.P.; Catalan, J.; Lecouturier, V.; Girerd-Chambaz, Y.; Diaz, F.; Maldonado-Arocho, F.; Gomila, R.C.; Bernard, M.-C.; Oomen, R.; et al. Chimeric yellow fever 17D-Zika virus (ChimeriVax-Zika) as a live-attenuated Zika virus vaccine. Sci. Rep. 2018, 8, 13206. [Google Scholar] [CrossRef] [PubMed]
- Bonaldo, M.C.; Mello, S.M.; Trindade, G.F.; Rangel, A.A.; Duarte, A.S.; Oliveira, P.J.; Freire, M.S.; Kubelka, C.F.; Galler, R. Construction and characterization of recombinant flaviviruses bearing insertions between E and NS1 genes. Virol. J. 2007, 4, 115. [Google Scholar] [CrossRef] [PubMed]
- Trindade, G.; de Santana, M.V.; Ribeiro, J.; Galler, R.; Bonaldo, M. Retention of a recombinant GFP protein expressed by the yellow fever 17D virus in the E/NS1 intergenic region in the endoplasmic reticulum. Mem. Inst. Oswaldo Cruz 2012, 107, 262–272. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martins, M.A.; Bonaldo, M.C.; Rudersdorf, R.A.; Piaskowski, S.M.; Rakasz, E.G.; Weisgrau, K.L.; Furlott, J.R.; Eernisse, C.M.; de Santana, M.G.V.; Hidalgo, B.; et al. Immunogenicity of Seven New Recombinant Yellow Fever Viruses 17D Expressing Fragments of SIVmac239 Gag, Nef, and Vif in Indian Rhesus Macaques. PLoS ONE 2013, 8, e54434. [Google Scholar] [CrossRef]
- Nogueira, R.T.; Nogueira, A.R.; Pereira, M.C.S.; Rodrigues, M.M.; da Costa Neves, P.C.; Galler, R.; Bonaldo, M.C. Recombinant yellow fever viruses elicit CD8+ T cell responses and protective immunity against Trypanosoma cruzi. PLoS ONE 2013, 8, e59347. [Google Scholar] [CrossRef]
- Franco, D.; Li, W.; Qing, F.; Stoyanov, C.T.; Moran, T.; Rice, C.M.; Ho, D.D. Evaluation of yellow fever virus 17D strain as a new vector for HIV-1 vaccine development. Vaccine 2010, 28, 5676–5685. [Google Scholar] [CrossRef]
- Jiang, X.; Dalebout, T.J.; Bredenbeek, P.J.; Carrion, R.; Brasky, K.; Patterson, J.; Goicochea, M.; Bryant, J.; Salvato, M.S.; Lukashevich, I.S.; et al. Yellow fever 17D-vectored vaccines expressing Lassa virus GP1 and GP2 glycoproteins provide protection against fatal disease in guinea pigs. Vaccine 2011, 29, 1248–1257. [Google Scholar] [CrossRef]
- Deas, T.S.; Binduga-Gajewska, I.; Tilgner, M.; Ren, P.; Stein, D.A.; Moulton, H.M.; Iversen, P.L.; Kauffman, E.B.; Kramer, L.D.; Shi, P.-Y. Inhibition of flavivirus infections by antisense oligomers specifically suppressing viral translation and RNA replication. J. Virol. 2005, 79, 4599–4609. [Google Scholar] [CrossRef]
- Zou, G.; Xu, H.Y.; Qing, M.; Wang, Q.-Y.; Shi, P.-Y. Development and characterization of a stable luciferase dengue virus for high-throughput screening. Antiviral Res. 2011, 91, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Bonaldo, M.C.; Sequeira, P.C.; Galler, R. The yellow fever 17D virus as a platform for new live attenuated vaccines. Hum. Vaccines Immunother. 2014, 10, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.J. Newcastle disease and other avian paramyxoviruses. OIE Rev. Sci. Tech. 2000. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, T.; Cros, J.; Park, M.S.; Nakaya, Y.; Zheng, H.; Sagrera, A.; Villar, E.; García-Sastre, A.; Palese, P. Recombinant Newcastle disease virus as a vaccine vector. J. Virol. 2001, 75, 11868–11873. [Google Scholar] [CrossRef]
- Kortekaas, J.; Dekker, A.; de Boer, S.M.; Weerdmeester, K.; Vloet, R.P.M.; de Wit, A.A.C.; Peeters, B.P.H.; Moormann, R.J.M. Intramuscular inoculation of calves with an experimental Newcastle disease virus-based vector vaccine elicits neutralizing antibodies against Rift Valley fever virus. Vaccine 2010, 28, 2271–2276. [Google Scholar] [CrossRef]
