Resident Memory T Cells and Their Effect on Cancer

 ,
,  , ,
, ,
Abstract
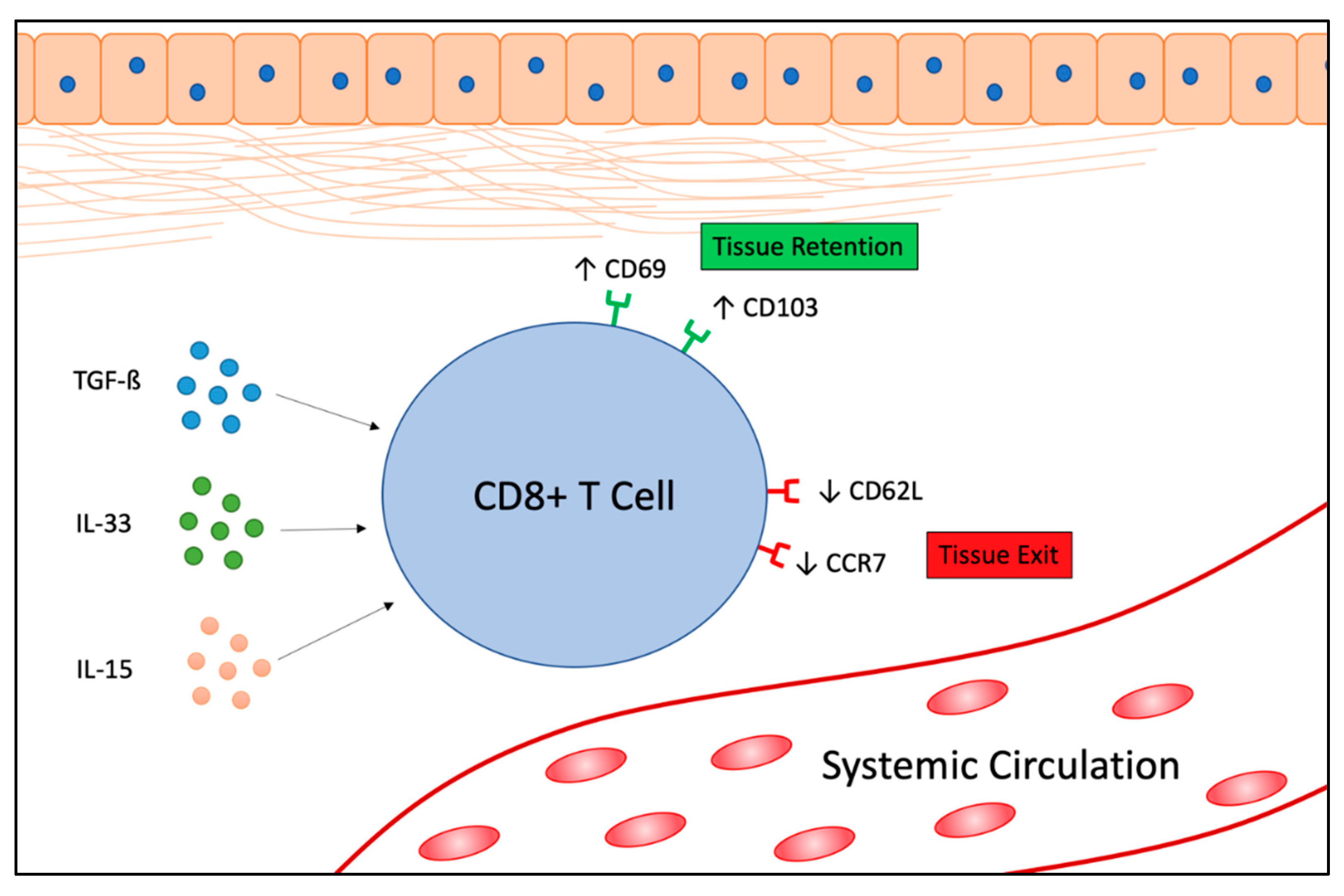
1. Introduction
2. TRM Cells in Cancer
2.1. Function of TRM Cells in Cancer
2.2. Identification of TRM Cells in Patient Samples
2.3. Improving Vaccine Efficacy
2.4. Improving Adoptive T Cell Therapy
2.5. TRM Cell Clinical Trials
3. Future Directions
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sallusto, F.; Lenig, D.; Förster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Jiang, A. Dendritic Cells and CD8 T Cell Immunity in Tumor Microenvironment. Front. Immunol. 2018, 9, 3059. [Google Scholar] [CrossRef] [PubMed]
- Masopust, D.; Thorpe, S.J.; Fabre-Thorpe, M. Preferential Localization of Effector Memory Cells in Nonlymphoid Tissue. Science 2001, 291, 2413–2417. [Google Scholar] [CrossRef] [PubMed]
- Ariotti, S.; Hogenbirk, M.A.; Dijkgraaf, F.E.; Visser, L.L.; Hoekstra, M.E.; Song, J.-Y.; Jacobs, H.; Haanen, J.B.; Schumacher, T.N. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science 2014, 346, 101–105. [Google Scholar] [CrossRef]
- Mueller, S.N.; Gebhardt, T.; Carbone, F.R.; Heath, W.R. Memory T Cell Subsets, Migration Patterns, and Tissue Residence. Annu. Rev. Immunol. 2013, 31, 137–161. [Google Scholar] [CrossRef]
- Schenkel, J.M.; Fraser, K.A.; Beura, L.K.; Pauken, K.E.; Vezys, V.; Masopust, D. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 2014, 346, 98–101. [Google Scholar] [CrossRef]
- Park, S.L.; Gebhardt, T.; Mackay, L.K. Tissue-Resident Memory T Cells in Cancer Immunosurveillance. Trends Immunol. 2019, 40, 735–747. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Milne, K.; DeRocher, H.; Webb, J.R.; Nelson, B.H.; Watson, P.H. CD103 and Intratumoral Immune Response in Breast Cancer. Clin. Cancer Res. 2016, 22, 6290–6297. [Google Scholar] [CrossRef]
- Djenidi, F.; Adam, J.; Goubar, A.; Durgeau, A.; Meurice, G.; De Montpreville, V.; Validire, P.; Besse, B.; Mami-Chouaib, F. CD8+CD103+ Tumor–Infiltrating Lymphocytes Are Tumor-Specific Tissue-Resident Memory T Cells and a Prognostic Factor for Survival in Lung Cancer Patients. J. Immunol. 2015, 194, 3475–3486. [Google Scholar] [CrossRef]
- Ganesan, A.-P.; Clarke, J.; Wood, O.; Garrido-Martin, E.M.; Chee, S.J.; Mellows, T.; Samaniego-Castruita, D.; Singh, D.; Seumois, G.; Alzetani, A.; et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat. Immunol. 2017, 18, 940–950. [Google Scholar] [CrossRef]
- Komdeur, F.L.; Prins, T.M.; Van De Wall, S.; Plat, A.; Wisman, G.B.A.; Hollema, H.; Daemen, T.; Church, D.N.; De Bruyn, M.; Nijman, H.W. CD103+ tumor-infiltrating lymphocytes are tumor-reactive intraepithelial CD8+ T cells associated with prognostic benefit and therapy response in cervical cancer. OncoImmunology 2017, 6, e1338230. [Google Scholar] [CrossRef] [PubMed]
- Comber, J.D.; Philip, R. MHC class I antigen presentation and implications for developing a new generation of therapeutic vaccines. Ther. Adv. Vaccines 2014, 2, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Minnich, M.; Kragten, N.A.M.; Liao, Y.; Nota, B.; Seillet, C.; Zaid, A.; Man, K.; Preston, S.; Freestone, D.; et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 2016, 352, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Dumauthioz, N.; Labiano, S.; Romero, P. Tumor Resident Memory T Cells: New Players in Immune Surveillance and Therapy. Front. Immunol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Mackay, L.K.; Rahimpour, A.; Ma, J.Z.; Collins, N.; Stock, A.T.; Hafon, M.-L.; Vega-Ramos, J.; Lauzurica, P.; Mueller, S.N.; Stefanovic, T.; et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013, 14, 1294–1301. [Google Scholar] [CrossRef]
- Mackay, L.K.; Braun, A.; MacLeod, B.L.; Collins, N.; Tebartz, C.; Bedoui, S.; Carbone, F.R.; Gebhardt, T. Cutting Edge: CD69 Interference with Sphingosine-1-Phosphate Receptor Function Regulates Peripheral T Cell Retention. J. Immunol. 2015, 194, 2059–2063. [Google Scholar] [CrossRef] [PubMed]
- Cepek, K.L.; Shaw, S.K.; Parker, C.M.; Russell, G.J.; Morrow, J.S.; Rimm, D.L.; Brenner, M.B. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the αEβ7 integrin. Nature 1994, 372, 190–193. [Google Scholar] [CrossRef]
- Mueller, S.N.; Mackay, L.K. Tissue-resident memory T cells: Local specialists in immune defence. Nat. Rev. Immunol. 2015, 16, 79–89. [Google Scholar] [CrossRef]
- Mami-Chouaib, F.; Blanc, C.; Corgnac, S.; Hans, S.; Malenica, I.; Granier, C.; Tihy, I.; Tartour, E. Resident memory T cells, critical components in tumor immunology. J. Immunother. Cancer 2018, 6, 87. [Google Scholar] [CrossRef]
- Schenkel, J.M.; Fraser, K.A.; Vezys, V.; Masopust, D. Sensing and alarm function of resident memory CD8+ T cells. Nat. Immunol. 2013, 14, 509–513. [Google Scholar] [CrossRef]
- Milner, J.J.; Toma, C.; Yu, B.; Zhang, K.; Omilusik, K.; Phan, A.T.; Wang, D.; Getzler, A.; Nguyen, T.; Crotty, S.; et al. Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature 2017, 552, 253–257. [Google Scholar] [CrossRef]
- Cruz-Guilloty, F.; Pipkin, M.E.; Djuretic, I.M.; Levanon, D.; Lotem, J.; Lichtenheld, M.G.; Groner, Y.; Rao, A. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J. Exp. Med. 2009, 206, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Wilmott, J.S.; Madore, J.; Gide, T.N.; Quek, C.Y.J.; Tasker, A.; Ferguson, A.L.; Chen, J.-B.; Hewavisenti, R.; Hersey, P.; et al. CD103+ Tumor-Resident CD8+ T Cells Are Associated with Improved Survival in Immunotherapy-Naïve Melanoma Patients and Expand Significantly During Anti-PD-1 Treatment. Clin. Cancer Res. 2018, 24, 3036–3045. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Gray, R.J.; DeMaria, S.; Goldstein, L.; Perez, E.A.; Shulman, L.N.; Martino, S.; Wang, M.; Jones, V.E.; Saphner, T.J.; et al. Prognostic Value of Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancers From Two Phase III Randomized Adjuvant Breast Cancer Trials: ECOG 2197 and ECOG 1199. J. Clin. Oncol. 2014, 32, 2959–2966. [Google Scholar] [CrossRef] [PubMed]
- Egelston, C.A.; Avalos, C.; Tu, T.Y.; Rosario, A.; Wang, R.; Solomon, S.; Srinivasan, G.; Nelson, M.S.; Huang, Y.; Lim, M.H.; et al. Resident memory CD8+ T cells within cancer islands mediate survival in breast cancer patients. JCI Insight 2019, 4, 130000. [Google Scholar] [CrossRef]
- Savas, P.; Kathleen Cuningham Foundation Consortium for research into Familial Breast cancer (kConFab); Virassamy, B.; Ye, C.; Salim, A.; Mintoff, C.P.; Caramia, F.; Salgado, R.; Byrne, D.J.; Teo, Z.L.; et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med. 2018, 24, 986–993. [Google Scholar] [CrossRef]
- Webb, J.R.; Milne, K.; Nelson, B.H. PD-1 and CD103 are widely co-expressed on prognostically favorable intraepithelial CD8 T cells in human ovarian cancer. Cancer Immunol. Res. 2015, 3, 926–935. [Google Scholar] [CrossRef]