- Khattar, S.K.; Collins, P.L.; Samal, S.K. Immunization of cattle with recombinant Newcastle disease virus expressing bovine herpesvirus-1 (BHV-1) glycoprotein D induces mucosal and serum antibody responses and provides partial protection against BHV-1. Vaccine 2010, 28, 3159–3170. [Google Scholar] [CrossRef]
- Kim, S.-H.; Paldurai, A.; Xiao, S.; Collins, P.L.; Samal, S.K. Modified Newcastle disease virus vectors expressing the H5 hemagglutinin induce enhanced protection against highly pathogenic H5N1 avian influenza virus in chickens. Vaccine 2014, 32, 4428–4435. [Google Scholar] [CrossRef]
- Khattar, S.K.; Nayak, B.; Kim, S.-H.; Xiao, S.; Samal, S.; Paldurai, A.; Buchholz, U.J.; Collins, P.L.; Samal, S.K. Evaluation of the replication, pathogenicity, and immunogenicity of avian paramyxovirus (APMV) serotypes 2, 3, 4, 5, 7, and 9 in rhesus macaques. PLoS ONE 2013, 8, e75456. [Google Scholar] [CrossRef]
- Yoshida, A.; Kim, S.-H.; Manoharan, V.K.; Varghese, B.P.; Paldurai, A.; Samal, S. Novel avian paramyxovirus-based vaccine vectors expressing the Ebola virus glycoprotein elicit mucosal and humoral immune responses in guinea pigs. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Yoshida, A.; Samal, S.K. Avian Paramyxovirus Type-3 as a Vaccine Vector: Identification of a Genome Location for High Level Expression of a Foreign Gene. Front. Microbiol. 2017, 8, 693. [Google Scholar] [CrossRef]
- Rose, J.; Schubert, M. Rhabdovirus Genomes and Their Products. In The Rhabdoviruses; Springer: New York, NY, USA, 1987; pp. 129–166. [Google Scholar]
- Schnell, M.J.; Foley, H.D.; Siler, C.A.; McGettigan, J.P.; Dietzschold, B.; Pomerantz, R.J. Recombinant rabies virus as potential live-viral vaccines for HIV-1. Proc. Natl. Acad. Sci. USA 2000, 97, 3544–3549. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.K.; Hendry, R.M.; Singh, V.; Rose, J.K.; Seligman, S.J.; Klug, B.; Kochhar, S.; Mac, L.M.; Carbery, B.; Chen, R.T.; et al. Live virus vaccines based on a vesicular stomatitis virus (VSV) backbone: Standardized template with key considerations for a risk/benefit assessment. Vaccine 2016, 34, 6597–6609. [Google Scholar] [CrossRef] [PubMed]
- Lévy, Y.; Lane, C.; Piot, P.; Beavogui, A.H.; Kieh, M.; Leigh, B.; Doumbia, S.; D’Ortenzio, E.; Lévy-Marchal, C.; Pierson, J.; et al. Prevention of Ebola virus disease through vaccination: Where we are in 2018. Lancet 2018, 392, P787–P790. [Google Scholar] [CrossRef]
- Longini, I.M.; Røttingen, J.-A.; Kieny, M.P.; Edmunds, W.J.; Henao-Restrepo, A.M. Questionable efficacy of the rVSV-ZEBOV Ebola vaccine—Authors’ reply. Lancet 2018, 391, 1021–1022. [Google Scholar] [CrossRef]
- Keusch, G.T.; McAdam, K.; Cuff, P.A.; Mancher, M.; Busta, E.R. Integrating Clinical Research into Epidemic Response: The Ebola Experience; National Academies Press: Washington, DC, USA, 2017; ISBN 9780309457798. [Google Scholar]
- Rechtien, A.; Richert, L.; Lorenzo, H.; Martrus, G.; Hejblum, B.; Dahlke, C.; Kasonta, R.; Zinser, M.; Stubbe, H.; Matschl, U.; et al. Systems Vaccinology Identifies an Early Innate Immune Signature as a Correlate of Antibody Responses to the Ebola Vaccine rVSV-ZEBOV. Cell Rep. 2017, 20, 2251–2261. [Google Scholar] [CrossRef]
- Emanuel, J.; Callison, J.; Dowd, K.A.; Pierson, T.C.; Feldmann, H.; Marzi, A. A VSV-based Zika virus vaccine protects mice from lethal challenge. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Racine, T.; Kobinger, G.P.; Arts, E.J. Development of an HIV vaccine using a vesicular stomatitis virus vector expressing designer HIV-1 envelope glycoproteins to enhance humoral responses. AIDS Res. Ther. 2017, 14, 55. [Google Scholar] [CrossRef]
- Bishnoi, S.; Tiwari, R.; Gupta, S.; Byrareddy, S.N.; Nayak, D. Oncotargeting by Vesicular Stomatitis Virus (VSV): Advances in Cancer Therapy. Viruses 2018, 10, 90. [Google Scholar] [CrossRef]
- Ugolini, G. Specificity of rabies virus as a transneuronal tracer of motor networks: Transfer from hypoglossal motoneurons to connected second-order and higher order central nervous system cell groups. J. Comp. Neurol. 1995, 356, 457–480. [Google Scholar] [CrossRef]
- Ugolini, G. Advances in viral transneuronal tracing. J. Neurosci. Methods 2010, 194, 2–20. [Google Scholar] [CrossRef]
- Osakada, F.; Callaway, E.M. Design and generation of recombinant rabies virus vectors. Nat. Protoc. 2013, 8, 1583–1601. [Google Scholar] [CrossRef]
- Wickersham, I.R.; Lyon, D.C.; Barnard, R.J.O.; Mori, T.; Finke, S.; Conzelmann, K.-K.; Young, J.A.T.; Callaway, E.M. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron 2007, 53, 639–647. [Google Scholar] [CrossRef] [PubMed]
- McGettigan, J.P.; Naper, K.; Orenstein, J.; Koser, M.; McKenna, P.M.; Schnell, M.J. Functional human immunodeficiency virus type 1 (HIV-1) Gag-Pol or HIV-1 Gag-Pol and env expressed from a single rhabdovirus-based vaccine vector genome. J. Virol. 2003, 77, 10889–10899. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mebatsion, T.; Conzelmann, K.K. Specific infection of CD4+ target cells by recombinant rabies virus pseudotypes carrying the HIV-1 envelope spike protein. Proc. Natl. Acad. Sci. USA 1996, 93, 11366–11370. [Google Scholar] [CrossRef]
- McGettigan, J.P.; Pomerantz, R.J.; Siler, C.A.; McKenna, P.M.; Foley, H.D.; Dietzschold, B.; Schnell, M.J. Second-generation rabies virus-based vaccine vectors expressing human immunodeficiency virus type 1 gag have greatly reduced pathogenicity but are highly immunogenic. J. Virol. 2003, 77, 237–244. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Keshwara, R.; Shiels, T.; Postnikova, E.; Kurup, D.; Wirblich, C.; Johnson, R.F.; Schnell, M.J. Rabies-based vaccine induces potent immune responses against Nipah virus. npj Vaccines 2019, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Wickersham, I.R.; Finke, S.; Conzelmann, K.-K.; Callaway, E.M. Retrograde neuronal tracing with a deletion-mutant rabies virus. Nat. Methods 2007, 4, 47–49. [Google Scholar] [CrossRef]
- Brun, A.; Albina, E.; Barret, T.; Chapman, D.A.G.; Czub, M.; Dixon, L.K.; Keil, G.M.; Klonjkowski, B.; Le Potier, M.-F.; Libeau, G.; et al. Antigen delivery systems for veterinary vaccine development. Viral-vector based delivery systems. Vaccine 2008, 26, 6508–6528. [Google Scholar] [CrossRef]
- Baron, M.D.; Iqbal, M.; Nair, V. Recent advances in viral vectors in veterinary vaccinology. Curr. Opin. Virol. 2018, 29, 1–7. [Google Scholar] [CrossRef]
- Ferreira, T.B.; Alves, P.M.; Aunins, J.G.; Carrondo, M.J.T. Use of adenoviral vectors as veterinary vaccines. Gene Ther. 2005, 12, S73–S83. [Google Scholar] [CrossRef]
- Veterinary Biological Products Licensees and Permittees. 2020. Available online: https://www.aphis.usda.gov/animal_health/vet_biologics/publications/currentprodcodebook.pdf (accessed on 13 November 2020).