- Malik, B.T.; Byrne, K.T.; Vella, J.L.; Zhang, P.; Shabaneh, T.B.; Steinberg, S.M.; Molodtsov, A.K.; Bowers, J.S.; Angeles, C.V.; Paulos, C.M.; et al. Resident memory T cells in the skin mediate durable immunity to melanoma. Sci. Immunol. 2017, 2, eaam6346. [Google Scholar] [CrossRef]
- Park, S.L.; Buzzai, A.; Rautela, J.; Hor, J.L.; Hochheiser, K.; Effern, M.; McBain, N.; Wagner, T.; Edwards, J.; McConville, R.; et al. Tissue-resident memory CD8+ T cells promote melanoma–immune equilibrium in skin. Nature 2018, 565, 366–371. [Google Scholar] [CrossRef]
- Nizard, M.; Roussel, H.; Diniz, M.O.; Karaki, S.; Tran, T.; Voron, T.; Dransart, E.; Sandoval, F.; Riquet, M.; Rance, B.; et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat. Commun. 2017, 8, 15221. [Google Scholar] [CrossRef]
- Gálvez-Cancino, F.; López, E.; Menares, E.; Díaz, X.; Flores, C.; Cáceres, P.; Hidalgo, S.; Chovar, O.; Alcántara-Hernández, M.; Borgna, V.; et al. Vaccination-induced skin-resident memory CD8+T cells mediate strong protection against cutaneous melanoma. OncoImmunology 2018, 7, e1442163-42. [Google Scholar] [CrossRef] [PubMed]
- Hombrink, P.; Helbig, C.A.; Backer, R.; Piet, B.E.; Oja, A.; Stark, R.; Brasser, G.; Jongejan, A.E.; Jonkers, R.; Nota, B.; et al. Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells. Nat. Immunol. 2016, 17, 1467–1478. [Google Scholar] [CrossRef] [PubMed]
- Hartana, C.A.; Bergman, E.A.; Broomé, A.; Berglund, S.; Johansson, M.; Alamdari, F.I.; Jakubczyk, T.; Huge, Y.; Aljabery, F.; Palmqvist, K.; et al. Tissue-resident memory T cells are epigenetically cytotoxic with signs of exhaustion in human urinary bladder cancer. Clin. Exp. Immunol. 2018, 194, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.R.; Milne, K.; Watson, P.; DeLeeuw, R.J.; Nelson, B.H. Tumor-Infiltrating Lymphocytes Expressing the Tissue Resident Memory Marker CD103 Are Associated with Increased Survival in High-Grade Serous Ovarian Cancer. Clin. Cancer Res. 2013, 20, 434–444. [Google Scholar] [CrossRef]
- Simoni, Y.; Becht, E.; Fehlings, M.G.; Loh, C.Y.; Koo, S.L.; Teng, K.W.W.; Yeong, J.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef]
- Boddupalli, C.S.; Bar, N.; Kadaveru, K.; Krauthammer, M.; Pornputtapong, N.; Mai, Z.; Ariyan, S.; Narayan, D.; Kluger, H.; Deng, Y.; et al. Interlesional diversity of T cell receptors in melanoma with immune checkpoints enriched in tissue-resident memory T cells. JCI Insight 2016, 1, e88955. [Google Scholar] [CrossRef]
- De La Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef]
- Pan, Y.; Tian, T.; Park, C.O.; Lofftus, S.Y.; Mei, S.; Liu, X.; Luo, C.; O’Malley, J.T.; Gehad, A.; Teague, J.E.; et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 2017, 543, 252–256. [Google Scholar] [CrossRef]
- Kumar, B.V.; Ma, W.; Miron, M.; Granot, T.; Guyer, R.S.; Carpenter, D.J.; Senda, T.; Sun, X.; Ho, S.-H.; Lerner, H.; et al. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep. 2017, 20, 2921–2934. [Google Scholar] [CrossRef]
- Enamorado, M.; Iborra, S.; Priego, E.; Cueto, F.J.; Quintana, J.A.; Martínez-Cano, S.; Mejías-Pérez, E.; Esteban, M.; Melero, I.; Hidalgo, A.; et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8+ T cells. Nat. Commun. 2017, 8, 16073. [Google Scholar] [CrossRef]