- Pardo, M.C.; Tanner, P.; Bauman, J.; Silver, K.; Fischer, L. Immunization of puppies in the presence of maternally derived antibodies against canine distemper virus. J. Comp. Pathol. 2007, 137, S72–S75. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, J.; Jarrett, O.; Neil, J.C.; Desmettre, P.; Paoletti, E. Protection of cats against feline leukemia virus by vaccination with a canarypox virus recombinant, ALVAC-FL. J. Virol. 1993, 67, 2370–2375. [Google Scholar] [CrossRef] [PubMed]
- Welter, J.; Taylor, J.; Tartaglia, J.; Paoletti, E.; Stephensen, C.B. Vaccination against canine distemper virus infection in infant ferrets with and without maternal antibody protection, using recombinant attenuated poxvirus vaccines. J. Virol. 2000, 74, 6358–6367. [Google Scholar] [CrossRef] [PubMed]
- Jas, D.; Coupier, C.; Toulemonde, C.E.; Guigal, P.M.; Poulet, H. Three-year duration of immunity in cats vaccinated with a canarypox-vectored recombinant rabies virus vaccine. Vaccine 2012, 30, 6991–6996. [Google Scholar] [CrossRef]
- Hendrick, M.J.; Goldschmidt, M.H.; Shofer, F.S.; Wang, Y.Y.; Somlyo, A.P. Postvaccinal sarcomas in the cat: Epidemiology and electron probe microanalytical identification of aluminum. Cancer Res. 1992, 52, 5391–5394. [Google Scholar]
- Hartmann, K.; Day, M.J.; Thiry, E.; Lloret, A.; Frymus, T.; Addie, D.; Boucraut-Baralon, C.; Egberink, H.; Gruffydd-Jones, T.; Horzinek, M.C.; et al. Feline injection-site sarcoma: ABCD guidelines on prevention and management. J. Feline Med. Surg. 2015, 17, 606–613. [Google Scholar] [CrossRef]
- Maki, J.; Guiot, A.-L.; Aubert, M.; Brochier, B.; Cliquet, F.; Hanlon, C.A.; King, R.; Oertli, E.H.; Rupprecht, C.E.; Schumacher, C.; et al. Oral vaccination of wildlife using a vaccinia-rabies-glycoprotein recombinant virus vaccine (RABORAL V-RG®): A global review. Vet. Res. 2017, 48, 57. [Google Scholar] [CrossRef]
- Rosatte, R. Evolution of wildlife rabies control tactics. Adv. Virus Res. 2011, 79, 397–419. [Google Scholar] [CrossRef]
- Greene, J.L. Update on the Highly-Pathogenic Avian Influenza Outbreak of 2014–2015; Congressional Research Service: Washington, DC, USA, 2014.
- Okazaki, W.; Purchase, H.G.; Burmester, B.R. Protection against Marek’s Disease by Vaccination with a Herpesvirus of Turkeys. Avian Dis. 1970, 14, 413–429. [Google Scholar] [CrossRef]
- Morgan, R.W.; Gelb, J.J.; Schreurs, C.S.; Lütticken, D.; Rosenberger, J.K.; Sondermeijer, P.J. Protection of chickens from Newcastle and Marek’s diseases with a recombinant herpesvirus of turkeys vaccine expressing the Newcastle disease virus fusion protein. Avian Dis. 1992, 36, 858–870. [Google Scholar] [CrossRef]
- Vagnozzi, A.; Zavala, G.; Riblet, S.M.; Mundt, A.; García, M. Protection induced by commercially available live-attenuated and recombinant viral vector vaccines against infectious laryngotracheitis virus in broiler chickens. Avian Pathol. 2012, 41, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Darteil, R.; Bublot, M.; Laplace, E.; Bouquet, J.F.; Audonnet, J.C.; Rivière, M.; Riviè, M. Herpesvirus of turkey recombinant viruses expressing infectious bursal disease virus (IBDV) VP2 immunogen induce protection against an IBDV virulent challenge in chickens. Virology 1995, 211, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Bublot, M.; Pritchard, N.; Le Gros, F.-X.; Goutebroze, S. Use of a vectored vaccine against infectious bursal disease of chickens in the face of high-titred maternally derived antibody. J. Comp. Pathol. 2007, 137, S81–S84. [Google Scholar] [CrossRef]
- Le Gros, F.X.; Dancer, A.; Giacomini, C.; Pizzoni, L.; Bublot, M.; Graziani, M.; Prandini, F. Field efficacy trial of a novel HVT-IBD vector vaccine for 1-day-old broilers. Vaccine 2009, 27, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Sadigh, Y.; Powers, C.; Spiro, S.; Pedrera, M.; Broadbent, A.; Nair, V. Gallid herpesvirus 3 SB-1 strain as a recombinant viral vector for poultry vaccination. npj Vaccines 2018, 3, 21. [Google Scholar] [CrossRef]
- Tang, N.; Zhang, Y.; Pedrera, M.; Chang, P.; Baigent, S.; Moffat, K.; Shen, Z.; Nair, V.; Yao, Y. A simple and rapid approach to develop recombinant avian herpesvirus vectored vaccines using CRISPR/Cas9 system. Vaccine 2018, 36, 716–722. [Google Scholar] [CrossRef]
- Tang, N.; Zhang, Y.; Sadigh, Y.; Moffat, K.; Shen, Z.; Nair, V.; Yao, Y. Generation of A Triple Insert Live Avian Herpesvirus Vectored Vaccine Using CRISPR/Cas9-Based Gene Editing. Vaccines 2020, 8, 97. [Google Scholar] [CrossRef]
- Brisse, M.; Vrba, S.M.; Kirk, N.; Liang, Y.; Ly, H. Emerging Concepts and Technologies in Vaccine Development. Front. Immunol. 2020, 11, 2578. [Google Scholar] [CrossRef]
- Fry, T.L.; Vandalen, K.K.; Shriner, S.A.; Moore, S.M.; Hanlon, C.A.; Vercauteren, K.C. Humoral immune response to oral rabies vaccination in raccoon kits: Problems and implications. Vaccine 2013, 31, 2811–2815. [Google Scholar] [CrossRef]
- Wang, Y.; Xiang, Z.; Pasquini, S.; Ertl, H.C. The use of an E1-deleted, replication-defective adenovirus recombinant expressing the rabies virus glycoprotein for early vaccination of mice against rabies virus. J. Virol. 1997, 71, 3677–3683. [Google Scholar] [CrossRef]
- Zakhartchouk, A.N.; Pyne, C.; Mutwiri, G.K.; Papp, Z.; Baca-Estrada, M.E.; Griebel, P.; Babiuk, L.A.; Tikoo, S.K. Mucosal immunization of calves with recombinant bovine adenovirus-3: Induction of protective immunity to bovine herpesvirus-1. J. Gen. Virol. 1999, 80, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.; Tronel, J.P.; Pardo-David, C.; Tanner, P.; Colombet, G.; Minke, J.; Audonnet, J.C. Vaccination of puppies born to immune dams with a canine adenovirus-based vaccine protects against a canine distemper virus challenge. Vaccine 2002, 20, 3485–3497. [Google Scholar] [CrossRef]