- Blanc, C.; Hans, S.; Tran, T.; Granier, C.; Saldman, A.; Anson, M.; Oudard, S.; Tartour, E. Targeting Resident Memory T Cells for Cancer Immunotherapy. Front. Immunol. 2018, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8+ T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 2017, 32, 377–391.e9. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.-A.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’Er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120–1133.e17. [Google Scholar] [CrossRef]
- Szabó, P.A.; Miron, M.; Farber, D.L. Location, location, location: Tissue resident memory T cells in mice and humans. Sci. Immunol. 2019, 4, eaas9673. [Google Scholar] [CrossRef] [PubMed]
- Booth, J.S.; Toapanta, F.R.; Salerno-Goncalves, R.; Patil, S.; Kader, H.A.; Safta, A.M.; Czinn, S.J.; Greenwald, B.D.; Sztein, M.B. Characterization and Functional Properties of Gastric Tissue-Resident Memory T Cells from Children, Adults, and the Elderly. Front. Immunol. 2014, 5, 294. [Google Scholar] [CrossRef]
- Okhrimenko, A.; Grün, J.R.; Westendorf, K.; Fang, Z.; Reinke, S.; Von Roth, P.; Wassilew, G.; Kühl, A.A.; Kudernatsch, R.; Demski, S.; et al. Human memory T cells from the bone marrow are resting and maintain long-lasting systemic memory. Proc. Natl. Acad. Sci. USA 2014, 111, 9229–9234. [Google Scholar] [CrossRef]
- Pallett, L.J.; Davies, J.; Colbeck, E.J.; Robertson, F.P.; Hansi, N.; Easom, N.J.; Burton, A.R.; Stegmann, K.A.; Schurich, A.; Swadling, L.; et al. IL-2high tissue-resident T cells in the human liver: Sentinels for hepatotropic infection. J. Exp. Med. 2017, 214, 1567–1580. [Google Scholar] [CrossRef]
- Wong, M.T.; Ong, D.E.H.; Lim, F.S.H.; Teng, K.W.W.; McGovern, N.; Narayanan, S.; Ho, W.Q.; Cerny, D.; Tan, H.K.K.; Anicete, R.; et al. A High-Dimensional Atlas of Human T Cell Diversity Reveals Tissue-Specific Trafficking and Cytokine Signatures. Immunity 2016, 45, 442–456. [Google Scholar] [CrossRef]
- Woon, H.G.; Braun, A.; Li, J.; Smith, C.; Edwards, J.; Sierro, F.; Feng, C.G.; Khanna, R.; Elliot, M.; Bell, A.I.; et al. Compartmentalization of Total and Virus-Specific Tissue-Resident Memory CD8+ T Cells in Human Lymphoid Organs. PLOS Pathog. 2016, 12, e1005799. [Google Scholar] [CrossRef] [PubMed]
- Swaims-Kohlmeier, A.; Haaland, R.E.; Haddad, L.B.; Sheth, A.N.; Evans-Strickfaden, T.; Lupo, L.D.; Cordes, S.; Aguirre, A.; Lupoli, K.; Chen, C.-Y.; et al. Progesterone Levels Associate with a Novel Population of CCR5+CD38+ CD4 T Cells Resident in the Genital Mucosa with Lymphoid Trafficking Potential. J. Immunol. 2016, 197, 368–376. [Google Scholar] [CrossRef]
- Smolders, J.; Heutinck, K.M.; Fransen, N.L.; Remmerswaal, E.B.M.; Hombrink, P.; Berge, I.J.M.T.; Van Lier, R.A.W.; Huitinga, I.; Hamann, J. Tissue-resident memory T cells populate the human brain. Nat. Commun. 2018, 9, 4593. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, K.; Vincenti, I.; Merkler, D. Resident-Memory T Cells in Tissue-Restricted Immune Responses: For Better or Worse? Front. Immunol. 2018, 9, 2827. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Masopust, D. Tissue-resident memory T cells. Immun. 2014, 41, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Klonowski, K.D.; Williams, K.J.; Marzo, A.L.A.; Blair, D.; Lingenheld, E.G.; Lefrançois, L. Dynamics of Blood-Borne CD8 Memory T Cell Migration In Vivo. Immunity 2004, 20, 551–562. [Google Scholar] [CrossRef]