- Van Rhijn, I.; Godfroid, J.; Michel, A.; Rutten, V. Bovine tuberculosis as a model for human tuberculosis: Advantages over small animal models. Microbes Infect. 2008, 10, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Vordermeier, H.M.; Villarreal-Ramos, B.; Cockle, P.J.; McAulay, M.; Rhodes, S.G.; Thacker, T.; Gilbert, S.C.; McShane, H.; Hill, A.V.S.; Xing, Z.; et al. Viral booster vaccines improve Mycobacterium bovis BCG-induced protection against bovine tuberculosis. Infect. Immun. 2009, 77, 3364–3373. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.E. Vaccination Against Tuberculosis: Revamping BCG by Molecular Genetics Guided by Immunology. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.N.; Everett, J.K.; Raymond, H.; Kafle, S.; Merricks, E.P.; Kazazian, H.H.; Nichols, T.C.; Bushman, F.D.; Sabatino, D.E. Long-Term AAV-Mediated Factor VIII Expression in Nine Hemophilia a Dogs: A 10 Year Follow-up Analysis on Durability, Safety and Vector Integration. Blood 2019, 134, 611. [Google Scholar] [CrossRef]
- Grubman, M.J.; Diaz-San Segundo, F.; Dias, C.C.A.; Moraes, M.P.; Perez-Martin, E.; De Los Santos, T. Use of replication-defective adenoviruses to develop vaccines and biotherapeutics against foot-and-mouth disease. Future Virol. 2012, 7, 767–778. [Google Scholar] [CrossRef]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2019, 20, 273–286. [Google Scholar] [CrossRef]
- Folegatti, P.M.; Ewer, K.J.; Aley, P.K.; Angus, B.; Becker, S.; Belij-Rammerstorfer, S.; Bellamy, D.; Bibi, S.; Bittaye, M.; Clutterbuck, E.A.; et al. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: A preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet 2020, 396, 467–478. [Google Scholar] [CrossRef]
- van Doremalen, N.; Lambe, T.; Spencer, A.; Belij-Rammerstorfer, S.; Purushotham, J.N.; Port, J.R.; Avanzato, V.A.; Bushmaker, T.; Flaxman, A.; Ulaszewska, M.; et al. ChAdOx1 nCoV-19 vaccine prevents SARS-CoV-2 pneumonia in rhesus macaques. Nature 2020, 1–8. [Google Scholar] [CrossRef]
- van Doremalen, N.; Haddock, E.; Feldmann, F.; Meade-White, K.; Bushmaker, T.; Fischer, R.J.; Okumura, A.; Hanley, P.W.; Saturday, G.; Edwards, N.J.; et al. A single dose of ChAdOx1 MERS provides protective immunity in rhesus macaques. Sci. Adv. 2020, 6, eaba8399. [Google Scholar] [CrossRef] [PubMed]
- COVID-19 Vaccine Trial Pauses After Adverse Reaction|The Scientist Magazine®. Available online: https://www.the-scientist.com/news-opinion/covid-19-vaccine-trial-pauses-after-adverse-reaction-67917 (accessed on 10 November 2020).
- Zhu, F.-C.C.; Li, Y.-H.H.; Guan, X.-H.H.; Hou, L.-H.H.; Wang, W.-J.W.W.J.; Li, J.-X.X.; Wu, S.-P.P.; Sen Wang, B.-S.; Wang, Z.; Wang, L.L.L.; et al. Safety, tolerability, and immunogenicity of a recombinant adenovirus type-5 vectored COVID-19 vaccine: A dose-escalation, open-label, non-randomised, first-in-human trial. Lancet 2020, 395, 1845–1854. [Google Scholar] [CrossRef]
- Mercado, N.B.; Zahn, R.; Wegmann, F.; Loos, C.; Chandrashekar, A.; Yu, J.; Liu, J.; Peter, L.; McMahan, K.; Tostanoski, L.H.; et al. Single-shot Ad26 vaccine protects against SARS-CoV-2 in rhesus macaques. Nature 2020, 586, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Johnson & Johnson Covid-19 Vaccine Study Paused Due to Illness. Available online: https://www.statnews.com/2020/10/12/johnson-johnson-covid-19-vaccine-study-paused-due-to-unexplained-illness-in-participant/ (accessed on 10 November 2020).
- Mahase, E. Covid-19: Russia approves vaccine without large scale testing or published results. BMJ 2020, 370, m3205. [Google Scholar] [CrossRef]
- Burki, T.K. The Russian vaccine for COVID-19. Lancet Respir. Med. 2020. [Google Scholar] [CrossRef]
- GRAd-COV2 Vaccine Against COVID-19—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04528641 (accessed on 10 November 2020).
- Safety and Immunogenicity Trial of an Oral SARS-CoV-2 Vaccine (VXA-CoV2-1) for Prevention of COVID-19 in Healthy Adults—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04563702 (accessed on 10 November 2020).
- Clinical Trial to Evaluate the Safety and Immunogenicitiy of the COVID-19 Vaccine—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04497298 (accessed on 6 September 2020).
- A Study to Assess Safety, Tolerability, and Immunogenicity of V591 (COVID-19 Vaccine) in Healthy Participants (V591-001)—Full Text View—ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04498247 (accessed on 6 September 2020).