- Steinert, E.M.; Schenkel, J.M.; Fraser, K.A.; Beura, L.K.; Manlove, L.S.; Igyártó, B.Z.; Southern, P.J.; Masopust, D. Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell 2015, 161, 737–749. [Google Scholar] [CrossRef]
- Bromley, S.K.; Yan, S.; Tomura, M.; Kanagawa, O.; Luster, A.D. Recirculating memory T cells are a unique subset of CD4+ T cells with a distinct phenotype and migratory pattern. J. Immunol. 2012, 190, 970–976. [Google Scholar] [CrossRef]
- Ugur, M.; Schulz, O.; Menon, M.B.; Krueger, A.; Pabst, O. Resident CD4+ T cells accumulate in lymphoid organs after prolonged antigen exposure. Nat. Commun. 2014, 5, 4821. [Google Scholar] [CrossRef][Green Version]
- Muruganandah, V.; Sathkumara, H.D.; Navarro, S.; Kupz, A. A Systematic Review: The Role of Resident Memory T Cells in Infectious Diseases and Their Relevance for Vaccine Development. Front. Immunol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Mullins, D.W.; Sheasley, S.L.; Ream, R.M.; Bullock, T.N.; Fu, Y.-X.; Engelhard, V.H. Route of Immunization with Peptide-pulsed Dendritic Cells Controls the Distribution of Memory and Effector T Cells in Lymphoid Tissues and Determines the Pattern of Regional Tumor Control. J. Exp. Med. 2003, 198, 1023–1034. [Google Scholar] [CrossRef]
- Sun, Y.-Y.; Peng, S.; Han, L.; Qiu, J.; Song, L.; Tsai, Y.; Yang, B.; Roden, R.B.; Trimble, C.L.; Hung, C.-F.; et al. Local HPV Recombinant Vaccinia Boost Following Priming with an HPV DNA Vaccine Enhances Local HPV-Specific CD8+ T-cell-Mediated Tumor Control in the Genital Tract. Clin. Cancer Res. 2015, 22, 657–669. [Google Scholar] [CrossRef]
- Morabito, K.M.; Ruckwardt, T.R.; Redwood, A.J.; Moin, S.M.; Price, D.A.; Graham, B.S. Intranasal administration of RSV antigen-expressing MCMV elicits robust tissue-resident effector and effector memory CD8+ T cells in the lung. Mucosal Immunol. 2016, 10, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Çuburu, N.; Wang, K.; Goodman, K.N.; Pang, Y.Y.; Thompson, C.D.; Lowy, D.R.; Cohen, J.I.; Schiller, J.T. Topical Herpes Simplex Virus 2 (HSV-2) Vaccination with Human Papillomavirus Vectors Expressing gB/gD Ectodomains Induces Genital-Tissue-Resident Memory CD8+T Cells and Reduces Genital Disease and Viral Shedding after HSV-2 Challenge. J. Virol. 2014, 89, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Calzascia, T.; Masson, F.; Di Berardino-Besson, W.; Contassot, E.; Wilmotte, R.; Aurrand-Lions, M.; Ruegg, C.; Dietrich, P.-Y.; Walker, P.R. Homing Phenotypes of Tumor-Specific CD8 T Cells Are Predetermined at the Tumor Site by Crosspresenting APCs. Immunity 2005, 22, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Çuburu, N.; Khan, S.; Thompson, C.D.; Kim, R.; Vellinga, J.; Zahn, R.; Lowy, D.R.; Scheper, G.; Schiller, J.T. Adenovirus vector-based prime-boost vaccination via heterologous routes induces cervicovaginal CD8+ T cell responses against HPV16 oncoproteins. Int. J. Cancer 2017, 142, 1467–1479. [Google Scholar] [CrossRef]
- Sandoval, F.; Terme, M.; Nizard, M.; Badoual, C.; Bureau, M.-F.; Freyburger, L.; Clément, O.; Marcheteau, E.; Gey, A.; Fraisse, G.; et al. Mucosal Imprinting of Vaccine-Induced CD8+ T Cells Is Crucial to Inhibit the Growth of Mucosal Tumors. Sci. Transl. Med. 2013, 5, 172ra20. [Google Scholar] [CrossRef]
- Maples, P.; Kumar, P.; Yü, Y.; Wang, Z.; Jay, C.; Pappen, B.; Rao, D.; Kuhn, J.; Nemunaitis, J.; Senzer, N.N. FANG Vaccine: Autologous Tumor Cell Vaccine Genetically Modified to Express GM-CSF and Block Production of Furin. BioProcess J. 2010, 8, 4–14. [Google Scholar] [CrossRef]
- Barve, M.; Kuhn, J.; Lamont, J.; Beitsch, P.; Manning, L.; Pappen, B.O.; Kumar, P.; Wallraven, G.; Senzer, N.N.; Nemunaitis, J. Follow-up of bi-shRNA furin/GM-CSF Engineered Autologous Tumor Cell (EATC) Immunotherapy Vigil® in patients with advanced melanoma. Biomed. Genet. Genom. 2016, 1. [Google Scholar] [CrossRef][Green Version]
- Ghisoli, M.; Barve, M.; Schneider, R.; Mennel, R.; Lenarsky, C.; Wallraven, G.O.; Pappen, B.; LaNoue, J.; Kumar, P.; Nemunaitis, D.; et al. Pilot Trial of FANG Immunotherapy in Ewing’s Sarcoma. Mol. Ther. 2015, 23, 1103–1109. [Google Scholar] [CrossRef]
- Senzer, N.; Barve, M.; Kuhn, J.; Melnyk, A.; Beitsch, P.; Lazar, M.; Lifshitz, S.; Magee, M.; Oh, J.; Mill, S.W.; et al. Phase I trial of “bi-shRNAi(furin)/GMCSF DNA/autologous tumor cell” vaccine (FANG) in advanced cancer. Mol. Ther. 2012, 20, 679–686. [Google Scholar] [CrossRef]
- Senzer, N.; Barve, M.; Nemunaitis, J.; Kuhn, J.; Melnyk, A.; Beitsch, P.; Magee, M.; Oh, J.; Bedell, C.; Kumar, P.; et al. Long Term Follow Up: Phase I Trial of “bi-shRNA furin/GMCSF DNA/Autologous Tumor Cell” Immunotherapy (FANG™) in Advanced Cancer. J. Vaccines Vaccine 2013, 4, 209. [Google Scholar] [CrossRef]
- Oh, J.; Barve, M.; Matthews, C.M.; Koon, E.C.; Heffernan, T.P.; Fine, B.; Grosen, E.; Bergman, M.K.; Fleming, E.L.; Demars, L.R.; et al. Phase II study of Vigil® DNA engineered immunotherapy as maintenance in advanced stage ovarian cancer. Gynecol. Oncol. 2016, 143, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Rocconi, R.P.; Grosen, E.A.; Ghamande, S.A.; Chan, J.C.-K.; Barve, M.A.; Oh, J.; Tewari, D.; Morris, P.C.; Stevens, E.E.; Bottsford-Miller, J.N.; et al. Randomized Double-Blind Placebo Controlled Trial of Primary Maintenance Vigil Immunotherapy (VITAL study) in Stage III/IV Ovarian Cancer: Efficacy Assessment in BRCA1/2-wt Patients (Late Breaking Oral Presentation). In Proceedings of the Society of Gynecologic Oncology Annual Meeting on Women’s Cancer, Toronto, ON, Canada, 28–31 March 2020. [Google Scholar]
- Herron, J.; Smith, N.; Stanbery, L.; Aaron, P.; Manning, L.; Bognar, E.; Wallraven, G.; Horvath, S.; Nemuanaitis, J. Vigil: Personalized Immunotherapy Generating Systemic Cytotoxic T cell Response. Cancer Sci. Res. 2020, 1, 210–221. [Google Scholar]
- Perica, K.; Varela, J.C.; Oelke, M.; Schneck, J. Adoptive T Cell Immunotherapy for Cancer. Rambam Maimonides Med. J. 2015, 6. [Google Scholar] [CrossRef]
- Wu, T.-C.; Xu, K.; Banchereau, R.; Marches, F.; Yu, C.I.; Martinek, J.; Anguiano, E.; Pedroza-Gonzalez, A.; Snipes, G.J.; O’Shaughnessy, J.; et al. Reprogramming tumor-infiltrating dendritic cells for CD103+ CD8+ mucosal T-cell differentiation and breast cancer rejection. Cancer Immunol. Res. 2014, 2, 487–500. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma and Indolent B-Cell Malignancies Can Be Effectively Treated With Autologous T Cells Expressing an Anti-CD19 Chimeric Antigen Receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Metzinger, M.N.; Verghese, C.; Hamouda, D.M.; Lenhard, A.; Choucair, K.; Senzer, N.; Brunicardi, F.C.; Dworkin, L.; Nemunaitis, J. Chimeric Antigen Receptor T-Cell Therapy: Reach to Solid Tumor Experience. Oncology 2019, 97, 59–74. [Google Scholar] [CrossRef]