- Dose Ranging Trial to Assess Safety and Immunogenicity of V590 (COVID-19 Vaccine) in Healthy Adults (V590-001)—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04569786 (accessed on 10 November 2020).
- Safety, Tolerability and Immunogenicity of the Candidate Vaccine MVA-SARS-2-S Against COVID-19—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04569383 (accessed on 10 November 2020).
- Mahase, E. Covid-19: Where are we on immunity and vaccines? BMJ 2020, 370, m3096. [Google Scholar] [CrossRef]


{kind=link}
| Virus Type | Retrovirus and Lentivirus | Adenovirus | Poxvirus | Alphavirus | Arenavirus | Herpesvirus | Flavivirus | Paramyxovirus | Rhabdovirus |
|---|---|---|---|---|---|---|---|---|---|
| Forms in development | Replication-defective Integrase-defective Single-cycle | Replication-competent Replication-defective | Replication-defective Replication-competent | Replication-competent Replication-defective Single-cycleReplicon | Reverse genetics system Replication-competent | Replication-defective Single-cycle | Replication-competent | Reverse genetics system Replication-competent | Replication-competent Single-cycle |
| Commonly used vectors | Moloney murine leukemia virus vector | RD-Ad5 SC-Ad6 | Modified vaccinia virus Ankara Fowlpox Canarypox | Sindbis virus (SIN) Semliki Forest virus (SFV) Venezuelan equine encephalitis (VEE) | Lymphocytic choriomeningitis virus Pichinde virus | Cytomegalovirus Turkey herpesvirus | YF-17D Yellow fever virus 17D (YF-17D) | Avian paramyxovirus serotype (APMV)-1 APMV-3 | Vesicular stomatitis virus (VSV) Rabies |
| Main advantages | Large packaging capacity Integrating ability Transducing non-dividing cells Ability to be pseudotyped | Broad tropism Variety of systemsdeveloped and tested Strong gene expression | Large packaging capacity Ability to induce a strong cellular immune response Broad host range | Broad tropism Ability to produce a large amount of heterologous protein while maintaining high titers | Low seroprevalence Ability to induce low antiviral immunity Targeting and infection of antigen presenting cells | Large packaging ability Capable of superinfecting a host Induces a long-lived T cell response Ease of manipulation | Ability to induce strong and long lasting adaptive immune response Relatively broad tropism | Does not undergo recombination so the vector is genetically stable Broad tropism Replication is generally limited to the respiratory tract | Ability to induce a robust humoral immune response Lack of preexisting immunity in generalpopulation Ability to be pseudotyped |
| Disadvantages | Concerns over insertional mutagenesis | Preexisting immunity to human adenoviral species like Ad5 | Inability to induce strong immune responses in clinical trials | Transient gene expression so not useful for diseases that require long-term therapeutics | Needs further testing to ensure safety in humans | Causes lifelong infections in hosts so needs to be attenuated | Low immunogenicity of recombinant vectors and vector instability | Needs further testing to ensure safety in humans | Potential of neuro-virulence for rabies virus vector |
| Insert capacity | 8 kB | 8 kB | >25 kB | 18 kB | 4 kB | >30 kB | 6 kB | 4.5 kB | 6 kB |
| References | [7,8,9] | [10,11,12,13,14] | [15,16,17,18] | [19,20] | [21,22,23] | [24,25,26,27] | [28,29] | [30,31] | [32,33,34,35] |
| Virus Vector | Phase I Clinical Trial |
| Poxviruses | |
| MVA (Modified vaccinia virus Ankara) | Ebola, HIV, Hepatitis C, MERS-CoV |
| FPV (Fowlpox vector) | HIV |
| ALVAC (canarypox vector) | HIV |