- Zhang, H.; Ye, Z.-L.; Yuan, Z.-G.; Luo, Z.-Q.; Jin, H.-J.; Qian, Q.-J. New Strategies for the Treatment of Solid Tumors with CAR-T Cells. Int. J. Boil. Sci. 2016, 12, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Wynne-Jones, E.; Freestone, D.; Pellicci, D.G.; Mielke, L.A.; Newman, D.M.; Braun, A.; Masson, F.; Kallies, A.; Belz, G.T.; et al. T-box Transcription Factors Combine with the Cytokines TGF-beta and IL-15 to Control Tissue-Resident Memory T Cell Fate. Immunity 2015, 43, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Komdeur, F.L.; Wouters, M.C.A.; Workel, H.H.; Tijans, A.M.; Terwindt, A.L.; Brunekreeft, K.L.; Plat, A.; Klip, H.G.A.; Eggink, F.; Leffers, N.; et al. CD103+ intraepithelial T cells in high-grade serous ovarian cancer are phenotypically diverse TCRαβ+ CD8αβ+ T cells that can be targeted for cancer immunotherapy. Oncotarget 2016, 7, 75130–75144. [Google Scholar] [CrossRef] [PubMed]
- Murray, T.; Marraco, S.A.F.; Baumgaertner, P.; Bordry, N.; Cagnon, L.; Donda, A.; Romero, P.; Verdeil, G.; Speiser, D.E. Very Late Antigen-1 Marks Functional Tumor-Resident CD8 T Cells and Correlates with Survival of Melanoma Patients. Front. Immunol. 2016, 7, 573. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.; Kim, S.; Kim, M.-Y.; Go, H.; Jeon, Y.K.; Chung, D.H. Prognostic implications of intratumoral CD103+ tumor-infiltrating lymphocytes in pulmonary squamous cell carcinoma. Oncotarget 2017, 8, 13762–13769. [Google Scholar] [CrossRef]
- Lohneis, P.; Sinn, M.; Bischoff, S.; Jühling, A.; Pelzer, U.; Wislocka, L.; Bahra, M.; Sinn, B.V.; Denkert, C.; Oettle, H.; et al. Cytotoxic tumour-infiltrating T lymphocytes influence outcome in resected pancreatic ductal adenocarcinoma. Eur. J. Cancer 2017, 83, 290–301. [Google Scholar] [CrossRef]
- Yau, C.; Wolf, D.; Campbell, M.; Savas, P.; Lin, S.; Brown-Swigart, L.; Hirst, G.; Asare, S.; Zhu, Z.; Loi, S.; et al. Abstract P3-10-06: Expression-based immune signatures as predictors of neoadjuvant targeted-/chemo-therapy response: Experience from the I-SPY 2 TRIAL of 1000 patients across 10 therapies. Poster Sess. Abstr. 2019, 79, P3–P10. [Google Scholar] [CrossRef]
- Adams, S.; Schmid, P.; Rugo, H.; Winer, E.; Loirat, D.; Awada, A.; Cescon, D.; Iwata, H.; Campone, M.; Nanda, R.; et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: Cohort A of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 397–404. [Google Scholar] [CrossRef]
- Adams, S.; Loi, S.M.; Toppmeyer, D.; Cescon, D.; De Laurentiis, M.; Nanda, R.; Winer, E.; Mukai, H.; Tamura, K.; Armstrong, A.; et al. Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: Cohort B of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 405–411. [Google Scholar] [CrossRef]
- Loi, S.; Schmid, P.; Cortés, J.; Cescon, D.W.; Winer, E.P.; Toppmeyer, D.; Rugo, H.S.; de Laurentiis, M.; Nanda, R.; Iwata, H.; et al. RNA molecular signatures as predictive biomarkers of response to monotherapy pembrolizumab in patients with metastatic triple negative breast cancer: KEYNOTE-086. Cancer Res. 2019, 79. [Google Scholar] [CrossRef]
- Choucair, K.; Morand, S.; Stanbery, L.; Edelman, G.; Dworkin, L.; Nemunaitis, J. TMB: A promising immune-response biomarker, and potential spearhead in advancing targeted therapy trials. Cancer Gene Ther. 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Taylor, M.H.; Kelly, K.; Beck, J.T.; Gordon, M.; Moore, K.M.; Patel, M.R.; Chaves, J.; Park, H.; Mita, A.C.; et al. Efficacy and Safety of Avelumab for Patients With Recurrent or Refractory Ovarian Cancer. JAMA Oncol. 2019, 5, 393. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Fujiwara, K.; Ledermann, J.; Oza, A.; Kristeleit, R.; Ray-Coquard, I.; Richardson, G.E.; Sessa, C.; Yonemori, K.; Banerjee, S.; et al. Avelumab alone or in combination with pegylted liposomal doxorubicin versus pegylated liposomal doxorubicin alone in platinum-resistant or refractory epithelial ovarian cancer: Primary and biomarker analysis of hte phase III JAVELIN Ovarian 200 trial. In Proceedings of the Society of Gynecologic Oncology Annual Meeting, Honolulu, HI, USA, 16–19 March 2019. [Google Scholar]
- Eskander, R.N.; Ledermann, J.A.; Birrer, M.J.; Fujiwara, K.; Gaillard, S.; Richardson, G.E.; Wei, C.; Baig, M.A.; Zohren, F.; Monk, B.J. JAVELIN ovarian PARP 100 study design: Phase III trial of avelumab + chemotherapy followed by avelumab + talazoparib maintenance in previously untreated epithelial ovarian cancer. J. Clin. Oncol. 2019, 37, TPS9. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Wada, S.; Jackson, C.M.; Yoshimura, K.; Yen, H.-R.; Getnet, D.; Harris, T.J.; Goldberg, M.V.; Bruno, T.C.; Grosso, J.F.; Durham, N.; et al. Sequencing CTLA-4 blockade with cell-based immunotherapy for prostate cancer. J. Transl. Med. 2013, 11, 89. [Google Scholar] [CrossRef]
- Loibl, S.; Untch, M.; Burchardi, N.; Huober, J.; Sinn, B.; Blohmer, J.-U.; Grischke, E.-M.; Furlanetto, J.; Tesch, H.; Hanusch, C.; et al. A randomised phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple-negative breast cancer: Clinical results and biomarker analysis of GeparNuevo study. Ann. Oncol. 2019, 30, 1279–1288. [Google Scholar] [CrossRef]


{kind=link}
| Tumor Type | TRM Markers | No. of Samples/Patients | Reference |
|---|---|---|---|
| Ovarian Cancer | CD103 | 489 | [27] |
| CD103 | 497 | [34] | |
| CD103, CD3, TCRαβ, CD8αβ, CD4 | 186 | [84] | |
| Cervical Cancer | CD103 | 460 | [11] |
| Melanoma | CD69, CD103, TNFRSF18, CD8 | 44 | [23] |
| CD8, CD103, CD69 | 18 | [85] | |
| Lung Cancers | CD8, CD103, CD3 | 101 | [9] |
| CD8, CD103 | 77 | [10] | |
| CD8, CD103 | 510 | [86] | |
| Pancreatic Cancer | CD8, CD103 | 136 | [87] |
| Breast Cancer | CD8, CD103 | 424 | [8] |
| CD8, T cell gene signatures | 989 | [88] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Craig, D.J.; Creeden, J.F.; Einloth, K.R.; Gillman, C.E.; Stanbery, L.; Hamouda, D.; Edelman, G.; Dworkin, L.; Nemunaitis, J.J. Resident Memory T Cells and Their Effect on Cancer. Vaccines 2020, 8, 562. https://doi.org/10.3390/vaccines8040562
Craig DJ, Creeden JF, Einloth KR, Gillman CE, Stanbery L, Hamouda D, Edelman G, Dworkin L, Nemunaitis JJ. Resident Memory T Cells and Their Effect on Cancer. Vaccines. 2020; 8(4):562. https://doi.org/10.3390/vaccines8040562
Chicago/Turabian StyleCraig, Daniel J., Justin F. Creeden, Katelyn R. Einloth, Cassidy E. Gillman, Laura Stanbery, Danae Hamouda, Gerald Edelman, Lance Dworkin, and John J. Nemunaitis. 2020. "Resident Memory T Cells and Their Effect on Cancer" Vaccines 8, no. 4: 562. https://doi.org/10.3390/vaccines8040562
APA StyleCraig, D. J., Creeden, J. F., Einloth, K. R., Gillman, C. E., Stanbery, L., Hamouda, D., Edelman, G., Dworkin, L., & Nemunaitis, J. J. (2020). Resident Memory T Cells and Their Effect on Cancer. Vaccines, 8(4), 562. https://doi.org/10.3390/vaccines8040562