| Adenoviruses (Ad) | |
| ChAd3 (Chimpanzee adenovirus) | Ebola Zaire, Hepatitis C, Ebola Sudan, Ebola Marburg |
| ChAdOx (Chimpanzee adenovirus) | Tuberculosis, Chikungunya, MERS-CoV |
| Ad5 (Adenovirus type 5) | Cystic fibrosis, HIV, Ebola Zaire |
| VXA (Replication-deficient Ad5) | Respiratory syncytial virus, Norovirus, Influenza |
| rAd26 (Recombinant Ad 26) | HIV, Ebola Zaire |
| Ad35 | Tuberculosis, HIV |
| Ad4 | HIV, Anthrax |
| Vesicular Stomatitis Virus (VSV) | |
| Replication-competent VSV | HIV |
| Alphaviruses | |
| VEE Replicon (Venezuelan equine encephalitis) | CMV |
| Virus Vector | Phase II Clinical Trial |
| Poxviruses | |
| MVA | CMV, Tuberculosis |
| Adenoviruses | |
| ChAdOx1 | Malaria, SARS-CoV-2 |
| ChAd63 (Chimpanzee adenovirus vector) | Malaria |
| VXA | Seasonal influenza |
| Ad5 | Ebola, HIV, Pandemic influenza |
| Ad35 | Malaria |
| Virus Vector | FDA-Approved |
| Replication-competent VSV | Ebola Zaire (ERVEBO®) |
| Species | Pathogen/Disease | Antigen | Product | Manufacturer |
|---|---|---|---|---|
| Canarypox Vector | ||||
| Dog | Canine distemper virus | HA and F glycoproteins | RECOMBITEK | Boehringer-Ingelheim |
| Cat | Feline leukemia virus (FeLV) Rabies virus | Env, gag, pol Glycoprotein G | PUREVAX FeLV PUREVAX Rabies | Boehringer-Ingelheim |
| Vaccinia Vector | ||||
| Raccoons, coyotes | Rabies virus | Glycoprotein G | Raboral V-RG | Boehringer-Ingelheim |
| Alphaherpesvirus (HVT) Vector | ||||
| Chicken | Infectious bursal (IBD), Marek’s, Newcastle disease (ND) ND and Marek’s disease IBD and Marek’s disease Marek’s disease and infectious laryngotracheitis (LT) | VP2 of IBDV, F glycoprotein of NDV F glycoprotein VP2 of NDV Glycoprotein B | VAXXITEK HVT + IBD + ND Ultifend IBD ND NEWXXITEK HVT + ND VAXXITEK HVT + IBD Vectormune HVT IBD Vectormune LT | Boehringer-Ingelheim Ceva Boehringer-Ingelheim Ceva Ceva |
| Vaccine Name | Vaccine Vector | Company and Country | Preliminary Results |
|---|---|---|---|
| AZD1222 (ChAdOx1 nCoV-19) | Adenovirus | Oxford University, UK |
|
| Ad5-nCoV | Adenovirus | CanSino Biologics, China |
|
| Ad26.COV2.S | Adenovirus | Johnson and Johnson, USA |
|
| Gam-COVID-Vac | Adenovirus | Gamaleya Research Institute of Epidemiology and Microbiology, Russia |
|
| GRAd-COV2 | Adenovirus | ReiTherra, Italy |
|
| VXA-CoV2-1 | Adenovirus | Vaxart, USA |
|
| TMV-083 | Measles | Institut Pasteur, France |
|
| V591 and V590 | Measles | Merck, USA |
|
| MVA-SARS-2-S | Vaccinia Ankara | Universitätsklinikum Hamburg-Eppendorf, Germany |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vrba, S.M.; Kirk, N.M.; Brisse, M.E.; Liang, Y.; Ly, H. Development and Applications of Viral Vectored Vaccines to Combat Zoonotic and Emerging Public Health Threats. Vaccines 2020, 8, 680. https://doi.org/10.3390/vaccines8040680
Vrba SM, Kirk NM, Brisse ME, Liang Y, Ly H. Development and Applications of Viral Vectored Vaccines to Combat Zoonotic and Emerging Public Health Threats. Vaccines. 2020; 8(4):680. https://doi.org/10.3390/vaccines8040680
Chicago/Turabian StyleVrba, Sophia M., Natalie M. Kirk, Morgan E. Brisse, Yuying Liang, and Hinh Ly. 2020. "Development and Applications of Viral Vectored Vaccines to Combat Zoonotic and Emerging Public Health Threats" Vaccines 8, no. 4: 680. https://doi.org/10.3390/vaccines8040680
APA StyleVrba, S. M., Kirk, N. M., Brisse, M. E., Liang, Y., & Ly, H. (2020). Development and Applications of Viral Vectored Vaccines to Combat Zoonotic and Emerging Public Health Threats. Vaccines, 8(4), 680. https://doi.org/10.3390/vaccines8040680